The Role of Altered Nucleotide Excision Repair and UVB-Induced DNA Damage in Melanomagenesis

Abstract
:1. Introduction
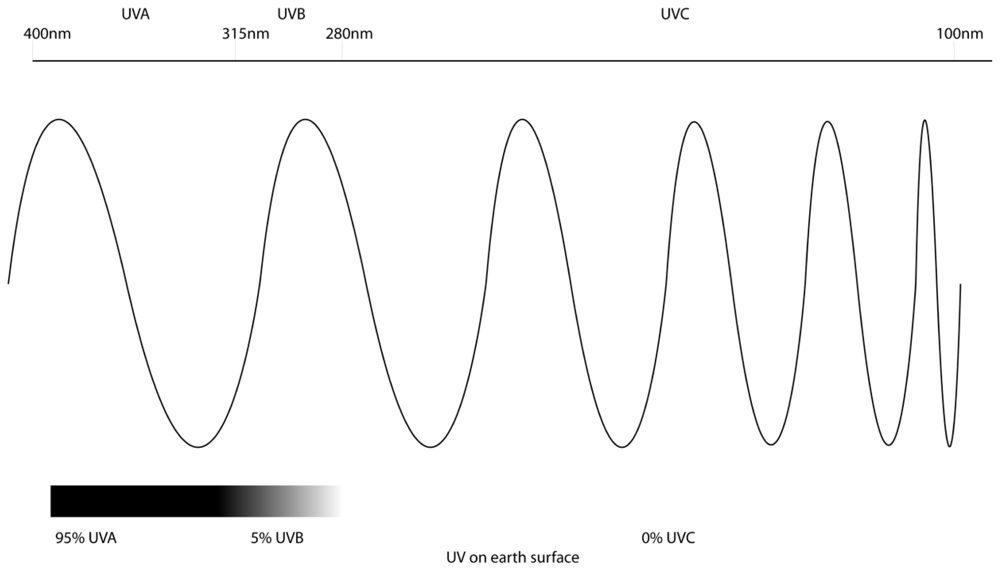
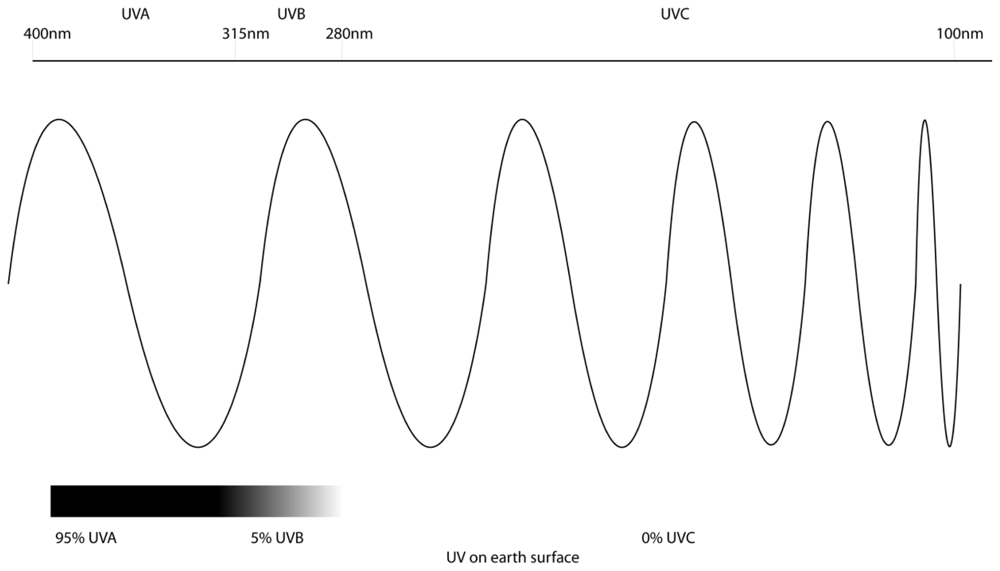
2. Ultraviolet Radiation and Melanoma
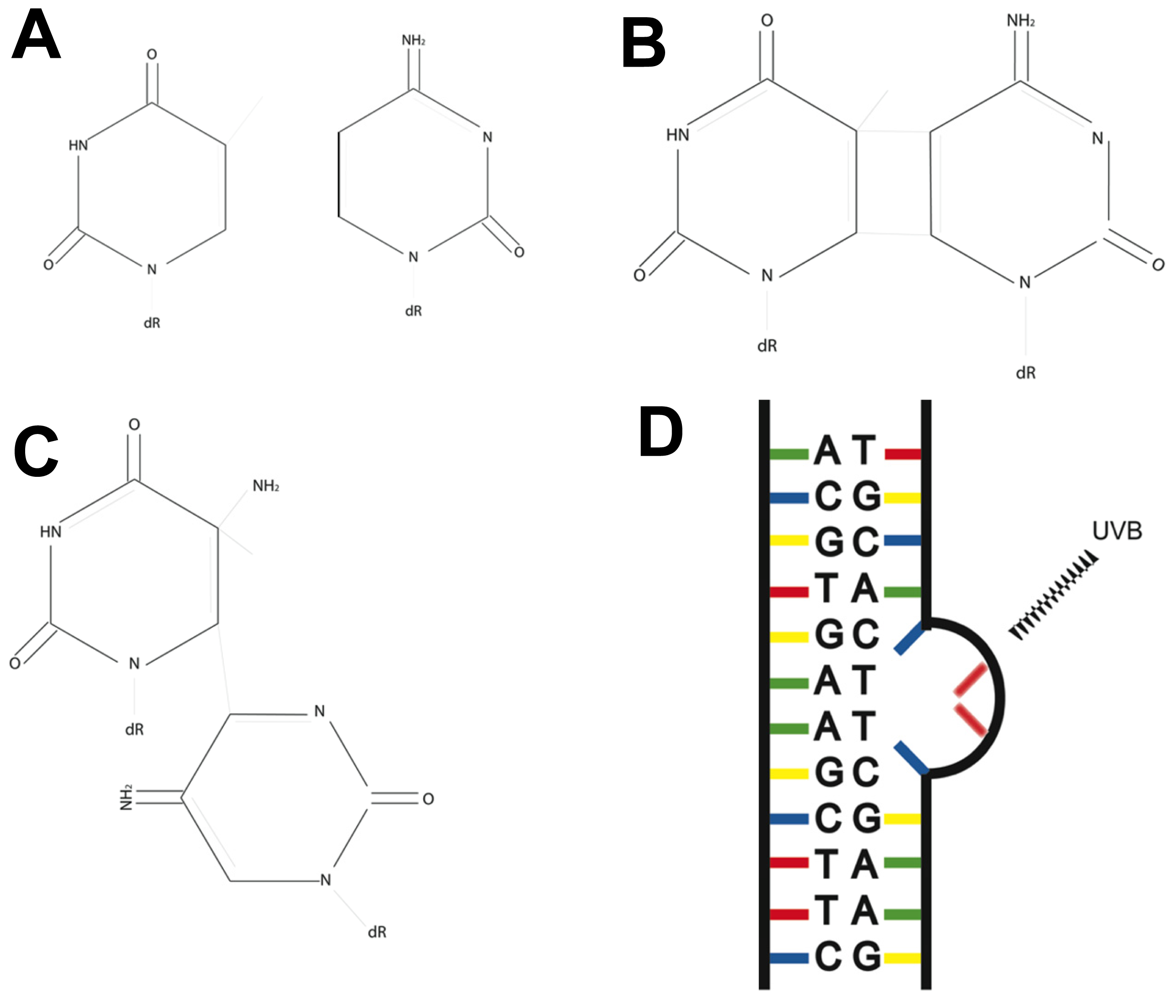
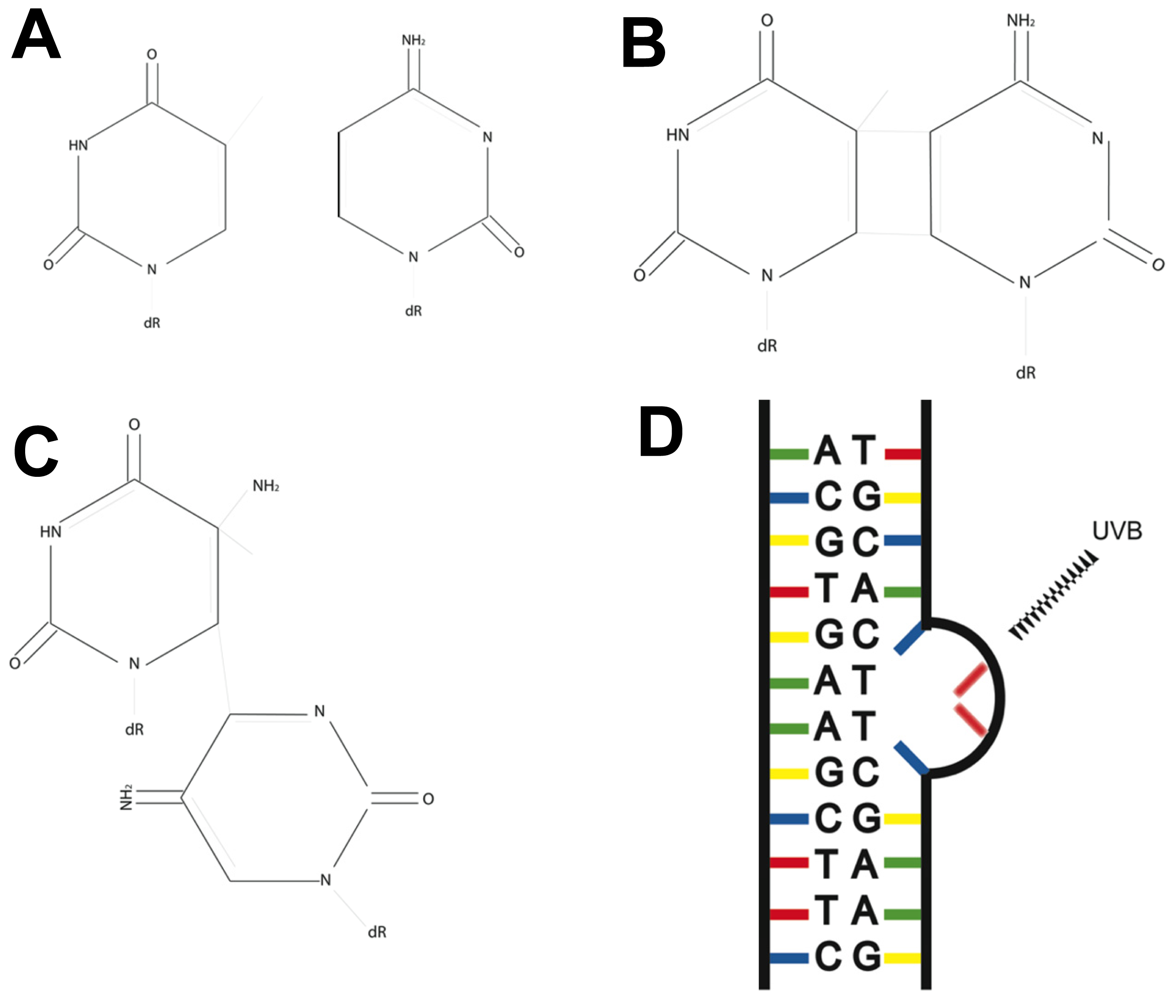
3. Fingerprint Mutations Suggest a Role for UVB in Melanoma
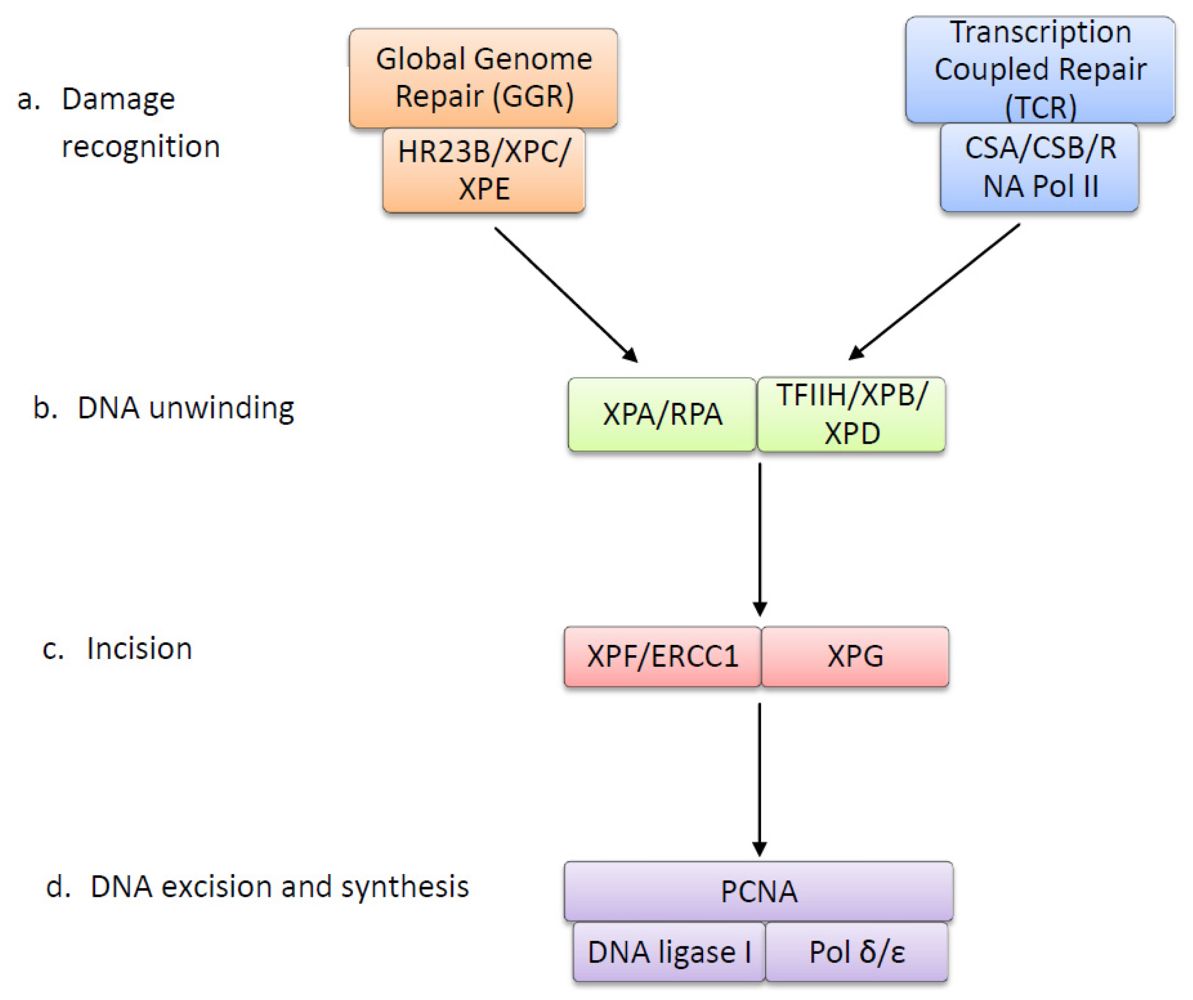
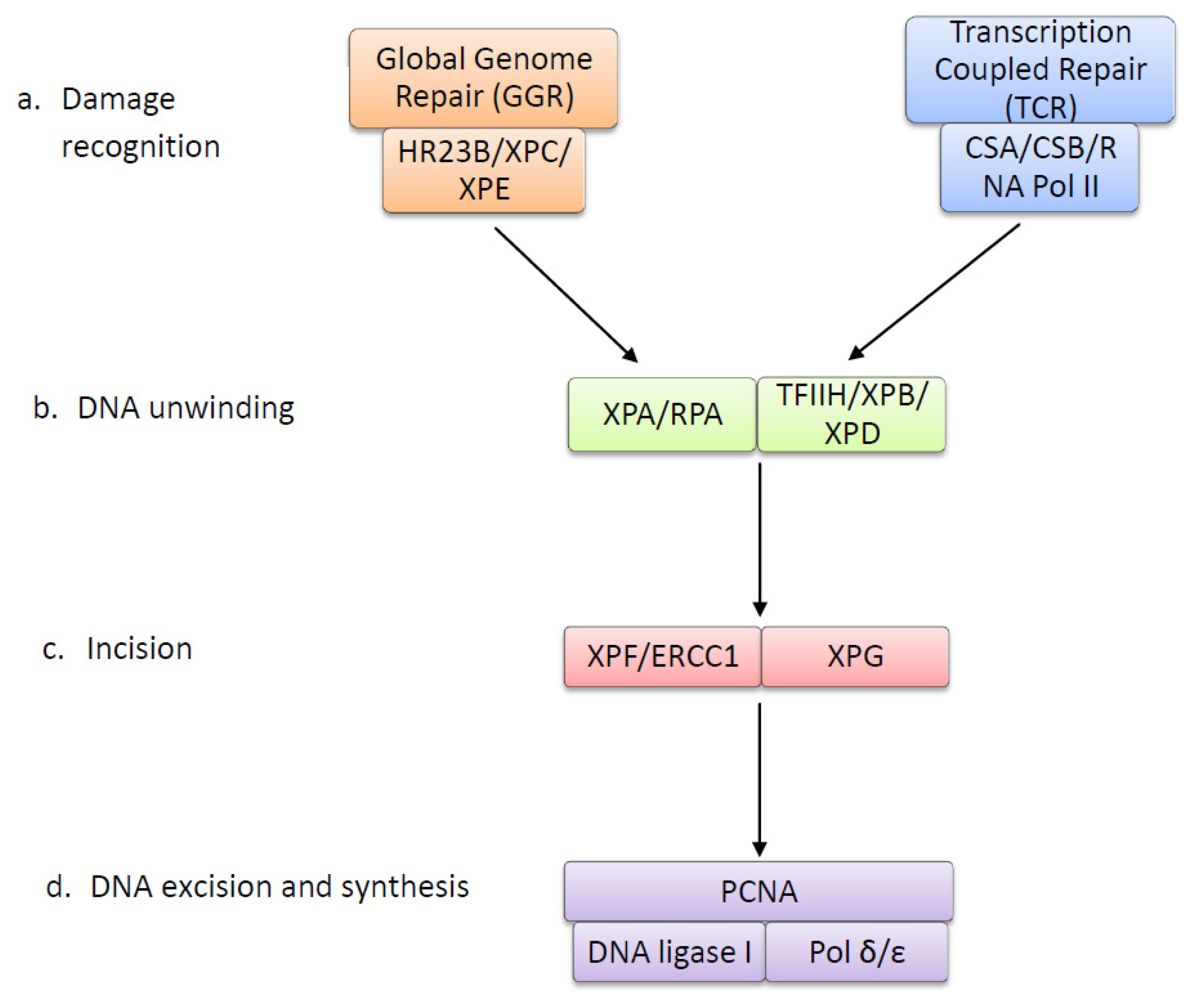
4. The Nucleotide Excision Repair Pathway Removes UV-Induced Lesions
5. Xeroderma Pigmentosum: A Link between Nucleotide Excision Repair and Melanoma
6. Research into NER and Melanoma
7. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar]
- Marrot, L.; Meunier, J.-R. Skin DNA photodamage and its biological consequences. J. Am. Acad. Dermatol 2008, 58, S139–S148. [Google Scholar]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br. J. Dermatol 2005, 153, 733–739. [Google Scholar]
