Identification of Recurrence Related microRNAs in Hepatocellular Carcinoma after Surgical Resection

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Patients and Samples
2.2. Identification of Recurrence-Related miRNAs in HCC Samples
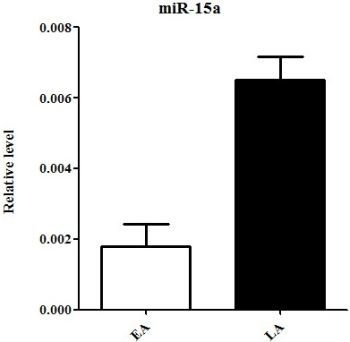
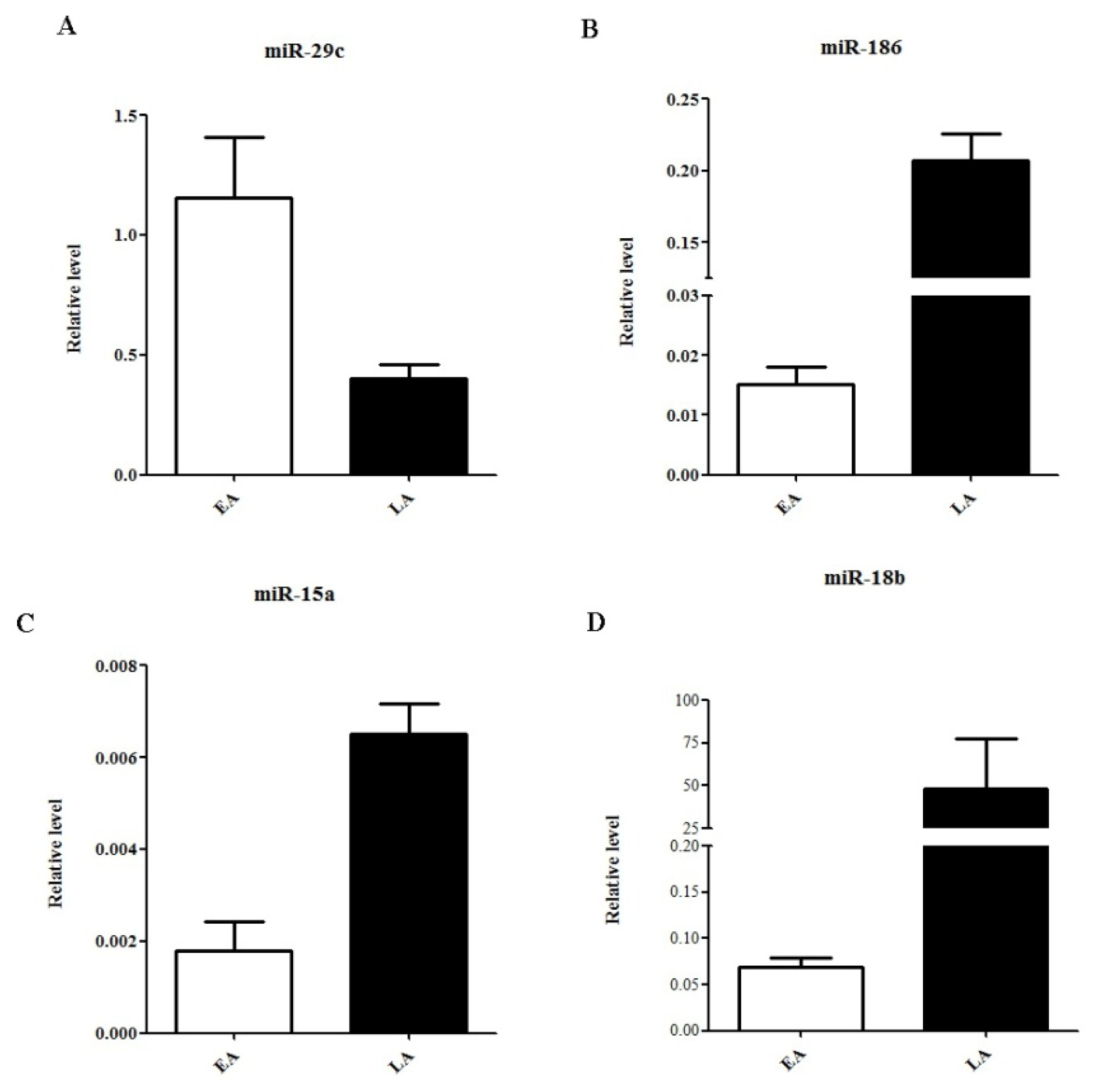
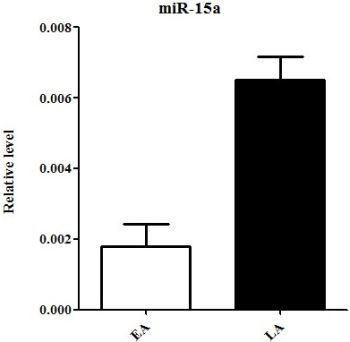
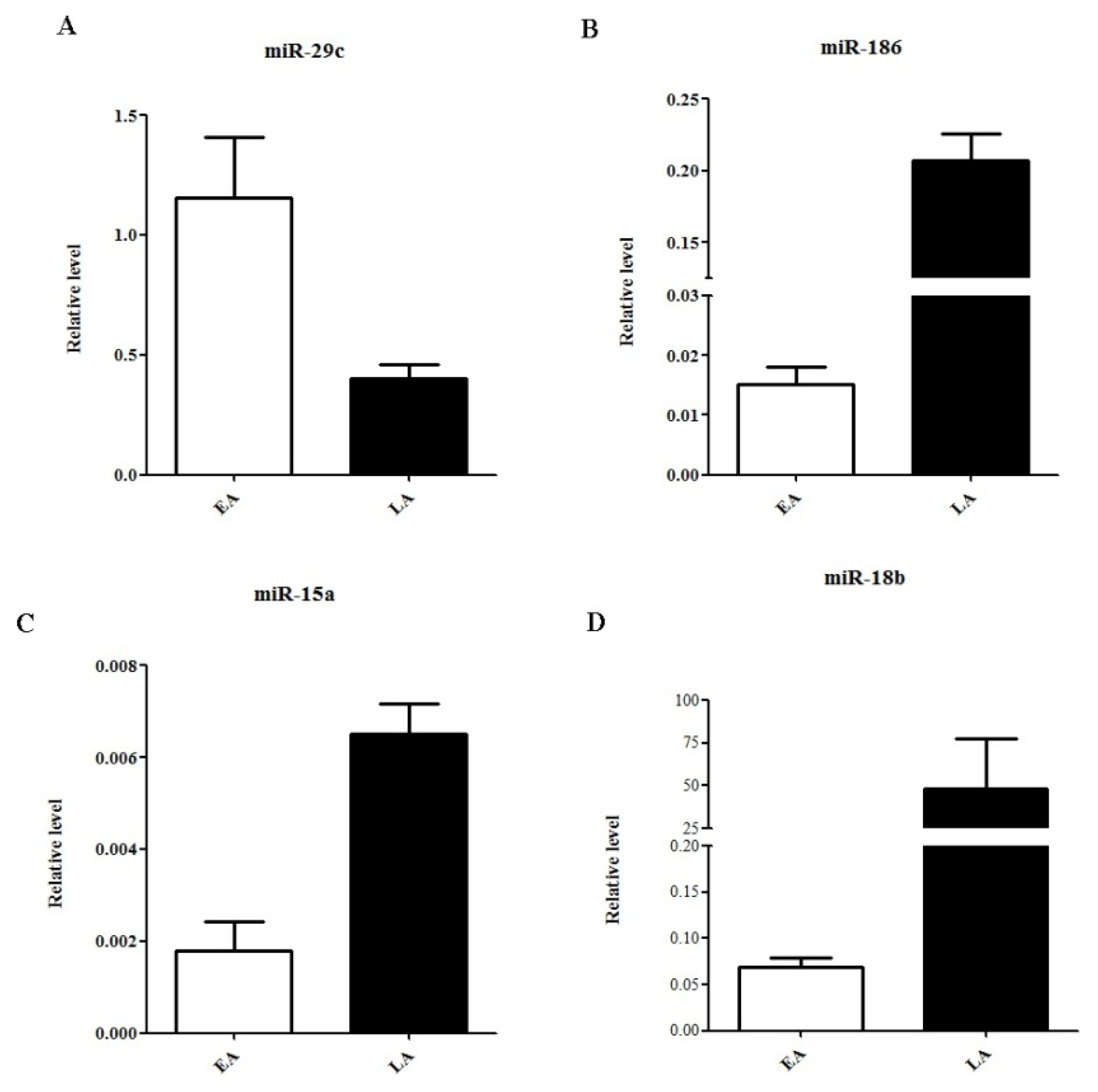
2.3. Quantitative Real-Time PCR Validation of miRNA Expression
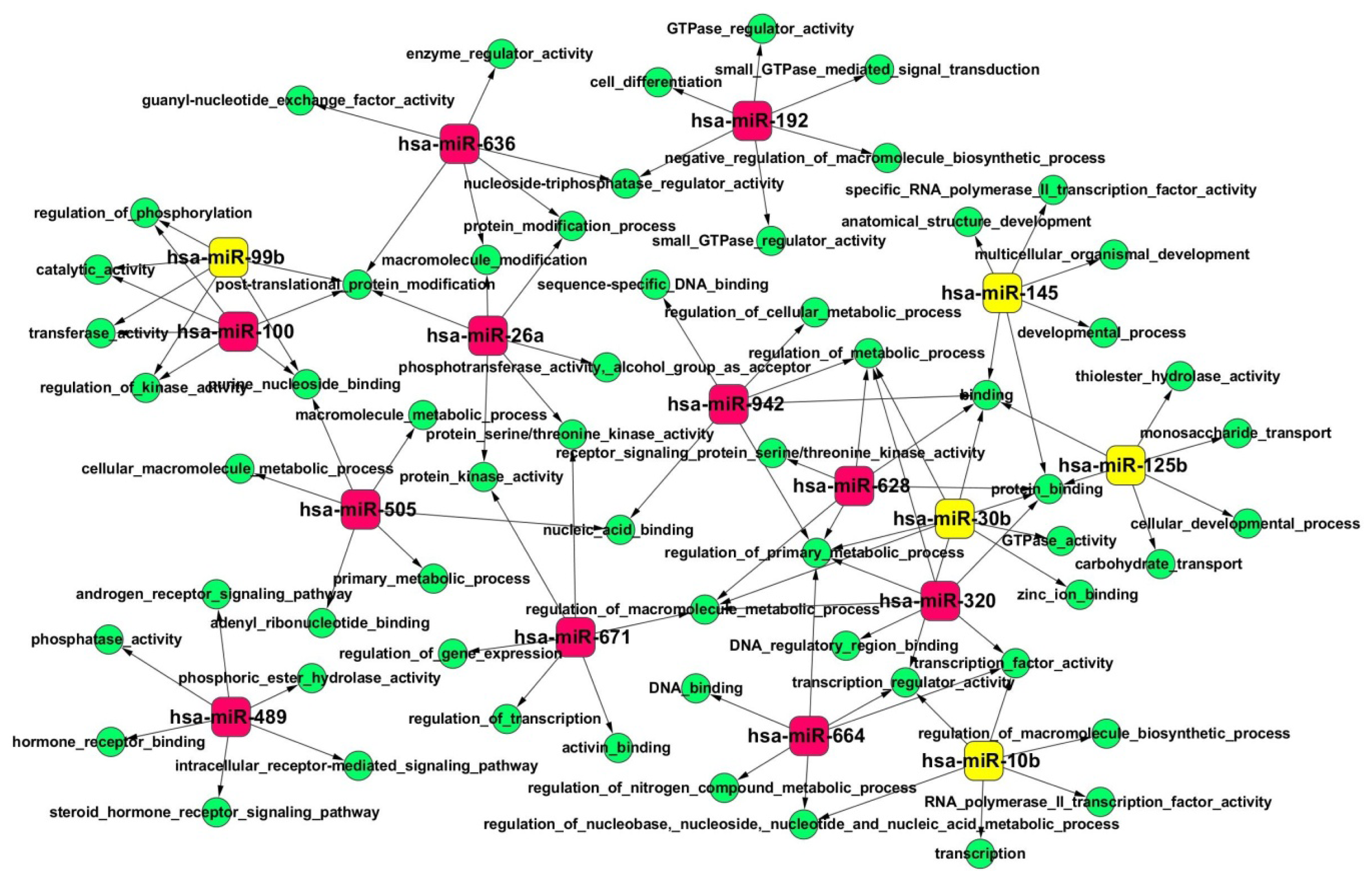
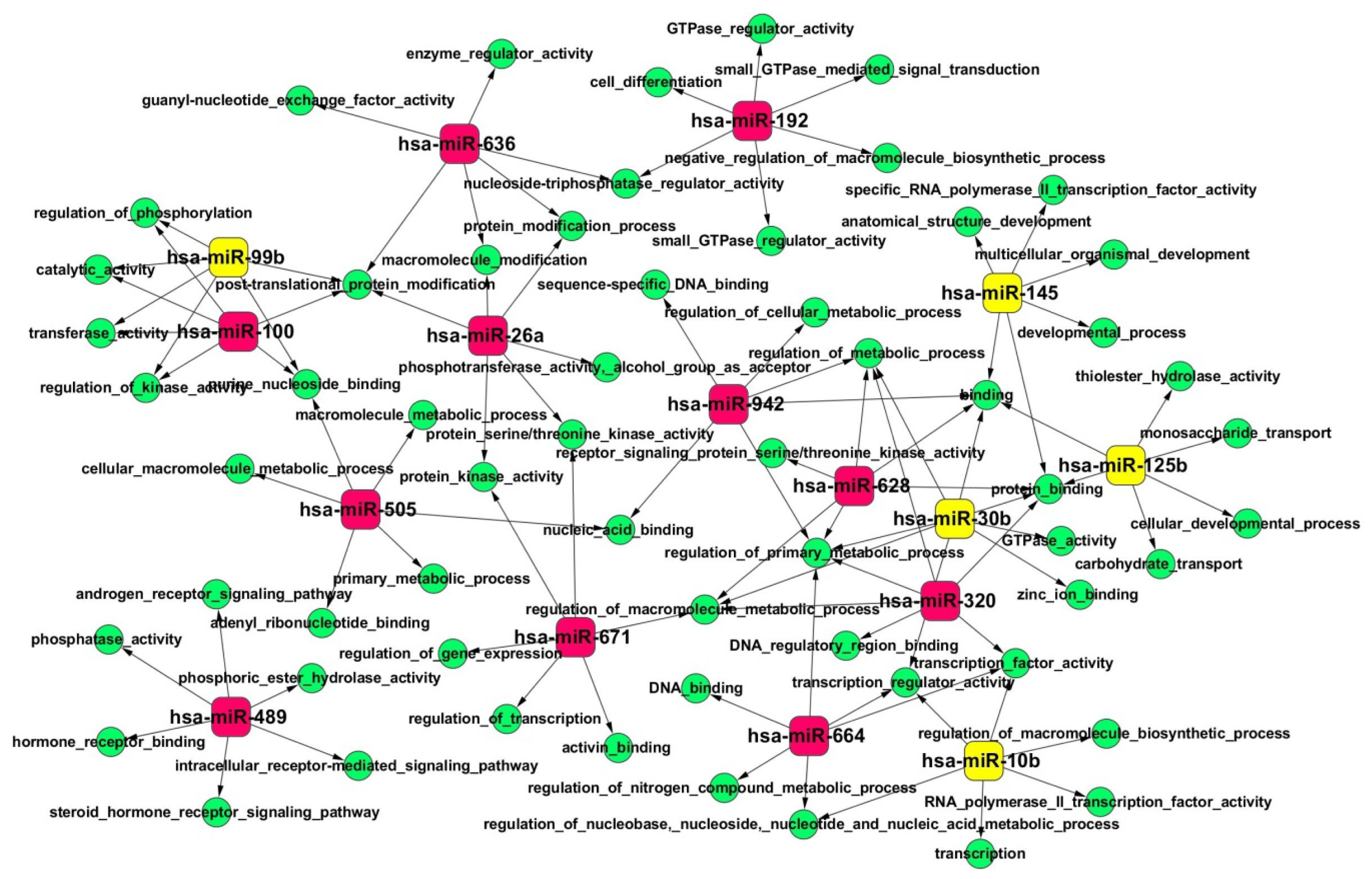
2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses of Predicted miRNA Targets
2.5. Discussion
3. Experimental Section
3.1. Sample Preparation and RNA Extraction
3.2. MicroRNA Expression Profiling
3.3. Statistical Analyses
3.4. Reverse Transcription and Quantitative RT-PCR Validation
3.5. miRNA Target Prediction and Functional Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar]
- Gomaa, A.I.; Khan, S.A.; Toledano, M.B.; Waked, I.; Taylor-Robinson, S.D. Hepatocellular carcinoma: Epidemiology, risk factors and pathogenesis. World J. Gastroenterol 2008, 14, 4300–4308. [Google Scholar]
- Blum, H.E. Hepatocellular carcinoma: Therapy and prevention. World J. Gastroenterol 2005, 11, 7391–7400. [Google Scholar]
- Figueras, J.; Jaurrieta, E.; Valls, C.; Ramos, E.; Serrano, T.; Rafecas, A.; Fabregat, J.; Torras, J. Resection or transplantation for hepatocellular carcinoma in cirrhotic patients: Outcomes based on indicated treatment strategy. J. Am. Coll. Surg 2000, 190, 580–587. [Google Scholar]
