Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance



Abstract
:1. Introduction
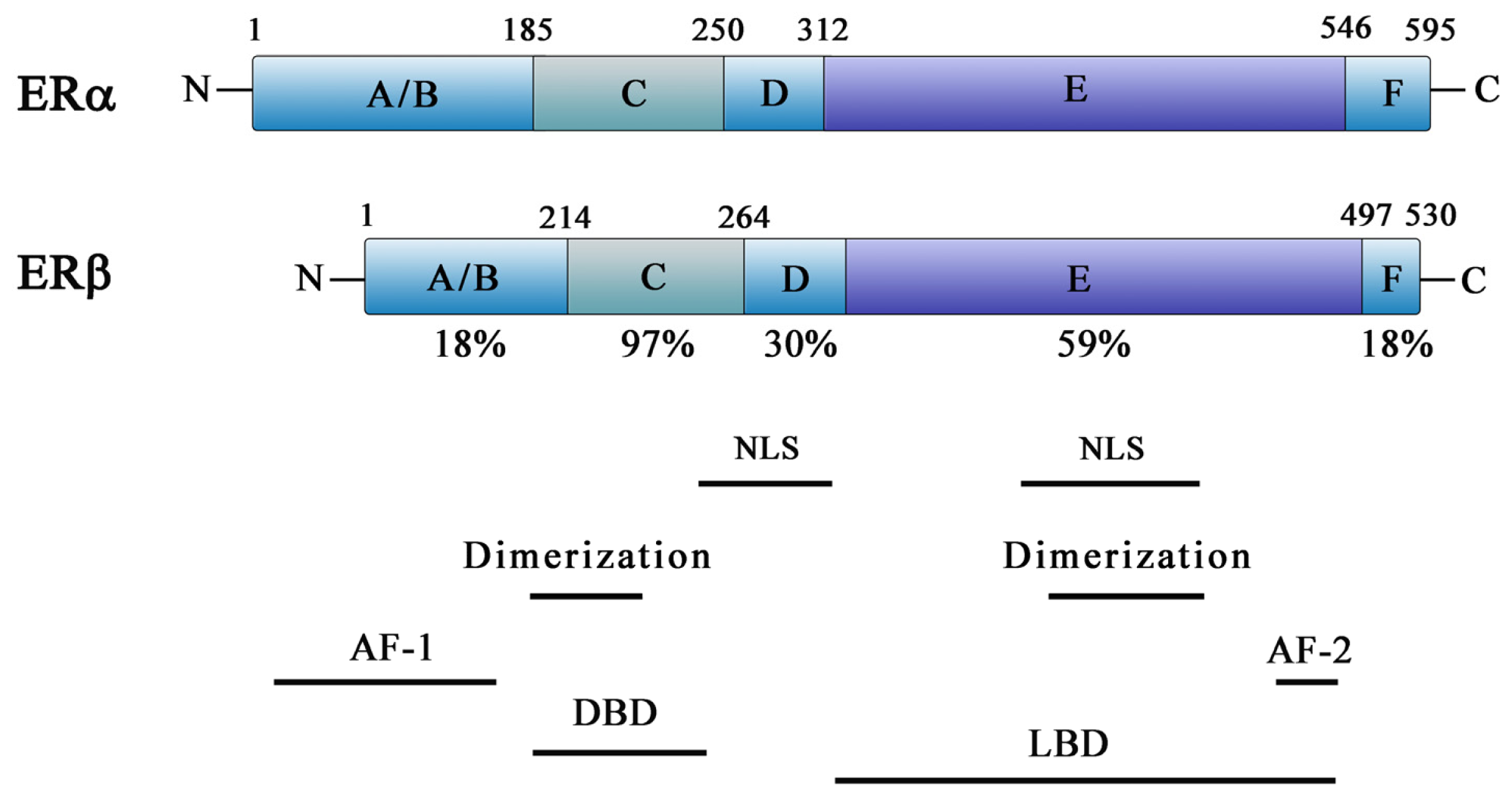
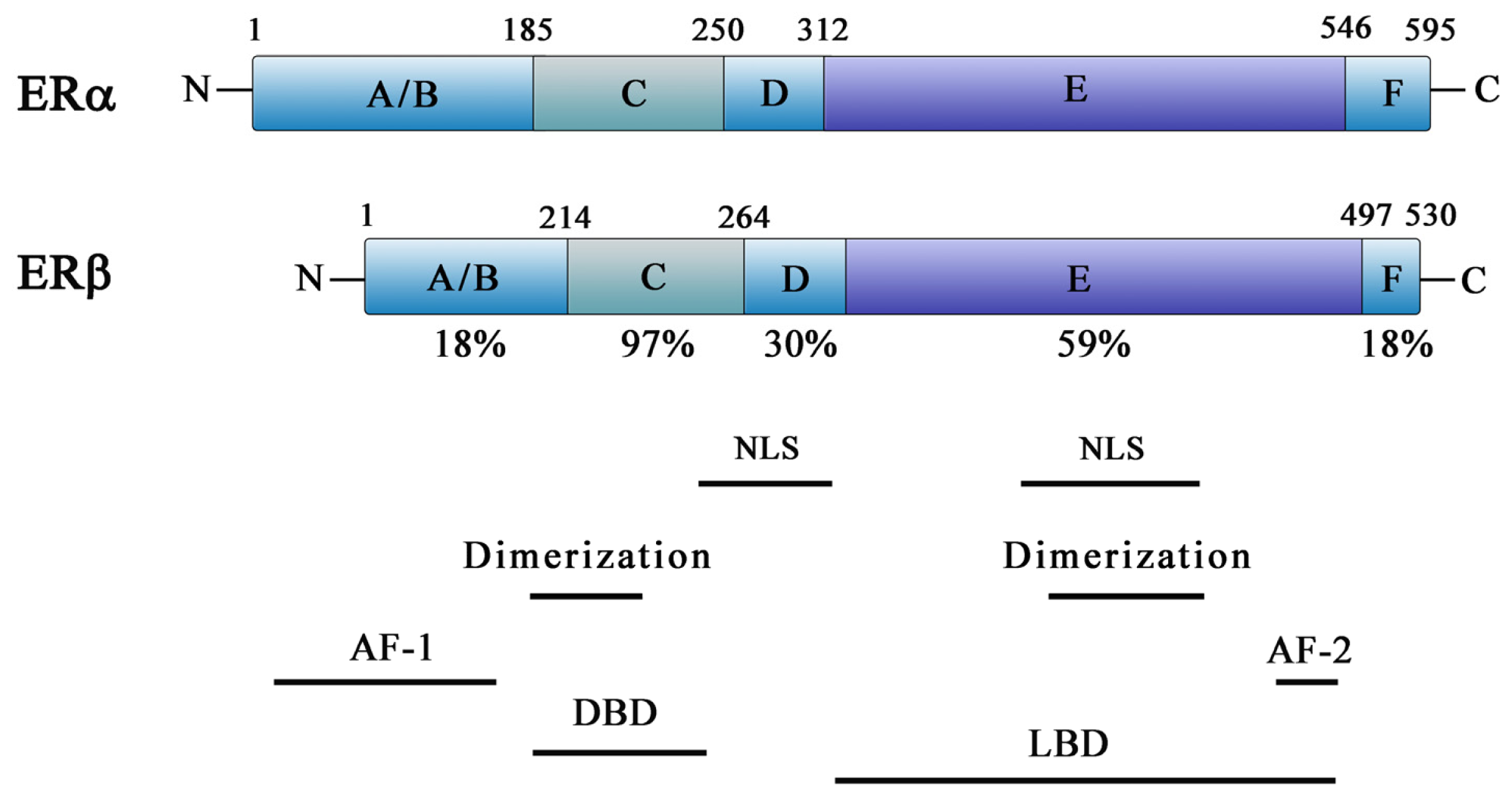
2. ER Action and Function
3. Mechanisms of Endocrine Resistance
3.1. Loss or Modification in the ER Expression
3.2. Epigenetics Mechanisms Regulating ER Expression
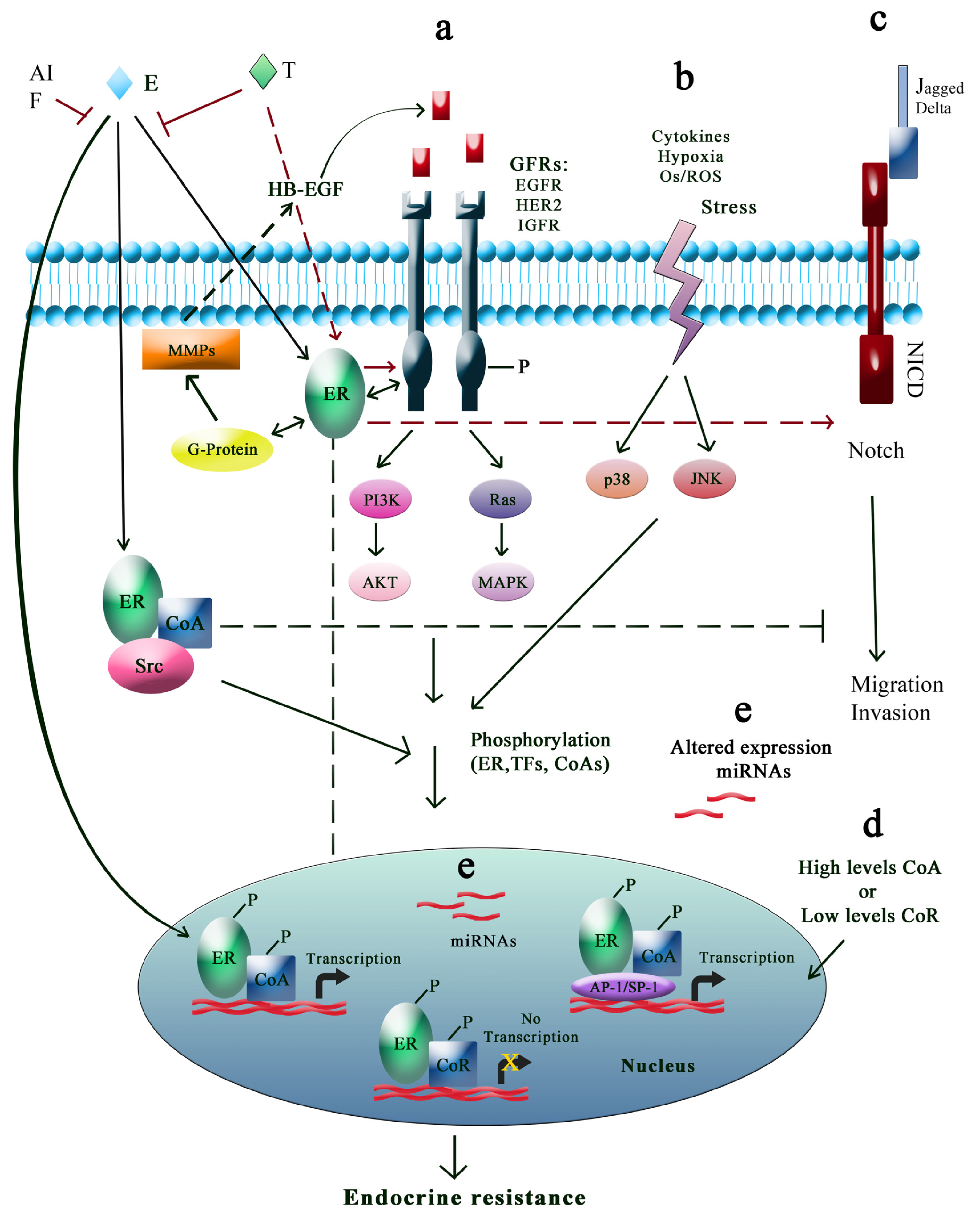
3.3. Regulation of Signal Transduction Pathways
3.3.1. Growth Factor Receptor Signaling Pathways
3.3.2. PI3K Cell Survival Pathways
3.3.3. Stress-Activated Protein Kinase/c-junNH2 Terminal Kinase Pathway
3.3.4. Other Signal Transduction Pathways
3.4. Altered Expression of Specific microRNAs
3.5. Coregulatory Proteins
3.6. Tamoxifen Metabolism and Genetically Based Resistance
4. Combination Therapy
5. Conclusions
- Conflict of InterestThe authors declare no conflict of interest.
References
- Clark, G.M.; Osborne, C.K.; McGuire, W.L. Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J. Clin. Oncol 1984, 2, 1102–1109. [Google Scholar]
- Miller, W.R.; Bartlett, J.M.; Canney, P.; Verrill, M. Hormonal therapy for postmenopausal breast cancer: the science of sequencing. Breast Cancer Res. Treat 2007, 103, 149–160. [Google Scholar]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med 1998, 339, 1609–1618. [Google Scholar]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar]
- Gradishar, W.J. Tamoxifen—What next? Oncologist 2004, 9, 378–384. [Google Scholar]
- Beelen, K.; Zwart, W.; Linn, S.C. Can predictive biomarkers in breast cancer guide adjuvant endocrine therapy? Nat. Rev. Clin. Oncol 2012, 9, 529–541. [Google Scholar]
- Hortobagyi, G.N. Toward individualized breast cancer therapy: Translating biological concepts to the bedside. Oncologist 2012, 17, 577–584. [Google Scholar]
- Brodie, A.; Sabnis, G. Adaptive changes result in activation of alternate signaling pathways and acquisition of resistance to aromatase inhibitors. Clin. Cancer Res 2011, 17, 4208–4213. [Google Scholar]
- Strasser-Weippl, K.; Goss, P.E. Advances in adjuvant hormonal therapy for postmenopausal women. J. Clin. Oncol 2005, 23, 1751–1759. [Google Scholar]
- Forbes, J.F.; Cuzick, J.; Buzdar, A.; Howell, A.; Tobias, J.S.; Baum, M. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-Month analysis of the ATAC trial. Lancet Oncol 2008, 9, 45–53. [Google Scholar]
- Howell, A.; Robertson, J.F.; Abram, P.; Lichinitser, M.R.; Elledge, R.; Bajetta, E.; Watanabe, T.; Morris, C.; Webster, A.; Dimery, I.; et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: A multinational, double-blind, randomized trial. J. Clin. Oncol 2004, 22, 1605–1613. [Google Scholar]
- Robertson, J.F.; Osborne, C.K.; Howell, A.; Jones, S.E.; Mauriac, L.; Ellis, M.; Kleeberg, U.R.; Come, S.E.; Vergote, I.; Gertler, S.; et al. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma in postmenopausal women: A prospective combined analysis of two multicenter trials. Cancer 2003, 98, 229–238. [Google Scholar]
- Weatherman, R.V.; Fletterick, R.J.; Scanlan, T.S. Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem 1999, 68, 559–581. [Google Scholar]
- Green, S.; Walter, P.; Greene, G.; Krust, A.; Goffin, C.; Jensen, E.; Scrace, G.; Waterfield, M.; Chambon, P. Cloning of the human oestrogen receptor cDNA. J. Steroid. Biochem 1986, 24, 77–83. [Google Scholar]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar]
- Osborne, C.K.; Schiff, R. Estrogen-receptor biology: Continuing progress and therapeutic implications. J. Clin. Oncol 2005, 23, 1616–1622. [Google Scholar]
- Osborne, C.K.; Schiff, R.; Fuqua, S.A.; Shou, J. Estrogen receptor: Current understanding of its activation and modulation. Clin. Cancer Res. 2001, 7, 4338s–4342s, discussion 4411s–4412s. [Google Scholar]
- Roger, P.; Sahla, M.E.; Makela, S.; Gustafsson, J.A.; Baldet, P.; Rochefort, H. Decreased expression of estrogen receptor β protein in proliferative preinvasive mammary tumors. Cancer Res 2001, 61, 2537–2541. [Google Scholar]
- Mann, S.; Laucirica, R.; Carlson, N.; Younes, P.S.; Ali, N.; Younes, A.; Li, Y.; Younes, M. Estrogen receptor beta expression in invasive breast cancer. Hum. Pathol 2001, 32, 113–118. [Google Scholar]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar]
- Shao, W.; Brown, M. Advances in estrogen receptor biology: Prospects for improvements in targeted breast cancer therapy. Breast Cancer Res 2004, 6, 39–52. [Google Scholar]
- Tora, L.; White, J.; Brou, C.; Tasset, D.; Webster, N.; Scheer, E.; Chambon, P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 1989, 59, 477–487. [Google Scholar]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar]
- Mader, S.; Chambon, P.; White, J.H. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res 1993, 21, 1125–1132. [Google Scholar]
- Kumar, V.; Green, S.; Staub, A.; Chambon, P. Localisation of the oestradiol-binding and putative DNA-binding domains of the human oestrogen receptor. EMBO J 1986, 5, 2231–2236. [Google Scholar]
- Kumar, V.; Green, S.; Stack, G.; Berry, M.; Jin, J.R.; Chambon, P. Functional domains of the human estrogen receptor. Cell 1987, 51, 941–951. [Google Scholar]
- Norris, J.D.; Fan, D.; Kerner, S.A.; McDonnell, D.P. Identification of a third autonomous activation domain within the human estrogen receptor. Mol. Endocrinol 1997, 11, 747–754. [Google Scholar]
- Picard, D.; Kumar, V.; Chambon, P.; Yamamoto, K.R. Signal transduction by steroid hormones: Nuclear localization is differentially regulated in estrogen and glucocorticoid receptors. Cell Regul 1990, 1, 291–299. [Google Scholar]
- Webster, N.J.; Green, S.; Tasset, D.; Ponglikitmongkol, M.; Chambon, P. The transcriptional activation function located in the hormone-binding domain of the human oestrogen receptor is not encoded in a single exon. EMBO J 1989, 8, 1441–1446. [Google Scholar]
- Berry, M.; Metzger, D.; Chambon, P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J 1990, 9, 2811–2818. [Google Scholar]