- Garland, C.F.; Garland, F.C.; Gorham, E.D. Epidemiologic evidence for different roles of ultraviolet A and B radiation in melanoma mortality rates. Ann. Epidemiol 2003, 13, 395–404. [Google Scholar]
- Tran, T.T.; Schulman, J.; Fisher, D.E. UV and pigmentation: Molecular mechanisms and social controversies. Pigment Cell Melanoma Res 2008, 21, 509–516. [Google Scholar]
- Tucker, M.A.; Goldstein, A.M. Melanoma etiology: Where are we? Oncogene 2003, 22, 3042–3052. [Google Scholar]
- Besaratinia, A.; Bates, S.E.; Synold, T.W.; Pfeifer, G.P. Similar mutagenicity of photoactivated porphyrins and ultraviolet A radiation in mouse embryonic fibroblasts: Involvement of oxidative DNA lesions in mutagenesis. Biochemistry 2004, 43, 15557–15566. [Google Scholar]
- Brozyna, A.; Zbytek, B.; Granese, J.; Carlson, A.; Ross, J.; Slominski, A. Mechanism of UV-related carcinogenesis and its contribution to nevi/melanoma. Expert Rev. Dermatol 2007, 2, 451–469. [Google Scholar]
- Slominski, A.; Pawelek, J. Animals under the sun: Effects of ultraviolet radiation on mammalian skin. Clin. Dermatol 1998, 16, 503–515. [Google Scholar]