- Sieghart, W.; Wang, X.; Schmid, K.; Pinter, M.; Konig, F.; Bodingbauer, M.; Wrba, F.; Rasoul-Rockenschaub, S.; Peck-Radosavljevic, M. Osteopontin expression predicts overall survival after liver transplantation for hepatocellular carcinoma in patients beyond the Milan criteria. J. Hepatol 2011, 54, 89–97. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet 2004, 5, 522–531. [Google Scholar]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004, 23, 4051–4060. [Google Scholar]
- Zeng, Y.; Yi, R.; Cullen, B.R. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 9779–9784. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011, 39, D152–D157. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Zhao, X.; Yang, Z.; Li, G.; Li, D.; Zhao, Y.; Wu, Y.; Robson, S.; He, L.; Xu, Y.; Miao, R.; Zhao, H. The role and clinical implications of microRNAs in hepatocellular carcinoma. Sci. China Life Sci 2012, 55, 906–919. [Google Scholar]
- Kong, G.; Zhang, J.; Zhang, S.; Shan, C.; Ye, L.; Zhang, X. Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTEN in cell culture model. PLoS One 2011, 6, e19518. [Google Scholar]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar]
- Budhu, A.; Jia, H.L.; Forgues, M.; Liu, C.G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.H.; Qin, L.X.; Croce, C.M.; et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar]