- Tzukerman, M.T.; Esty, A.; Santiso-Mere, D.; Danielian, P.; Parker, M.G.; Stein, R.B.; Pike, J.W.; McDonnell, D.P. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol. Endocrinol 1994, 8, 21–30. [Google Scholar]
- Enmark, E.; Gustafsson, J.A. Oestrogen receptors—An overview. J. Intern. Med 1999, 246, 133–138. [Google Scholar]
- Klinge, C.M. Estrogen receptor interaction with co-activators and co-repressors. Steroids 2000, 65, 227–251. [Google Scholar]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev 1999, 20, 321–344. [Google Scholar]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Invest 2006, 116, 561–570. [Google Scholar]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar]
- Razandi, M.; Oh, P.; Pedram, A.; Schnitzer, J.; Levin, E.R. ERs associate with and regulate the production of caveolin: Implications for signaling and cellular actions. Mol. Endocrinol 2002, 16, 100–115. [Google Scholar]
- Razandi, M.; Pedram, A.; Park, S.T.; Levin, E.R. Proximal events in signaling by plasma membrane estrogen receptors. J. Biol. Chem 2003, 278, 2701–2712. [Google Scholar]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor α and β influences 17β-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858. [Google Scholar]
- Migliaccio, A.; di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 1996, 15, 1292–1300. [Google Scholar]
- Migliaccio, A.; Piccolo, D.; Castoria, G.; Di Domenico, M.; Bilancio, A.; Lombardi, M.; Gong, W.; Beato, M.; Auricchio, F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J 1998, 17, 2008–2018. [Google Scholar]
- Kahlert, S.; Nuedling, S.; van Eickels, M.; Vetter, H.; Meyer, R.; Grohe, C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J. Biol. Chem 2000, 275, 18447–18453. [Google Scholar]
- Wong, C.W.; McNally, C.; Nickbarg, E.; Komm, B.S.; Cheskis, B.J. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc. Natl. Acad. Sci. USA 2002, 99, 14783–14788. [Google Scholar]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res 2004, 10, S331–S336. [Google Scholar]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar]
- Thomas, P.; Alyea, R.; Pang, Y.; Peyton, C.; Dong, J.; Berg, A.H. Conserved estrogen binding and signaling functions of the G protein-coupled estrogen receptor 1 (GPER) in mammals and fish. Steroids 2010, 75, 595–602. [Google Scholar]
- Alexander, S.P.; Mathie, A.; Peters, J.A. Guide to Receptors and Channels (GRAC), 5th edition. Br. J. Pharmacol 2011, 164, S1–S324. [Google Scholar]
- Barton, M. Position paper: The membrane estrogen receptor GPER—Clues and questions. Steroids 2012, 77, 935–942. [Google Scholar]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 1996, 15, 2174–2183. [Google Scholar]
- Joel, P.B.; Smith, J.; Sturgill, T.W.; Fisher, T.L.; Blenis, J.; Lannigan, D.A. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell. Biol 1998, 18, 1978–1984. [Google Scholar]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J. Biol. Chem 2001, 276, 9817–9824. [Google Scholar]
- Sun, M.; Paciga, J.E.; Feldman, R.I.; Yuan, Z.; Coppola, D.; Lu, Y.Y.; Shelley, S.A.; Nicosia, S.V.; Cheng, J.Q. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor α (ERα) via interaction between ERα and PI3K. Cancer Res 2001, 61, 5985–5991. [Google Scholar]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res 2006, 12, 6359–6366. [Google Scholar]
- Chang, J.; Fan, W. Endocrine therapy resistance: Current status, possible mechanisms and overcoming strategies. Anticancer Agents Med. Chem. 2012. [Epub ahead of print]. [Google Scholar]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res 2001, 61, 7025–7029. [Google Scholar]
- Parl, F.F. Multiple mechanisms of estrogen receptor gene repression contribute to ER-negative breast cancer. Pharmacogenomics J 2003, 3, 251–253. [Google Scholar]
- Weigel, R.J.; deConinck, E.C. Transcriptional control of estrogen receptor in estrogen receptor-negative breast carcinoma. Cancer Res 1993, 53, 3472–3474. [Google Scholar]
- Ottaviano, Y.L.; Issa, J.P.; Parl, F.F.; Smith, H.S.; Baylin, S.B.; Davidson, N.E. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res 1994, 54, 2552–2555. [Google Scholar]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet 2000, 25, 338–342. [Google Scholar]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet 2000, 25, 269–277. [Google Scholar]
- Fan, J.; Yin, W.J.; Lu, J.S.; Wang, L.; Wu, J.; Wu, F.Y.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol 2008, 134, 883–890. [Google Scholar]
- Marks, P.A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 534–535. [Google Scholar]
- Zhou, Q.; Shaw, P.G.; Davidson, N.E. Inhibition of histone deacetylase suppresses EGF signaling pathways by destabilizing EGFR mRNA in ER-negative human breast cancer cells. Breast Cancer Res. Treat 2009, 117, 443–451. [Google Scholar]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M. Functional activation of the estrogen receptor-α and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer Res 2011, 71, 1893–1903. [Google Scholar]
- Keen, J.C.; Yan, L.; Mack, K.M.; Pettit, C.; Smith, D.; Sharma, D.; Davidson, N.E. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res. Treat 2003, 81, 177–186. [Google Scholar]
- Giacinti, L.; Giacinti, C.; Gabellini, C.; Rizzuto, E.; Lopez, M.; Giordano, A. Scriptaid effects on breast cancer cell lines. J. Cell Physiol 2012, 227, 3426–3433. [Google Scholar]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res 1995, 55, 3331–3338. [Google Scholar]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol 2005, 23, 2469–2476. [Google Scholar]
- Dowsett, M. Overexpression of HER-2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr. Relat. Cancer 2001, 8, 191–195. [Google Scholar]
- Osborne, C.K.; Pippen, J.; Jones, S.E.; Parker, L.M.; Ellis, M.; Come, S.; Gertler, S.Z.; May, J.T.; Burton, G.; Dimery, I.; et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J. Clin. Oncol 2002, 20, 3386–3395. [Google Scholar]