- Bennett, D.C. Ultraviolet wavebands and melanoma initiation. Pigment Cell Melanoma Res 2008, 21, 520–524. [Google Scholar]
- Hussein, M.R. Ultraviolet radiation and skin cancer: Molecular mechanisms. J. Cutan. Pathol 2005, 32, 191–205. [Google Scholar]
- Pfeifer, G.P. Formation and processing of UV photoproducts: Effects of DNA sequence and chromatin environment. Photochem. Photobiol 1997, 65, 270–283. [Google Scholar]
- Courdavault, S.; Baudouin, C.; Charveron, M.; Canguilhem, B.; Favier, A.; Cadet, J.; Douki, T. Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations. DNA Repair 2005, 4, 836–844. [Google Scholar]
- Kim, J.-K.; Patel, D.; Choi, B.-S. Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: Implication in mutagenesis and repair activity. Photochem. Photobiol 1995, 62, 44–50. [Google Scholar]
- You, Y.H.; Lee, D.H.; Yoon, J.H.; Nakajima, S.; Yasui, A.; Pfeifer, G.P. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J. Biol. Chem 2001, 276, 44688–44694. [Google Scholar]
- Brash, D.E. Roles of the transcription factor p53 in keratinocyte carcinomas. Br. J. Dermatol 2006, 154, S8–S10. [Google Scholar]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science 1999, 283, 1001–1004. [Google Scholar]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar]
- Drouin, R.; Therrien, J.-P. UVB-induced cyclobutane pyrimidine dimer frequency correlates with skin cancer mutational hotspots in p53. Photochem. Photobiol 1997, 66, 719–726. [Google Scholar]
- Tommasi, S.; Denissenko, M.F.; Pfeifer, G.P. Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res 1997, 57, 4727–4730. [Google Scholar]
- Tu, Y.; Dammann, R.; Pfeifer, G.P. Sequence and time-dependent deamination of cytosine bases in UVB-induced cyclobutane pyrimidine dimers in vivo. J. Mol. Biol 1998, 284, 297–311. [Google Scholar]
- Garibyan, L.; Fisher, D.E. How sunlight causes melanoma. Curr. Oncol. Rep 2010, 12, 319–326. [Google Scholar]
- World Health Organisation (WHO), The Global Burden of Disease: 2004 Update; WHO: Geneva, Switzerland, 2008.