- Si, M.L.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y.Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799–2803. [Google Scholar]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet 2007, 39, 1278–1284. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000, 25, 25–29. [Google Scholar]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005, 33, W741–W748. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008, 36, D480–D484. [Google Scholar]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar]
- Yang, C.; Wei, W. The miRNA expression profile of the uveal melanoma. Sci. China Life Sci 2011, 54, 351–358. [Google Scholar]
- Bandyopadhyay, S.; Mitra, R.; Maulik, U.; Zhang, M.Q. Development of the human cancer microRNA network. Silence 2010, 1, 6. [Google Scholar]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005, 65, 7065–7070. [Google Scholar]
- Li, X.; Zhang, Y.; Ding, J.; Wu, K.; Fan, D. Survival prediction of gastric cancer by a seven-microRNA signature. Gut 2010, 59, 579–585. [Google Scholar]
- Huang, Y.S.; Dai, Y.; Yu, X.F.; Bao, S.Y.; Yin, Y.B.; Tang, M.; Hu, C.X. Microarray analysis of microRNA expression in hepatocellular carcinoma and non-tumorous tissues without viral hepatitis. J. Gastroenterol. Hepatol 2008, 23, 87–94. [Google Scholar]
- Huang, X.H.; Wang, Q.; Chen, J.S.; Fu, X.H.; Chen, X.L.; Chen, L.Z.; Li, W.; Bi, J.; Zhang, L.J.; Fu, Q.; et al. Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: miR-338 is downregulated. Hepatol Res 2009, 39, 786–794. [Google Scholar]