- Howell, A.; Pippen, J.; Elledge, R.M.; Mauriac, L.; Vergote, I.; Jones, S.E.; Come, S.E.; Osborne, C.K.; Robertson, J.F. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma: A prospectively planned combined survival analysis of two multicenter trials. Cancer 2005, 104, 236–239. [Google Scholar]
- Stoner, M.; Saville, B.; Wormke, M.; Dean, D.; Burghardt, R.; Safe, S. Hypoxia induces proteasome-dependent degradation of estrogen receptor alpha in ZR-75 breast cancer cells. Mol. Endocrinol 2002, 16, 2231–2242. [Google Scholar]
- Creighton, C.J.; Hilger, A.M.; Murthy, S.; Rae, J.M.; Chinnaiyan, A.M.; El-Ashry, D. Activation of mitogen-activated protein kinase in estrogen receptor α-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor α-negative human breast tumors. Cancer Res. 2006, 66, 3903–3911. [Google Scholar]
- Stoica, A.; Saceda, M.; Doraiswamy, V.L.; Coleman, C.; Martin, M.B. Regulation of estrogen receptor-α gene expression by epidermal growth factor. J. Endocrinol 2000, 165, 371–378. [Google Scholar]
- Angeloni, S.V.; Martin, M.B.; Garcia-Morales, P.; Castro-Galache, M.D.; Ferragut, J.A.; Saceda, M. Regulation of estrogen receptor-α expression by the tumor suppressor gene p53 in MCF-7 cells. J. Endocrinol 2004, 180, 497–504. [Google Scholar]
- Martin, M.B.; Angeloni, S.V.; Garcia-Morales, P.; Sholler, P.F.; Castro-Galache, M.D.; Ferragut, J.A.; Saceda, M. Regulation of estrogen receptor-α expression in MCF-7 cells by taxol. J. Endocrinol 2004, 180, 487–496. [Google Scholar]
- Macaluso, M.; Cinti, C.; Russo, G.; Russo, A.; Giordano, A. pRb2/p130-E2F4/5-HDAC1- SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene 2003, 22, 3511–3517. [Google Scholar]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 1997, 57, 1244–1249. [Google Scholar]
- Wolf, D.M.; Jordan, V.C. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res. Treat 1994, 31, 129–138. [Google Scholar]
- Fuqua, S.A.; Wiltschke, C.; Zhang, Q.X.; Borg, A.; Castles, C.G.; Friedrichs, W.E.; Hopp, T.; Hilsenbeck, S.; Mohsin, S.; O’Connell, P.; et al. A hypersensitive estrogen receptor-α mutation in premalignant breast lesions. Cancer Res 2000, 60, 4026–4029. [Google Scholar]
- Herynk, M.H.; Fuqua, S.A. Estrogen receptor mutations in human disease. Endocr Rev 2004, 25, 869–898. [Google Scholar]
- Speirs, V.; Parkes, A.T.; Kerin, M.J.; Walton, D.S.; Carleton, P.J.; Fox, J.N.; Atkin, S.L. Coexpression of estrogen receptor α and β: Poor prognostic factors in human breast cancer? Cancer Res 1999, 59, 525–528. [Google Scholar]
- Hopp, T.A.; Weiss, H.L.; Parra, I.S.; Cui, Y.; Osborne, C.K.; Fuqua, S.A. Low levels of estrogen receptor β protein predict resistance to tamoxifen therapy in breast cancer. Clin. Cancer Res 2004, 10, 7490–7499. [Google Scholar]
- Borgquist, S.; Holm, C.; Stendahl, M.; Anagnostaki, L.; Landberg, G.; Jirstrom, K. Oestrogen receptors α and β show different associations to clinicopathological parameters and their co-expression might predict a better response to endocrine treatment in breast cancer. J. Clin. Pathol 2008, 61, 197–203. [Google Scholar]
- Trimarchi, M.P.; Mouangsavanh, M.; Huang, T.H. Cancer epigenetics: A perspective on the role of DNA methylation in acquired endocrine resistance. Chin. J. Cancer 2011, 30, 749–756. [Google Scholar]
- Widschwendter, M.; Siegmund, K.D.; Muller, H.M.; Fiegl, H.; Marth, C.; Muller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res 2004, 64, 3807–3813. [Google Scholar]
- Lo, P.K.; Sukumar, S. Epigenomics and breast cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar]
- Badia, E.; Duchesne, M.J.; Semlali, A.; Fuentes, M.; Giamarchi, C.; Richard-Foy, H.; Nicolas, J.C.; Pons, M. Long-term hydroxytamoxifen treatment of an MCF-7-derived breast cancer cell line irreversibly inhibits the expression of estrogenic genes through chromatin remodeling. Cancer Res 2000, 60, 4130–4138. [Google Scholar]
- Jansen, M.P.; Sieuwerts, A.M.; Look, M.P.; Ritstier, K.; Meijer-van Gelder, M.E.; van Staveren, I.L.; Klijn, J.G.; Foekens, J.A.; Berns, E.M. HOXB13-to-IL17BR expression ratio is related with tumor aggressiveness and response to tamoxifen of recurrent breast cancer: A retrospective study. J. Clin. Oncol 2007, 25, 662–668. [Google Scholar]
- Iorns, E.; Turner, N.C.; Elliott, R.; Syed, N.; Garrone, O.; Gasco, M.; Tutt, A.N.; Crook, T.; Lord, C.J.; Ashworth, A. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell 2008, 13, 91–104. [Google Scholar]
- Heller, G.; Ziegler, B.; Brandstetter, A.; Novak, S.; Rudas, M.; Hennig, G.; Gehrmann, M.; Acht, T.; Zochbauer-Muller, S.; Filipits, M. CDK10 is not a target for aberrant DNA methylation in breast cancer. Anticancer Res 2009, 29, 3939–3944. [Google Scholar]
- Pathiraja, T.N.; Stearns, V.; Oesterreich, S. Epigenetic regulation in estrogen receptor positive breast cancer—Role in treatment response. J. Mammary Gland Biol. Neoplasia 2010, 15, 35–47. [Google Scholar]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst 2004, 96, 926–935. [Google Scholar]
- Joel, P.B.; Traish, A.M.; Lannigan, D.A. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J. Biol. Chem 1998, 273, 13317–13323. [Google Scholar]
- Britton, D.J.; Hutcheson, I.R.; Knowlden, J.M.; Barrow, D.; Giles, M.; McClelland, R.A.; Gee, J.M.; Nicholson, R.I. Bidirectional cross talk between ERα and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res. Treat 2006, 96, 131–146. [Google Scholar]
- De Mora, J.F.; Brown, M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol. Cell. Biol 2000, 20, 5041–5047. [Google Scholar]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst 2003, 95, 353–361. [Google Scholar]
- Nicholson, R.I.; Hutcheson, I.R.; Jones, H.E.; Hiscox, S.E.; Giles, M.; Taylor, K.M.; Gee, J.M. Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev. Endocr. Metab. Disord 2007, 8, 241–253. [Google Scholar]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat 1992, 24, 85–95. [Google Scholar]
- Chung, Y.L.; Sheu, M.L.; Yang, S.C.; Lin, C.H.; Yen, S.H. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int. J. Cancer 2002, 97, 306–312. [Google Scholar]