- AIHW, Cancer in Australia: An overview, 2008; Cancer series no. 46; Cat. no. CAN 42; AIHW: Canberra, Australia, 2008.
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar]
- Fayolle, C.; Pourchet, J.; de Fromentel, C.C.; Puisieux, A.; Dore, J.F.; Voeltzel, T. Gadd45a activation protects melanoma cells from ultraviolet B-induced apoptosis. J. Investig. Dermatol 2008, 128, 196–202. [Google Scholar]
- Tracey, E.; Kerr, T.; Dobrovic, A.; Currow, D. Cancer in New South Wales: Incidence and Mortality Report 2008; Cancer Institute NSW: Sydney, Australia, 2008. [Google Scholar]
- Jhappan, C.; Noonan, F.P.; Merlino, G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 2003, 22, 3099–3112. [Google Scholar]
- De Gruijl, F.R. Skin cancer and solar UV radiation. Eur. J. Cancer 1999, 35, 2003–2009. [Google Scholar]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet 2012, 44, 1006–1014. [Google Scholar]
- Stark, M.S.; Woods, S.L.; Gartside, M.G.; Bonazzi, V.F.; Dutton-Regester, K.; Aoude, L.G.; Chow, D.; Sereduk, C.; Niemi, N.M.; Tang, N.; et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet 2012, 44, 165–169. [Google Scholar]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med 2010, 363, 809–819. [Google Scholar]
- Gailani, M.R.; Stahle-Backdahl, M.; Leffell, D.J.; Glynn, M.; Zaphiropoulos, P.G.; Pressman, C.; Unden, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet 1996, 14, 78–81. [Google Scholar]
- Boukamp, P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis 2005, 26, 1657–1667. [Google Scholar]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360. [Google Scholar]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Human Mutat 2003, 21, 217–228. [Google Scholar]
- Ziegler, A.; Jonason, A.; Leffell, D.; Simon, J.; Sharma, H.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar]
- Balint, E.; Vousden, K.H. Activation and activities of the p53 tumour suppressor protein. Br. J. Cancer 2001, 85, 1813–1823. [Google Scholar]
- Zerp, S.F.; van Elsas, A.; Poltenburg, L.T.C.; Schrier, P.I. p53 mutations in human cutaneous melanoma correlate with sun exposure, but are not always involved in melanomagenesis. Br. J. Cancer 1999, 79, 921. [Google Scholar]
- Piepkorn, M. Melanoma genetics: An update with focus on the CDKN2A(p16)/ARF tumor suppressors. J. Am. Acad. Dermatol. 2000, 42, 705–722, quiz 723–706. [Google Scholar]
- Recio, J.A.; Noonan, F.P.; Takayama, H.; Anver, M.R.; Duray, P.; Rush, W.L.; Lindner, G.; de Fabo, E.C.; DePinho, R.A.; Merlino, G.; et al. Ink4a/arf deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer Res 2002, 62, 6724–6730. [Google Scholar]
- Hocker, T.; Tsao, H. Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants. Human Mutat 2007, 28, 578–588. [Google Scholar]
- Ming, M.; Feng, L.; Shea, C.R.; Soltani, K.; Zhao, B.; Han, W.; Smart, R.C.; Trempus, C.S.; He, Y.-Y. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res 2011, 71, 5287–5295. [Google Scholar]
- Guldberg, P.; thor Straten, P.; Birck, A.; Ahrenkiel, V.; Kirkin, A.F.; Zeuthen, J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res 1997, 57, 3660–3663. [Google Scholar]
- Tsao, H.; Zhang, X.; Benoit, E.; Haluska, F.G. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 1998, 16, 3397–3402. [Google Scholar]
- Wang, Y.; Digiovanna, J.J.; Stern, J.B.; Hornyak, T.J.; Raffeld, M.; Khan, S.G.; Oh, K.S.; Hollander, M.C.; Dennis, P.A.; Kraemer, K.H.; et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc. Nat. Acad. Sci. USA 2009, 106, 6279–6284. [Google Scholar]
- Hakoshima, T.; Shimizu, T.; Maesaki, R. Structural basis of the Rho GTPase signaling. J. Biochem 2003, 134, 327–331. [Google Scholar]
- Jaffe, A.B.; Hall, A. RHO GTPASES: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol 2005, 21, 247–269. [Google Scholar]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci 2005, 118, 843–846. [Google Scholar]
- Batista, L.F.Z.; Kaina, B.; Meneghini, R.; Menck, C.F.M. How DNA lesions are turned into powerful killing structures: Insights from UV-induced apoptosis. Mutat. Res 2009, 681, 197–208. [Google Scholar]
- Friedberg, E.C. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 2001, 1, 22–33. [Google Scholar]
- Marini, F.; Nardo, T.; Giannattasio, M.; Minuzzo, M.; Stefanini, M.; Plevani, P.; Falconi, M.M. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc. Natl. Acad. Sci. USA 2006, 103, 17325–17330. [Google Scholar]
- Aboussekhra, A.; Al-Sharif, I.S. Homologous recombination is involved in transcription-coupled repair of UV damage in Saccharomyces cerevisiae. EMBO J 2005, 24, 1999–2010. [Google Scholar]