- Xi, Y.; Formentini, A.; Chien, M.; Weir, D.B.; Russo, J.J.; Ju, J.; Kornmann, M. Prognostic values of microRNAs in colorectal cancer. Biomark Insights 2006, 2, 113–121. [Google Scholar]
- Sand, M.; Skrygan, M.; Georgas, D.; Sand, D.; Hahn, S.A.; Gambichler, T.; Altmeyer, P.; Bechara, F.G. Microarray analysis of microRNA expression in cutaneous squamous cell carcinoma. J. Dermatol. Sci 2012, 68, 119–126. [Google Scholar]
- Xie, Q.H.; He, X.X.; Chang, Y.; Sun, S.Z.; Jiang, X.; Li, P.Y.; Lin, J.S. MiR-192 inhibits nucleotide excision repair by targeting ERCC3 and ERCC4 in HepG2.2.15 cells. Biochem. Biophys. Res. Commun 2011, 410, 440–445. [Google Scholar]
- Sand, M.; Skrygan, M.; Sand, D.; Georgas, D.; Hahn, S.A.; Gambichler, T.; Altmeyer, P.; Bechara, F.G. Expression of microRNAs in basal cell carcinoma. Br. J. Dermatol 2012, 167, 847–855. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar]
- TargetScanHuman 6.2. Available online: http://www.targetscan.org/ accessed on 1 June 2012.
- PITA Catalog version 6. Available online: http://genie.weizmann.ac.il/pubs/mir07/mir07_data.html accessed on 31 August 2008.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Early recurrent cohort (n = 8) | Late recurrent cohort (n = 7) | p-Value |
|---|---|---|---|
| Age (year) | |||
| mean | 53.5 ± 7.3 | 65.0 ± 6.6 | 0.007 a |
| range | |||
| Gender (num) | |||
| Male | 8 | 6 | 0.268 b |
| Female | 0 | 1 | |