- Kaufman, B.; Mackey, J.R.; Clemens, M.R.; Bapsy, P.P.; Vaid, A.; Wardley, A.; Tjulandin, S.; Jahn, M.; Lehle, M.; Feyereislova, A.; et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J. Clin. Oncol 2009, 27, 5529–5537. [Google Scholar]
- Massarweh, S.; Osborne, C.K.; Jiang, S.; Wakeling, A.E.; Rimawi, M.; Mohsin, S.K.; Hilsenbeck, S.; Schiff, R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res 2006, 66, 8266–8273. [Google Scholar]
- Wang, X.; Masri, S.; Phung, S.; Chen, S. The role of amphiregulin in exemestane-resistant breast cancer cells: Evidence of an autocrine loop. Cancer Res 2008, 68, 2259–2265. [Google Scholar]
- Chen, A.C.; Migliaccio, I.; Rimawi, M.; Lopez-Tarruella, S.; Creighton, C.J.; Massarweh, S.; Huang, C.; Wang, Y.C.; Batra, S.K.; Gutierrez, M.C.; et al. Upregulation of mucin4 in ER-positive/HER2-overexpressing breast cancer xenografts with acquired resistance to endocrine and HER2-targeted therapies. Breast Cancer Res. Treat 2012, 134, 583–593. [Google Scholar]
- Johnston, S.; Pippen, J., Jr; Pivot, X.; Lichinitser, M.; Sadeghi, S.; Dieras, V.; Gomez, H.L.; Romieu, G.; Manikhas, A.; Kennedy, M.J.; et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2009, 27, 5538–5546. [Google Scholar]
- Evans, A.H.; Pancholi, S.; Farmer, I.; Thornhill, A.; Evans, D.B.; Johnston, S.R.; Dowsett, M.; Martin, L.A. EGFR/HER2 inhibitor AEE788 increases ER-mediated transcription in HER2/ER-positive breast cancer cells but functions synergistically with endocrine therapy. Br. J. Cancer 2010, 102, 1235–1243. [Google Scholar]
- Surmacz, E. Function of the IGF-I receptor in breast cancer. J. Mammary Gland Biol. Neoplasia 2000, 5, 95–105. [Google Scholar]
- Knowlden, J.M.; Hutcheson, I.R.; Barrow, D.; Gee, J.M.; Nicholson, R.I. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: A supporting role to the epidermal growth factor receptor. Endocrinology 2005, 146, 4609–4618. [Google Scholar]
- Hurbin, A.; Dubrez, L.; Coll, J.L.; Favrot, M.C. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J. Biol. Chem 2002, 277, 49127–49133. [Google Scholar]
- Zhang, Y.; Moerkens, M.; Ramaiahgari, S.; de Bont, H.; Price, L.; Meerman, J.; van de Water, B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res 2011, 13, R52. [Google Scholar]
- Chong, K.; Subramanian, A.; Sharma, A.; Mokbel, K. Measuring IGF-1, ER-α and EGFR expression can predict tamoxifen-resistance in ER-positive breast cancer. Anticancer Res 2011, 31, 23–32. [Google Scholar]
- Fagan, D.H.; Uselman, R.R.; Sachdev, D.; Yee, D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: Implications for breast cancer treatment. Cancer Res 2012, 72, 3372–3380. [Google Scholar]
- Cui, J.; Germer, K.; Wu, T.; Wang, J.; Luo, J.; Wang, S.C.; Wang, Q.; Zhang, X. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res 2012, 72, 5625–5634. [Google Scholar]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev 1999, 13, 2905–2927. [Google Scholar]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther 2002, 1, 707–717. [Google Scholar]
- Martin, M.B.; Franke, T.F.; Stoica, G.E.; Chambon, P.; Katzenellenbogen, B.S.; Stoica, B.A.; McLemore, M.S.; Olivo, S.E.; Stoica, A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology 2000, 141, 4503–4511. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Miller, T.W.; Perez-Torres, M.; Narasanna, A.; Guix, M.; Stal, O.; Perez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res 2009, 69, 4192–4201. [Google Scholar]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res 2011, 13, 224. [Google Scholar]
- Ma, C.X.; Crowder, R.J.; Ellis, M.J. Importance of PI3-kinase pathway in response/resistance to aromatase inhibitors. Steroids 2011, 76, 750–752. [Google Scholar]
- Creighton, C.J.; Fu, X.; Hennessy, B.T.; Casa, A.J.; Zhang, Y.; Gonzalez-Angulo, A.M.; Lluch, A.; Gray, J.W.; Brown, P.H.; Hilsenbeck, S.G.; et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res 2010, 12, R40. [Google Scholar]
- Miller, T.W.; Balko, J.M.; Arteaga, C.L. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J. Clin. Oncol 2011, 29, 4452–4461. [Google Scholar]
- Barnadas, A.; Estevez, L.G.; Lluch-Hernandez, A.; Rodriguez-Lescure, A.; Rodriguez-Sanchez, C.; Sanchez-Rovira, P. An overview of letrozole in postmenopausal women with hormone-responsive breast cancer. Adv. Ther 2011, 28, 1045–1058. [Google Scholar]
- Cavazzoni, A.; Bonelli, M.A.; Fumarola, C.; la Monica, S.; Airoud, K.; Bertoni, R.; Alfieri, R.R.; Galetti, M.; Tramonti, S.; Galvani, E.; et al. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett 2012, 323, 77–87. [Google Scholar]
- Chan, C.M.; Martin, L.A.; Johnston, S.R.; Ali, S.; Dowsett, M. Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J. Steroid Biochem. Mol. Biol 2002, 81, 333–341. [Google Scholar]
- Santen, R.J.; Song, R.X.; Masamura, S.; Yue, W.; Fan, P.; Sogon, T.; Hayashi, S.; Nakachi, K.; Eguchi, H. Adaptation to estradiol deprivation causes up-regulation of growth factor pathways and hypersensitivity to estradiol in breast cancer cells. Adv. Exp. Med. Biol 2008, 630, 19–34. [Google Scholar]
- Santen, R.J.; Song, R.X.; Zhang, Z.; Kumar, R.; Jeng, M.H.; Masamura, A.; Lawrence, J., Jr; Berstein, L.; Yue, W. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer 2005, 12, S61–S73. [Google Scholar]
- Masamura, S.; Santner, S.J.; Heitjan, D.F.; Santen, R.J. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J. Clin. Endocrinol. Metab 1995, 80, 2918–2925. [Google Scholar]
- Hurvitz, S.A.; Pietras, R.J. Rational management of endocrine resistance in breast cancer: A comprehensive review of estrogen receptor biology, treatment options, and future directions. Cancer 2008, 113, 2385–2397. [Google Scholar]
- Masri, S.; Phung, S.; Wang, X.; Wu, X.; Yuan, Y.C.; Wagman, L.; Chen, S. Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer Res 2008, 68, 4910–4918. [Google Scholar]