- Van Oosten, M.; Rebel, H.; Friedberg, E.C.; van Steeg, H.; van der Horst, G.T.J.; van Kranen, H.J.; Westerman, A.; van Zeeland, A.A.; Mullenders, L.H.F.; de Gruijl, F.R. Differential role of transcription-coupled repair in UVB-induced G2 arrest and apoptosis in mouse epidermis. Proc. Natl. Acad. Sci. USA 2000, 97, 11268–11273. [Google Scholar]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res 2008, 18, 73–84. [Google Scholar]
- Van Hoffen, A.; Venema, J.; Meschini, R.; van Zeeland, A.A.; Mullenders, L.H. Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J 1995, 14, 360–367. [Google Scholar]
- Sugasawa, K. UV-induced ubiquitylation of XPC complex, the UV-DDB-ubiquitin ligase complex and DNA repair. J. Mol. Histol 2006, 37, 189–202. [Google Scholar]
- Compe, E.; Egly, J.-M. TFIIH: When transcription met DNA repair. Nat. Rev. Mol. Cell Biol 2012, 13, 343–354. [Google Scholar]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010, 1–32. [Google Scholar]
- Sugasawa, K.; Ng, J.M.; Masutani, C.; Iwai, S.; van der Spek, P.J.; Eker, A.P.; Hanaoka, F.; Bootsma, D.; Hoeijmakers, J.H. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 1998, 2, 223–232. [Google Scholar]
- Fitch, M.E.; Nakajima, S.; Yasui, A.; Ford, J.M. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem 2003, 278, 46906–46910. [Google Scholar]
- Scrima, A.; Konickova, R.; Czyzewski, B.K.; Kawasaki, Y.; Jeffrey, P.D.; Groisman, R.; Nakatani, Y.; Iwai, S.; Pavletich, N.P.; Thomä, N.H. Structural Basis of UV DNA-Damage Recognition by the DDB1/DDB2 Complex. Cell 2008, 135, 1213–1223. [Google Scholar]
- Li, J.; Bhat, A.; Xiao, W. Regulation of nucleotide excision repair through ubiquitination. Acta Biochim. Biophys. Sin 2011, 43, 919–929. [Google Scholar]
- Xie, Z.; Liu, S.; Zhang, Y.; Wang, Z. Roles of Rad23 protein in yeast nucleotide excision repair. Nucleic Acids Res 2004, 32, 5981–5990. [Google Scholar]
- Ng, J.M.Y.; Vermeulen, W.; van der Horst, G.T.J.; Bergink, S.; Sugasawa, K.; Vrieling, H.; Hoeijmakers, J.H.J. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 2003, 17, 1630–1645. [Google Scholar]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci 2003, 116, 447–451. [Google Scholar]
- Bregman, D.B.; Halaban, R.; van Gool, A.J.; Henning, K.A.; Friedberg, E.C.; Warren, S.L. UV-induced ubiquitination of RNA polymerase II: A novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11586–11590. [Google Scholar]
- Ratner, J.N.; Balasubramanian, B.; Corden, J.; Warren, S.L.; Bregman, D.B. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA Polymerase II. J. Biol. Chem 1998, 273, 5184–5189. [Google Scholar]
- Bradsher, J.; Coin, F.; Egly, J.M. Distinct roles for the helicases of TFIIH in transcript initiation and promoter escape. J. Biol. Chem 2000, 275, 2532–2538. [Google Scholar]
- Oksenych, V.; Coin, F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle 2010, 9, 90–96. [Google Scholar]
- Volker, M.; Mone, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.; van Driel, R.; van Zeeland, A.A.; Mullenders, L.H. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar]
- He, Z.; Henricksen, L.A.; Wold, M.S.; Ingles, C.J. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 1995, 374, 566–569. [Google Scholar]
- Matsuda, T.; Saijo, M.; Kuraoka, I.; Kobayashi, T.; Nakatsu, Y.; Nagai, A.; Enjoji, T.; Masutani, C.; Sugasawa, K.; Hanaoka, F.; et al. DNA repair protein XPA binds replication protein A (RPA). J. Biol. Chem 1995, 270, 4152–4157. [Google Scholar]
- Clarkson, S.G. The XPG story. Biochimie 2003, 85, 1113–1121. [Google Scholar]
- Gaillard, P.H.; Wood, R.D. Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res 2001, 29, 872–879. [Google Scholar]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.-F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 1996, 86, 811–822. [Google Scholar]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Scharer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J 2007, 26, 4768–4776. [Google Scholar]
- Popanda, O.; Thielmann, H.W. The function of DNA polymerases in DNA repair synthesis of ultraviolet-irradiated human fibroblasts. Biochim. Biophys. Acta 1992, 1129, 155–160. [Google Scholar]
- Shivji, K.K.; Kenny, M.K.; Wood, R.D. Proliferating cell nuclear antigen is required for DNA excision repair. Cell 1992, 69, 367–374. [Google Scholar]
- Nocentini, S. Rejoining kinetics of DNA single- and double-strand breaks in normal and DNA ligase-deficient cells after exposure to ultraviolet C and gamma radiation: An evaluation of ligating activities involved in different DNA repair processes. Radiat. Res 1999, 151, 423–432. [Google Scholar]
- Smith, M.L.; Ford, J.M.; Hollander, M.C.; Bortnick, R.A.; Amundson, S.A.; Seo, Y.R.; Deng, C.-X.; Hanawalt, P.C.; Fornace, A.J. p53-Mediated DNA repair responses to UV radiation: Studies of mouse cells lacking p53, p21 and/orgadd45 genes. Mol. Cell. Biol 2000, 20, 3705–3714. [Google Scholar]