| Tumor size(cm) | |||
| ≤5 | 5 | 6 | 0.31 b |
| >5 | 3 | 1 | |
| Child-Pugh class | |||
| A | 8 | 3 | 0.013 b |
| B | 0 | 4 | |
| Cirrhosis | |||
| yes | 7 | 5 | 0.438 b |
| no | 1 | 2 | |
| Tumor grade (pathology) | |||
| High | 3 | 2 | 0.218 c |
| Medium | 5 | 3 | |
| Low | 0 | 2 | |
| HBV | |||
| (+) | 7 | 6 | 0.919 b |
| (−) | 1 | 1 | |
| Multinodular | |||
| yes | 5 | 4 | 0.833 b |
| no | 3 | 3 | |
| Micro-vascular invasion | |||
| yes | 4 | 7 | 0.029 b |
| no | 4 | 0 | |
| Serum AFP level (ng/mL) | 1336 (18.5, 71775) | 135 (2.35, 591.8) | 0.029 b |
| Mean recurrence-free survival | 2.27 ± 1.69 | 44.76 ± 21.91 | 0.000 a |
| miRNA ID | Chromosome coordinates | Score(d) | Fold Change | Status |
|---|---|---|---|---|
| hsa-miR-636 | chr17: 74732532-74732630 [−] | 0.773301691 | 28.96554407 | up |
| hsa-miR-671 | chr7: 150935507-150935624 [+] | 0.617872345 | 27.76690648 | up |
| hsa-miR-489 | chr7: 93113248-93113331 [−] | 0.8255668 | 17.98873011 | up |
| hsa-miR-26a | chr3: 38010895-38010971 [+] | 0.473722595 | 12.24271937 | up |
| hsa-miR-320 | chr8: 22102475-22102556 [−] | 0.67291134 | 9.39225375 | up |
| hsa-miR-628 | chr15: 55665138-55665232 [−] | 0.814261793 | 7.507041018 | up |
| hsa-miR-505 | chrX: 139006307-139006390 [−] | 0.64190506 | 7.495093696 | up |
| hsa-miR-100 | chr11: 122022937-122023016 [−] | 0.424532207 | 7.089941626 | up |
| hsa-miR-664 | chr1: 220373880-220373961 [−] | 0.950787576 | 6.755908146 | up |