- Berstein, L.M.; Zheng, H.; Yue, W.; Wang, J.P.; Lykkesfeldt, A.E.; Naftolin, F.; Harada, H.; Shanabrough, M.; Santen, R.J. New approaches to the understanding of tamoxifen action and resistance. Endocr. Relat. Cancer 2003, 10, 267–277. [Google Scholar]
- Miller, T.W.; Hennessy, B.T.; Gonzalez-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; Garcia-Echeverria, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J. Clin. Invest 2010, 120, 2406–2413. [Google Scholar]
- Sanchez, C.G.; Ma, C.X.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 2011, 13, R21. [Google Scholar]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar]
- Dumont, J.A.; Bitonti, A.J.; Wallace, C.D.; Baumann, R.J.; Cashman, E.A.; Cross-Doersen, D.E. Progression of MCF-7 breast cancer cells to antiestrogen-resistant phenotype is accompanied by elevated levels of AP-1 DNA-binding activity. Cell Growth Differ 1996, 7, 351–359. [Google Scholar]
- Johnston, S.R.; Lu, B.; Scott, G.K.; Kushner, P.J.; Smith, I.E.; Dowsett, M.; Benz, C.C. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin. Cancer Res 1999, 5, 251–256. [Google Scholar]
- Schiff, R.; Reddy, P.; Ahotupa, M.; Coronado-Heinsohn, E.; Grim, M.; Hilsenbeck, S.G.; Lawrence, R.; Deneke, S.; Herrera, R.; Chamness, G.C.; et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J. Natl. Cancer Inst 2000, 92, 1926–1934. [Google Scholar]
- Clarke, R.; Skaar, T.C.; Bouker, K.B.; Davis, N.; Lee, Y.R.; Welch, J.N.; Leonessa, F. Molecular and pharmacological aspects of antiestrogen resistance. J. Steroid Biochem. Mol. Biol 2001, 76, 71–84. [Google Scholar]
- Webb, P.; Lopez, G.N.; Uht, R.M.; Kushner, P.J. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol 1995, 9, 443–456. [Google Scholar]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β). J. Biol. Chem 1996, 271, 17920–17926. [Google Scholar]
- New, L.; Han, J. The p38 MAP kinase pathway and its biological function. Trends Cardiovasc. Med 1998, 8, 220–228. [Google Scholar]
- Lee, H.; Bai, W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol. Cell. Biol 2002, 22, 5835–5845. [Google Scholar]
- Antoon, J.W.; Bratton, M.R.; Guillot, L.M.; Wadsworth, S.; Salvo, V.A.; Elliott, S.; McLachlan, J.A.; Burow, M.E. Pharmacology and anti-tumor activity of RWJ67657, a novel inhibitor of p38 mitogen activated protein kinase. Am. J. Cancer Res 2012, 2, 446–458. [Google Scholar]
- Aesoy, R.; Sanchez, B.C.; Norum, J.H.; Lewensohn, R.; Viktorsson, K.; Linderholm, B. An autocrine VEGF/VEGFR2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol. Cancer Res 2008, 6, 1630–1638. [Google Scholar]
- Al Saleh, S.; Sharaf, L.H.; Luqmani, Y.A. Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int. J. Oncol 2011, 38, 1197–1217. [Google Scholar]
- Weigel, M.T.; Ghazoui, Z.; Dunbier, A.; Pancholi, S.; Dowsett, M.; Martin, L.A. Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast Cancer Res 2012, 14, R78. [Google Scholar]
- Zhou, Y.; Yau, C.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Eppenberger, U.; Eppenberger-Castori, S.; Benz, C.C. Enhanced NF κB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007, 7, 59. [Google Scholar]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr; Sledge, G.W., Jr. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar]
- Sovak, M.A.; Bellas, R.E.; Kim, D.W.; Zanieski, G.J.; Rogers, A.E.; Traish, A.M.; Sonenshein, G.E. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J. Clin. Invest 1997, 100, 2952–2960. [Google Scholar]
- Biswas, D.K.; Singh, S.; Shi, Q.; Pardee, A.B.; Iglehart, J.D. Crossroads of estrogen receptor and NF-κB signaling. Sci STKE 2005, 2005, e27. [Google Scholar]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; van dam, P.; Pauwels, P.; Dirix, L.Y.; van Laere, S.J. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res 2012, 14, 212. [Google Scholar]
- Chang, H.L.; Sugimoto, Y.; Liu, S.; Ye, W.; Wang, L.S.; Huang, Y.W.; Lin, Y.C. Keratinocyte growth factor (KGF) induces tamoxifen (Tam) resistance in human breast cancer MCF-7 cells. Anticancer Res 2006, 26, 1773–1784. [Google Scholar]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res 2004, 6, R605–R615. [Google Scholar]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res 2006, 66, 1517–1525. [Google Scholar]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res 2008, 68, 5226–5235. [Google Scholar]
- Dibb, N.J.; Dilworth, S.M.; Mol, C.D. Switching on kinases: Oncogenic activation of BRAF and the PDGFR family. Nat. Rev. Cancer 2004, 4, 718–727. [Google Scholar]
- Yuan, Z.M.; Huang, Y.; Whang, Y.; Sawyers, C.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature 1996, 382, 272–274. [Google Scholar]
- Brasher, B.B.; van Etten, R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem 2000, 275, 35631–35637. [Google Scholar]
- Sikora, M.J.; Strumba, V.; Lippman, M.E.; Johnson, M.D.; Rae, J.M. Mechanisms of estrogen-independent breast cancer growth driven by low estrogen concentrations are unique versus complete estrogen deprivation. Breast Cancer Res. Treat 2012, 134, 1027–1039. [Google Scholar]
- Li, M.; Li, J.; Ding, X.; He, M.; Cheng, S.Y. microRNA and cancer. AAPS J 2010, 12, 309–317. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci 2010, 101, 2087–2092. [Google Scholar]
- Cho, W.C. MicroRNAs as therapeutic targets and their potential applications in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 747–759. [Google Scholar]
- Iorio, M.V.; Casalini, P.; Piovan, C.; Braccioli, L.; Tagliabue, E. Breast cancer and microRNAs: Therapeutic impact. Breast 2011, 20, S63–S70. [Google Scholar]
- Xin, F.; Li, M.; Balch, C.; Thomson, M.; Fan, M.; Liu, Y.; Hammond, S.M.; Kim, S.; Nephew, K.P. Computational analysis of microRNA profiles and their target genes suggests significant involvement in breast cancer antiestrogen resistance. Bioinformatics 2009, 25, 430–434. [Google Scholar]
- Lyng, M.B.; Laenkholm, A.V.; Sokilde, R.; Gravgaard, K.H.; Litman, T.; Ditzel, H.J. Global microRNA expression profiling of high-risk ER+ breast cancers from patients receiving adjuvant tamoxifen mono-therapy: A DBCG study. PLoS One 2012, 7, e36170. [Google Scholar]