- Ford, J.M.; Hanawalt, P.C. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J. Biol. Chem 1997, 272, 28073–28080. [Google Scholar]
- Mathonnet, G.; Leger, C.; Desnoyers, J.; Drouin, R.; Therrien, J.-P.; Drobetsky, E.A. UV wavelength-dependent regulation of transcription-coupled nucleotide excision repair in p53-deficient human cells. Proc. Natl. Acad. Sci 2003, 100, 7219–7224. [Google Scholar]
- Adimoolam, S.; Ford, J.M. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. USA 2002, 99, 12985–12990. [Google Scholar]
- Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 1999, 96, 424–428. [Google Scholar]
- Tan, T.; Chu, G. p53 binds and activates the xeroderma pigmentosum DDB2 gene in humans, but not mice. Mol. Cell. Biol 2002, 22, 3247–3254. [Google Scholar]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci 2009, 66, 994–1009. [Google Scholar]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet 1992, 42, 68–84. [Google Scholar]
- Frontini, M.; Proietti-de-Santis, L. Cockayne syndrome B protein (CSB): Linking p53, HIF-1 and p300 to robustness, lifespan, cancer and cell fate decisions. Cell Cycle 2009, 8, 693–696. [Google Scholar]
- Hoeijmakers, J.H.J. DNA damage, aging and cancer. N. Engl. J. Med 2009, 361, 1475–1485. [Google Scholar]
- Chavanne, F.; Broughton, B.C.; Pietra, D.; Nardo, T.; Browitt, A.; Lehmann, A.R.; Stefanini, M. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein and transcript levels. Cancer Res 2000, 60, 1974–1982. [Google Scholar]
- Lehmann, A.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis 2011, 6, 70. [Google Scholar]
- Stary, A.; Sarasin, A. The genetics of the hereditary xeroderma pigmentosum syndrome. Biochimie 2002, 84, 49–60. [Google Scholar]
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.-S.; Imoto, K.; Inui, H.; Moriwaki, S.-I.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet 2011, 48, 168–176. [Google Scholar]
- Kraemer, K.H.; Lee, M.M.; Scotto, J. Xeroderma pigmentosum: Cutaneous, ocular and neurologic abnormalities in 830 published cases. Arch. Dermatol 1987, 123, 241–250. [Google Scholar]
- Cleaver, J.E.; Thompson, L.H.; Richardson, A.S.; States, J.C. A summary of mutations in the UV-sensitive disorders: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Human Mutation 1999, 14, 9–22. [Google Scholar]
- Queille, S.; Drougard, C.; Sarasin, A.; Daya-Grosjean, L. Effects of XPD mutations on ultraviolet-induced apoptosis in relation to skin cancer-proneness in repair-deficient syndromes. J. Invest. Dermatol 2001, 117, 1162–1170. [Google Scholar]
- Van der Wees, C.; Jansen, J.; Vrieling, H.; van der Laarse, A.; van Zeeland, A.; Mullenders, L.H. Nucleotide excision repair in differentiated cells. Mutat. Res 2007, 614, 16–23. [Google Scholar]
- Hanawalt, P.C.; Ford, J.M.; Lloyd, D.R. Functional characterization of global genomic DNA repair and its implications for cancer. Mutat. Res 2003, 544, 107–114. [Google Scholar]
- Daya-Grosjean, L.; Robert, C.; Drougard, C.; Suarez, H.; Sarasin, A. High mutation frequency in ras genes of skin tumors isolated from DNA repair deficient xeroderma pigmentosum patients. Cancer Res 1993, 53, 1625–1629. [Google Scholar]
- Dumaz, N.; Drougard, C.; Sarasin, A.; Daya-Grosjean, L. Specific UV-induced mutation spectrum in the p53 gene of skin tumors from DNA-repair-deficient xeroderma pigmentosum patients. Proc. Natl. Acad. Sci 1993, 90, 10529–10533. [Google Scholar]
- Soufir, N.; Daya-Grosjean, L.; de La Salmoniere, P.; Moles, J.-P.; Dubertret, L.; Sarasin, A.; Basset-Seguin, N. Association between INK4a-ARF and p53 mutations in skin carcinomas of xeroderma pigmentosum patients. J. Nat. Cancer Inst 2000, 92, 1841–1847. [Google Scholar]
- Law, M.H.; Macgregor, S.; Hayward, N.K. Melanoma genetics: Recent findings take us beyond well-traveled pathways. J. Invest. Dermatol 2012, 132, 1763–1774. [Google Scholar]
- Blankenburg, S.; Konig, I.R.; Moessner, R.; Laspe, P.; Thoms, K.M.; Krueger, U.; Khan, S.G.; Westphal, G.; Berking, C.; Volkenandt, M.; et al. Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: A case-control study. Carcinogenesis 2005, 26, 1085–1090. [Google Scholar]
- Blankenburg, S.; Konig, I.R.; Moessner, R.; Laspe, P.; Thoms, K.M.; Krueger, U.; Khan, S.G.; Westphal, G.; Volkenandt, M.; Neumann, C.; et al. No association between three xeroderma pigmentosum group C and one group G gene polymorphisms and risk of cutaneous melanoma. Eur. J. Human Genet 2005, 13, 253–255. [Google Scholar]
- Povey, J.E.; Darakhshan, F.; Robertson, K.; Bisset, Y.; Mekky, M.; Rees, J.; Doherty, V.; Kavanagh, G.; Anderson, N.; Campbell, H.; et al. DNA repair gene polymorphisms and genetic predisposition to cutaneous melanoma. Carcinogenesis 2007, 28, 1087–1093. [Google Scholar]