| hsa-miR-942 | chr1: 117637265-117637350 [+] | 0.525349883 | 6.459339163 | up |
| hsa-miR-192 | chr11: 64658609-64658718 [−] | 0.31306747 | 6.351855536 | up |
| hsa-miR-339 | chr7: 1062569-1062662 [−] | 0.665577861 | 4.870026318 | up |
| hsa-miR-29c | chr1: 207975197-207975284 [−] | 0.458932548 | 4.169859654 | up |
| hsa-miR-191 | chr3: 49058051-49058142 [−] | 0.385561147 | 3.935817957 | up |
| hsa-miR-196b | chr7: 27209099-27209182 [−] | 0.44164008 | 3.877894721 | up |
| hsa-miR-190b | chr1: 154166141-154166219 [−] | 0.500700227 | 2.874169033 | up |
| hsa-miR-18b | chrX: 133304071-133304141 [−] | −0.057442249 | −2.12894251 | down |
| hsa-miR-106b | chr7: 99691616-99691697 [−] | −0.506823027 | −2.190671054 | down |
| hsa-miR-92a | chr13: 92003568-92003645 [+] | −0.528714339 | −2.351269626 | down |
| hsa-miR-186 | chr1: 71533314-71533399 [−] | −0.59646555 | −2.658452545 | down |
| hsa-miR-204 | chr9: 73424891-73425000 [−] | −0.48889237 | −2.960625198 | down |
| hsa-miR-1180 | chr17: 19247819-19247887 [−] | −0.699507876 | −3.031757322 | down |
| hsa-miR-193a-5p | chr17: 29887015-29887102 [+] | −0.811044599 | −3.042132652 | down |
| hsa-miR-23b | chr9: 97847490-97847586 [+] | −0.726168543 | −3.324757638 | down |
| hsa-miR-376c | chr14: 101506027-101506092 [+] | −0.78522436 | −3.845047161 | down |
| hsa-miR-15a | chr3: 160122376-160122473 [+] | −1.704080648 | −3.895068364 | down |
| hsa-miR-769 | chr19: 46522190-46522307 [+] | −0.860296951 | −4.785376714 | down |