- Manavalan, T.T.; Teng, Y.; Appana, S.N.; Datta, S.; Kalbfleisch, T.S.; Li, Y.; Klinge, C.M. Differential expression of microRNA expression in tamoxifen-sensitive MCF-7 versus tamoxifen-resistant LY2 human breast cancer cells. Cancer Lett 2011, 313, 26–43. [Google Scholar]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem 2008, 283, 29897–29903. [Google Scholar]
- Cittelly, D.M.; Das, P.M.; Spoelstra, N.S.; Edgerton, S.M.; Richer, J.K.; Thor, A.D.; Jones, F.E. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol. Cancer 2010, 9, 317. [Google Scholar]
- Klinge, C.M.; Riggs, K.A.; Wickramasinghe, N.S.; Emberts, C.G.; McConda, D.B.; Barry, P.N.; Magnusen, J.E. Estrogen receptor α 46 is reduced in tamoxifen resistant breast cancer cells and re-expression inhibits cell proliferation and estrogen receptor α 66-regulated target gene transcription. Mol. Cell. Endocrinol 2010, 323, 268–276. [Google Scholar]
- Bergamaschi, A.; Katzenellenbogen, B.S. Tamoxifen downregulation of miR-451 increases 14-3-3ζ and promotes breast cancer cell survival and endocrine resistance. Oncogene 2012, 31, 39–47. [Google Scholar]
- Carlomagno, C.; Perrone, F.; Gallo, C.; de Laurentiis, M.; Lauria, R.; Morabito, A.; Pettinato, G.; Panico, L.; D’Antonio, A.; Bianco, A.R.; et al. c-erb B2 overexpression decreases the benefit of adjuvant tamoxifen in early-stage breast cancer without axillary lymph node metastases. J. Clin. Oncol 1996, 14, 2702–2708. [Google Scholar]
- Giacinti, L.; Claudio, P.P.; Lopez, M.; Giordano, A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist 2006, 11, 1–8. [Google Scholar]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar]
- Rao, X.; di Leva, G.; Li, M.; Fang, F.; Devlin, C.; Hartman-Frey, C.; Burow, M.E.; Ivan, M.; Croce, C.M.; Nephew, K.P. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011, 30, 1082–1097. [Google Scholar]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem 2008, 283, 31079–31086. [Google Scholar]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-α (ERα) and represses ERα messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol 2007, 21, 1132–1147. [Google Scholar]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 Expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Res 2008, 68, 5004–5008. [Google Scholar]
- Tessel, M.A.; Krett, N.L.; Rosen, S.T. Steroid receptor and microRNA regulation in cancer. Curr. Opin. Oncol 2010, 22, 592–597. [Google Scholar]
- Xiong, J.; Yu, D.; Wei, N.; Fu, H.; Cai, T.; Huang, Y.; Wu, C.; Zheng, X.; Du, Q.; Lin, D.; Liang, Z. An estrogen receptor α suppressor, microRNA-22, is downregulated in estrogen receptor α-positive human breast cancer cell lines and clinical samples. FEBS J 2010, 277, 1684–1694. [Google Scholar]
- Mitra, D.; Brumlik, M.J.; Okamgba, S.U.; Zhu, Y.; Duplessis, T.T.; Parvani, J.G.; Lesko, S.M.; Brogi, E.; Jones, F.E. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer Ther 2009, 8, 2152–2162. [Google Scholar]
- Cittelly, D.M.; Das, P.M.; Salvo, V.A.; Fonseca, J.P.; Burow, M.E.; Jones, F.E. Oncogenic HER2δ16 suppresses miR-15a/16 and deregulates BCL-2 to promote endocrine resistance of breast tumors. Carcinogenesis 2010, 31, 2049–2057. [Google Scholar]
- Neal, C.L.; Yao, J.; Yang, W.; Zhou, X.; Nguyen, N.T.; Lu, J.; Danes, C.G.; Guo, H.; Lan, K.H.; Ensor, J.; Hittelman, W.; Hung, M.C.; Yu, D. 14-3-3ζ overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res 2009, 69, 3425–3432. [Google Scholar]
- Frasor, J.; Chang, E.C.; Komm, B.; Lin, C.Y.; Vega, V.B.; Liu, E.T.; Miller, L.D.; Smeds, J.; Bergh, J.; Katzenellenbogen, B.S. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res 2006, 66, 7334–7340. [Google Scholar]
- Bergamaschi, A.; Christensen, B.L.; Katzenellenbogen, B.S. Reversal of endocrine resistance in breast cancer: Interrelationships among 14-3-3ζ, FOXM1, and a gene signature associated with mitosis. Breast Cancer Res 2011, 13, R70. [Google Scholar]
- Rothe, F.; Ignatiadis, M.; Chaboteaux, C.; Haibe-Kains, B.; Kheddoumi, N.; Majjaj, S.; Badran, B.; Fayyad-Kazan, H.; Desmedt, C.; Harris, A.L.; et al. Global microRNA expression profiling identifies MiR-210 associated with tumor proliferation, invasion and poor clinical outcome in breast cancer. PLoS One 2011, 6, e20980. [Google Scholar]
- Shi, W.; Gerster, K.; Alajez, N.M.; Tsang, J.; Waldron, L.; Pintilie, M.; Hui, A.B.; Sykes, J.; P’ng, C.; Miller, N.; et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res 2011, 71, 2926–2937. [Google Scholar]
- Sachdeva, M.; Wu, H.; Ru, P.; Hwang, L.; Trieu, V.; Mo, Y.Y. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 2011, 30, 822–831. [Google Scholar]
- Knabbe, C.; Zugmaier, G.; Schmahl, M.; Dietel, M.; Lippman, M.E.; Dickson, R.B. Induction of transforming growth factor β by the antiestrogens droloxifene, tamoxifen, and toremifene in MCF-7 cells. Am. J. Clin. Oncol 1991, 14, S15–S20. [Google Scholar]
- Knabbe, C.; Kopp, A.; Hilgers, W.; Lang, D.; Muller, V.; Zugmaier, G.; Jonat, W. Regulation and role of TGF β production in breast cancer. Ann. N. Y. Acad. Sci 1996, 784, 263–276. [Google Scholar]
- Buck, M.B.; Pfizenmaier, K.; Knabbe, C. Antiestrogens induce growth inhibition by sequential activation of p38 mitogen-activated protein kinase and transforming growth factor-β pathways in human breast cancer cells. Mol. Endocrinol 2004, 18, 1643–1657. [Google Scholar]
- Perry, R.R.; Kang, Y.; Greaves, B.R. Relationship between tamoxifen-induced transforming growth factor β 1 expression, cytostasis and apoptosis in human breast cancer cells. Br. J. Cancer 1995, 72, 1441–1446. [Google Scholar]
- Yoo, Y.A.; Kim, Y.H.; Kim, J.S.; Seo, J.H. The functional implications of Akt activity and TGF-β signaling in tamoxifen-resistant breast cancer. Biochim. Biophys. Acta 2008, 1783, 438–447. [Google Scholar]