- Fernandez, A.A.; Garcia, R.; Paniker, L.; Trono, D.; Mitchell, D.L. An experimental population study of nucleotide excision repair as a risk factor for UVB-induced melanoma. Photochem. Photobiol 2011, 87, 335–341. [Google Scholar]
- Patton, E.E.; Mitchell, D.L.; Nairn, R.S. Genetic and environmental melanoma models in fish. Pigment Cell Melanoma Res 2010, 23, 314–337. [Google Scholar]
- Gaddameedhi, S.; Kemp, M.G.; Reardon, J.T.; Shields, J.M.; Smith-Roe, S.L.; Kaufmann, W.K.; Sancar, A. Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res 2010, 70, 4922–4930. [Google Scholar]
- Wang, H.-T.; Choi, B.; Tang, M.-s. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc. Natl. Acad. Sci. USA 2010, 107, 12180–12185. [Google Scholar]
- Bradbury, P.A.; Middleton, M.R. DNA repair pathways in drug resistance in melanoma. Anti-Cancer Drugs 2004, 15, 421–426. [Google Scholar]
- Bowden, N.A.; Ashton, K.A.; Avery-Kiejda, K.A.; Zhang, X.D.; Hersey, P.; Scott, R.J. Nucleotide excision repair gene expression after Cisplatin treatment in melanoma. Cancer Res 2010, 70, 7918–7926. [Google Scholar]
- Li, W.; Melton, D.W. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene 2012, 31, 2412–2422. [Google Scholar]
- Song, L.; Ritchie, A.-M.; McNeil, E.M.; Li, W.; Melton, D.W. Identification of DNA repair gene Ercc1 as a novel target in melanoma. Pigment Cell Melanoma Res 2011, 24, 966–971. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Protein | Function |
|---|---|
| XPC | DNA-binding protein that recognizes UV lesions in global genome repair and recruits subsequent repair proteins. Can easily detect 6-4 photoproducts (6-4 PPs), but requires XPE to recognize and bind to CPDs [63,64]. |
| XPE | Composed of subunits DDB1 and DDB2. Detects and binds to both CPDs and 6-4 PPs via DDB2 subunit. Forms ubiquitin ligase complex, which polyubiquitinates XPC to increase affinity to UV damage, allowing recognition of CPDs [65,66]. |
| HR23B | Forms a complex with XPC and prevents it from being degraded after polyubiquitination [67,68]. |
| RNA Polymerase II | Stalled RNA polymerase II at the site of a UV lesion is the damage recognition step in transcription couples repair [69]. |
| CSA (ERCC8) | Together with CSB, displaces stalled RNA polymerase and acts to recruit repair proteins [70]. |
| CSB (ERCC6) | Together with CSA, displaces stalled RNA polymerase and acts to recruit repair proteins [70]. May ubiquitinate RNA polymerase II to enhance this process [71]. |
| TFIIH Complex | Ten subunit protein ring complex, including XPB and XPD, that unwinds DNA around UV lesion to form denaturation bubble [72]. |
| XPB (ERCC3) | DNA helicase subunit of TFIIH that unwinds the DNA damage site in the 3′ to 5′ direction [73]. |
| XPD (ERCC2) | DNA helicase subunit of TFIIH that unwinds the DNA damage site in the 5′ to 3′ direction [73]. |
| XPA | DNA damage verification by binding to the DNA damage that is marked by XPE and XPC and unwound by TFIIH. Allows for binding of XPF-ERCC1 complex [74]. |
| RPA | Single-stranded DNA binding protein that binds to the undamaged strand opposite UV lesion and allows for binding of XPG [75,76]. |
| XPG (ERCC5) | Endonuclease that makes the first incision 3′ to UV lesion [77]. |
| XPF (ERCC4) | Forms an endonuclease complex with ERCC1 that makes the incision 5′ to UV lesion [78]. The XPF subunit of the XPF-ERCC1 complex contains the nuclease activity [79]. |
| ERCC1 | Forms an endonuclease complex with XPF that makes the incision 5′ to UV lesion [78]. ERCC1 subunit is required for binding to DNA [80]. |
| DNA Polymerase δ/ɛ | Synthesises a new strand of DNA to replace the excised DNA containing the UV lesion [81]. |
| PCNA | Required for DNA synthesis by acting with DNA polymerase δ/ɛ to form short repair patches [82]. |
| DNA Ligase | Seals the nicks between the newly synthesised DNA strands [83]. |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Budden, T.; Bowden, N.A. The Role of Altered Nucleotide Excision Repair and UVB-Induced DNA Damage in Melanomagenesis. Int. J. Mol. Sci. 2013, 14, 1132-1151. https://doi.org/10.3390/ijms14011132
Budden T, Bowden NA. The Role of Altered Nucleotide Excision Repair and UVB-Induced DNA Damage in Melanomagenesis. International Journal of Molecular Sciences. 2013; 14(1):1132-1151. https://doi.org/10.3390/ijms14011132
Chicago/Turabian StyleBudden, Timothy, and Nikola A. Bowden. 2013. "The Role of Altered Nucleotide Excision Repair and UVB-Induced DNA Damage in Melanomagenesis" International Journal of Molecular Sciences 14, no. 1: 1132-1151. https://doi.org/10.3390/ijms14011132