| hsa-miR-99b | chr19: 52195865-52195934 [+] | −1.068532726 | −5.370680926 | down |
| hsa-miR-125b | chr11: 121970465-121970552 [−] | −0.571324674 | −6.378593023 | down |
| hsa-miR-10b | chr2: 177015031-177015140 [+] | −0.49959367 | −6.668372154 | down |
| hsa-miR-30b | chr8: 135812763-135812850 [−] | −0.474034967 | −11.45095534 | down |
| hsa-miR-145 | chr5: 148810209-148810296 [+] | −0.601999659 | −23.69139215 | down |
| KEGG pathway | Number of genes | Entrez gene ID | p-Value |
|---|---|---|---|
| Focal adhesion | 15 | 1284 331 5578 5156 7057 3725 595 7791 5880 1398 10000 896 5062 80310 2932 | 0.0000616 |
| Pathways in cancer | 13 | 2246 2261 5925 5728 8322 2475 8325 1871 3845 6932 1857 867 7423 | 0.0001 |
| MAPK signaling pathway | 9 | 2246 2261 6195 23162 4296 2872 3845 3552 6197 | 0.0002 |
| Bladder cancer | 5 | 3845 2261 5925 7423 1871 | 0.0003 |
| Prostate cancer | 6 | 3845 6932 5925 5728 2475 1871 | 0.0005 |
| mTOR signaling pathway | 5 | 6198 2475 6195 6197 7423 | 0.0005 |
| Tight junction | 7 | 3845 9223 71 2771 5728 5580 2770 | 0.0007 |
| Wnt signaling pathway | 7 | 56998 6932 5529 1857 8322 8325 8945 | 0.0011 |
| Colorectal cancer | 5 | 3845 6932 1857 8322 8325 | 0.0021 |
| Gap junction | 5 | 3845 2771 1453 2770 107 | 0.0027 |
| Basal cell carcinoma | 4 | 6932 1857 8322 8325 | 0.0027 |
| Long-term potentiation | 4 | 3845 6195 6197 107 | 0.0052 |
| Pancreatic cancer | 4 | 3845 5925 7423 1871 | 0.0054 |
| ErbB signaling pathway | 4 | 3845 6198 867 2475 | 0.0099 |