- Arteaga, C.L.; Koli, K.M.; Dugger, T.C.; Clarke, R. Reversal of tamoxifen resistance of human breast carcinomas in vivo by neutralizing antibodies to transforming growth factor-β. J. Natl. Cancer Inst 1999, 91, 46–53. [Google Scholar]
- Foekens, J.A.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; de Weerd, V.; Boersma, A.W.; Klijn, J.G.; Wiemer, E.A.; Martens, J.W. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 13021–13026. [Google Scholar]
- Masri, S.; Liu, Z.; Phung, S.; Wang, E.; Yuan, Y.C.; Chen, S. The role of microRNA-128a in regulating TGFβ signaling in letrozole-resistant breast cancer cells. Breast Cancer Res. Treat 2010, 124, 89–99. [Google Scholar]
- Sommer, S.; Fuqua, S.A. Estrogen receptor and breast cancer. Semin. Cancer Biol 2001, 11, 339–352. [Google Scholar]
- Smith, C.L.; Nawaz, Z.; O’Malley, B.W. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol. Endocrinol 1997, 11, 657–666. [Google Scholar]
- Jordan, V.C.; O’Malley, B.W. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol 2007, 25, 5815–5824. [Google Scholar]
- Lydon, J.P.; O’Malley, B.W. Minireview: Steroid receptor coactivator-3: A multifarious coregulator in mammary gland metastasis. Endocrinology 2011, 152, 19–25. [Google Scholar]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965–968. [Google Scholar]
- Murphy, L.C.; Simon, S.L.; Parkes, A.; Leygue, E.; Dotzlaw, H.; Snell, L.; Troup, S.; Adeyinka, A.; Watson, P.H. Altered expression of estrogen receptor coregulators during human breast tumorigenesis. Cancer Res 2000, 60, 6266–6271. [Google Scholar]
- List, H.J.; Reiter, R.; Singh, B.; Wellstein, A.; Riegel, A.T. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res. Treat 2001, 68, 21–28. [Google Scholar]
- Kressler, D.; Hock, M.B.; Kralli, A. Coactivators PGC-1β and SRC-1 interact functionally to promote the agonist activity of the selective estrogen receptor modulator tamoxifen. J. Biol. Chem 2007, 282, 26897–26907. [Google Scholar]
- Harigopal, M.; Heymann, J.; Ghosh, S.; Anagnostou, V.; Camp, R.L.; Rimm, D.L. Estrogen receptor co-activator (AIB1) protein expression by automated quantitative analysis (AQUA) in a breast cancer tissue microarray and association with patient outcome. Breast Cancer Res. Treat 2009, 115, 77–85. [Google Scholar]
- McIlroy, M.; Fleming, F.J.; Buggy, Y.; Hill, A.D.; Young, L.S. Tamoxifen-induced ER-α-SRC-3 interaction in HER2 positive human breast cancer; a possible mechanism for ER isoform specific recurrence. Endocr. Relat. Cancer 2006, 13, 1135–1145. [Google Scholar]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; An, P.; Kim, H.J.; Huang, J.; Clarke, R.; Osborne, C.K.; Inman, J.K.; Appella, E.; et al. Disruption of estrogen receptor DNA-binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell 2006, 10, 487–499. [Google Scholar]
- Kirkegaard, T.; McGlynn, L.M.; Campbell, F.M.; Muller, S.; Tovey, S.M.; Dunne, B.; Nielsen, K.V.; Cooke, T.G.; Bartlett, J.M. Amplified in breast cancer 1 in human epidermal growth factor receptor-positive tumors of tamoxifen-treated breast cancer patients. Clin. Cancer Res 2007, 13, 1405–1411. [Google Scholar]
- Haugan Moi, L.L.; Hauglid Flageng, M.; Gandini, S.; Guerrieri-Gonzaga, A.; Bonanni, B.; Lazzeroni, M.; Gjerde, J.; Lien, E.A.; DeCensi, A.; Mellgren, G. Effect of low-dose tamoxifen on steroid receptor coactivator 3/amplified in breast cancer 1 in normal and malignant human breast tissue. Clin. Cancer Res 2010, 16, 2176–2186. [Google Scholar]
- Redmond, A.M.; Bane, F.T.; Stafford, A.T.; McIlroy, M.; Dillon, M.F.; Crotty, T.B.; Hill, A.D.; Young, L.S. Coassociation of estrogen receptor and p160 proteins predicts resistance to endocrine treatment; SRC-1 is an independent predictor of breast cancer recurrence. Clin. Cancer Res 2009, 15, 2098–2106. [Google Scholar]
- Haugan Moi, L.L.; Flageng, M.H.; Gjerde, J.; Madsen, A.; Rost, T.H.; Gudbrandsen, O.A.; Lien, E.A.; Mellgren, G. Steroid receptor coactivators, HER-2 and HER-3 expression is stimulated by tamoxifen treatment in DMBA-induced breast cancer. BMC Cancer 2012, 12, 247. [Google Scholar]
- Bates, N.P.; Hurst, H.C. An intron 1 enhancer element mediates oestrogen-induced suppression of ERBB2 expression. Oncogene 1997, 15, 473–481. [Google Scholar]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res 2004, 64, 1522–1533. [Google Scholar]
- Hurtado, A.; Holmes, K.A.; Geistlinger, T.R.; Hutcheson, I.R.; Nicholson, R.I.; Brown, M.; Jiang, J.; Howat, W.J.; Ali, S.; Carroll, J.S. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 2008, 456, 663–666. [Google Scholar]
- McCartan, D.; Bolger, J.C.; Fagan, A.; Byrne, C.; Hao, Y.; Qin, L.; McIlroy, M.; Xu, J.; Hill, A.D.; Gaora, P.O.; et al. Global characterization of the SRC-1 transcriptome identifies ADAM22 as an ER-independent mediator of endocrine-resistant breast cancer. Cancer Res 2012, 72, 220–229. [Google Scholar]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in cancer formation and progression. Clin. Cancer Res 2009, 15, 1140–1144. [Google Scholar]
- Flageng, M.H.; Moi, L.L.; Dixon, J.M.; Geisler, J.; Lien, E.A.; Miller, W.R.; Lonning, P.E.; Mellgren, G. Nuclear receptor co-activators and HER-2/neu are upregulated in breast cancer patients during neo-adjuvant treatment with aromatase inhibitors. Br. J. Cancer 2009, 101, 1253–1260. [Google Scholar]
- O’Hara, J.; Vareslija, D.; McBryan, J.; Bane, F.; Tibbitts, P.; Byrne, C.; Conroy, R.M.; Hao, Y.; Gaora, P.O.; Hill, A.D.; et al. AIB1:ERα transcriptional activity is selectively enhanced in aromatase inhibitor-resistant breast cancer cells. Clin. Cancer Res 2012, 18, 3305–3315. [Google Scholar]
- McBryan, J.; Theissen, S.M.; Byrne, C.; Hughes, E.; Cocchiglia, S.; Sande, S.; O’Hara, J.; Tibbitts, P.; Hill, A.D.; Young, L.S. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res 2012, 72, 548–559. [Google Scholar]