| Fc gamma R-mediated phagocytosis | 4 | 6198 382 10092 5580 | 0.0137 |
| Leukocyte transendothelial migration | 4 | 71 4478 2771 2770 | 0.0254 |
| Neurotrophin signaling pathway | 4 | 3845 6195 6197 5580 | 0.0302 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, Z.; Miao, R.; Li, G.; Wu, Y.; Robson, S.C.; Yang, X.; Zhao, Y.; Zhao, H.; Zhong, Y. Identification of Recurrence Related microRNAs in Hepatocellular Carcinoma after Surgical Resection. Int. J. Mol. Sci. 2013, 14, 1105-1118. https://doi.org/10.3390/ijms14011105
Yang Z, Miao R, Li G, Wu Y, Robson SC, Yang X, Zhao Y, Zhao H, Zhong Y. Identification of Recurrence Related microRNAs in Hepatocellular Carcinoma after Surgical Resection. International Journal of Molecular Sciences. 2013; 14(1):1105-1118. https://doi.org/10.3390/ijms14011105
Chicago/Turabian StyleYang, Zhen, Ruoyu Miao, Guangbing Li, Yan Wu, Simon C. Robson, Xiaobo Yang, Yi Zhao, Haitao Zhao, and Yang Zhong. 2013. "Identification of Recurrence Related microRNAs in Hepatocellular Carcinoma after Surgical Resection" International Journal of Molecular Sciences 14, no. 1: 1105-1118. https://doi.org/10.3390/ijms14011105
APA StyleYang, Z., Miao, R., Li, G., Wu, Y., Robson, S. C., Yang, X., Zhao, Y., Zhao, H., & Zhong, Y. (2013). Identification of Recurrence Related microRNAs in Hepatocellular Carcinoma after Surgical Resection. International Journal of Molecular Sciences, 14(1), 1105-1118. https://doi.org/10.3390/ijms14011105