- Cottone, E.; Orso, F.; Biglia, N.; Sismondi, P.; de Bortoli, M. Role of coactivators and corepressors in steroid and nuclear receptor signaling: Potential markers of tumor growth and drug sensitivity. Int. J. Biol. Markers 2001, 16, 151–166. [Google Scholar]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bieche, I. Expression analysis of estrogen receptor α coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res 2003, 9, 1259–1266. [Google Scholar]
- Lavinsky, R.M.; Jepsen, K.; Heinzel, T.; Torchia, J.; Mullen, T.M.; Schiff, R.; Del-Rio, A.L.; Ricote, M.; Ngo, S.; Gemsch, J.; et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 2920–2925. [Google Scholar]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res 2000, 60, 5887–5894. [Google Scholar]
- Kurebayashi, J.; Otsuki, T.; Kunisue, H.; Tanaka, K.; Yamamoto, S.; Sonoo, H. Expression levels of estrogen receptor-α, estrogen receptor-β, coactivators, and corepressors in breast cancer. Clin. Cancer Res 2000, 6, 512–518. [Google Scholar]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell 2002, 110, 55–67. [Google Scholar]
- Cutrupi, S.; Reineri, S.; Panetto, A.; Grosso, E.; Caizzi, L.; Ricci, L.; Friard, O.; Agati, S.; Scatolini, M.; Chiorino, G.; et al. Targeting of the adaptor protein Tab2 as a novel approach to revert tamoxifen resistance in breast cancer cells. Oncogene 2012, 31, 4353–4361. [Google Scholar]
- Zhu, P.; Baek, S.H.; Bourk, E.M.; Ohgi, K.A.; Garcia-Bassets, I.; Sanjo, H.; Akira, S.; Kotol, P.F.; Glass, C.K.; Rosenfeld, M.G.; et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006, 124, 615–629. [Google Scholar]
- Weinshilboum, R. Inheritance and drug response. N. Engl. J. Med 2003, 348, 529–537. [Google Scholar]
- Desta, Z.; Ward, B.A.; Soukhova, N.V.; Flockhart, D.A. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. J. Pharmacol. Exp. Ther 2004, 310, 1062–1075. [Google Scholar]
- Kiyotani, K.; Mushiroda, T.; Nakamura, Y.; Zembutsu, H. Pharmacogenomics of tamoxifen: roles of drug metabolizing enzymes and transporters. Drug Metab. Pharmacokinet 2012, 27, 122–131. [Google Scholar]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar]
- Jin, Y.; Desta, Z.; Stearns, V.; Ward, B.; Ho, H.; Lee, K.H.; Skaar, T.; Storniolo, A.M.; Li, L.; Araba, A.; et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst 2005, 97, 30–39. [Google Scholar]
- Hoskins, J.M.; Carey, L.A.; McLeod, H.L. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat. Rev. Cancer 2009, 9, 576–586. [Google Scholar]
- Gaedigk, A.; Simon, S.D.; Pearce, R.E.; Bradford, L.D.; Kennedy, M.J.; Leeder, J.S. The CYP2D6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther 2008, 83, 234–242. [Google Scholar]
- O’Donnell, P.H.; Ratain, M.J. Germline pharmacogenomics in oncology: Decoding the patient for targeting therapy. Mol. Oncol 2012, 6, 251–259. [Google Scholar]
- Nowell, S.; Sweeney, C.; Winters, M.; Stone, A.; Lang, N.P.; Hutchins, L.F.; Kadlubar, F.F.; Ambrosone, C.B. Association between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapy. J. Natl. Cancer Inst 2002, 94, 1635–1640. [Google Scholar]
- Nowell, S.A.; Ahn, J.; Rae, J.M.; Scheys, J.O.; Trovato, A.; Sweeney, C.; MacLeod, S.L.; Kadlubar, F.F.; Ambrosone, C.B. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Res. Treat 2005, 91, 249–258. [Google Scholar]
- Tengstrom, M.; Mannermaa, A.; Kosma, V.M.; Hirvonen, A.; Kataja, V. SULT1A1 rs9282861 polymorphism-a potential modifier of efficacy of the systemic adjuvant therapy in breast cancer? BMC Cancer 2012, 12, 257. [Google Scholar]
- Chen, L.; Waxman, D.J. Cytochrome P450 gene-directed enzyme prodrug therapy (GDEPT) for cancer. Curr. Pharm. Des 2002, 8, 1405–1416. [Google Scholar]
- Jounaidi, Y. Cytochrome P450-based gene therapy for cancer treatment: From concept to the clinic. Curr. Drug Metab 2002, 3, 609–622. [Google Scholar]
- Braybrooke, J.P.; Slade, A.; Deplanque, G.; Harrop, R.; Madhusudan, S.; Forster, M.D.; Gibson, R.; Makris, A.; Talbot, D.C.; Steiner, J.; et al. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin. Cancer Res 2005, 11, 1512–1520. [Google Scholar]
- Miyoshi, Y.; Ando, A.; Takamura, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by CYP3A4 mRNA expression in breast cancer tissues. Int. J. Cancer 2002, 97, 129–132. [Google Scholar]
- Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Tamaki, Y.; Noguchi, S. Prediction of response to docetaxel by immunohistochemical analysis of CYP3A4 expression in human breast cancers. Breast Cancer 2005, 12, 11–15. [Google Scholar]
- Dhaini, H.R.; Thomas, D.G.; Giordano, T.J.; Johnson, T.D.; Biermann, J.S.; Leu, K.; Hollenberg, P.F.; Baker, L.H. Cytochrome P450 CYP3A4/5 expression as a biomarker of outcome in osteosarcoma. J. Clin. Oncol 2003, 21, 2481–2485. [Google Scholar]
- Schiavon, G.; Tonini, G. Hormone-biological therapy in breast cancer: Preclinical evidences, clinical studies and future directions. Curr. Cancer Drug Targets 2010, 10, 3–18. [Google Scholar]
- Mokbel, K. The evolving role of aromatase inhibitors in breast cancer. Int. J. Clin. Oncol 2002, 7, 279–283. [Google Scholar]
- Bareschino, M.A.; Schettino, C.; Colantuoni, G.; Rossi, E.; Rossi, A.; Maione, P.; Ciardiello, F.; Gridelli, C. The role of antiangiogenetic agents in the treatment of breast cancer. Curr. Med. Chem 18, 5022–5032.
- Coxon, A.; Bush, T.; Saffran, D.; Kaufman, S.; Belmontes, B.; Rex, K.; Hughes, P.; Caenepeel, S.; Rottman, J.B.; Tasker, A.; et al. Broad antitumor activity in breast cancer xenografts by motesanib, a highly selective, oral inhibitor of vascular endothelial growth factor, platelet-derived growth factor, and Kit receptors. Clin. Cancer Res 2009, 15, 110–118. [Google Scholar]
- Sabnis, G.; Schayowitz, A.; Goloubeva, O.; Macedo, L.; Brodie, A. Trastuzumab reverses letrozole resistance and amplifies the sensitivity of breast cancer cells to estrogen. Cancer Res 2009, 69, 1416–1428. [Google Scholar]
- Leary, A.F.; Drury, S.; Detre, S.; Pancholi, S.; Lykkesfeldt, A.E.; Martin, L.A.; Dowsett, M.; Johnston, S.R. Lapatinib restores hormone sensitivity with differential effects on estrogen receptor signaling in cell models of human epidermal growth factor receptor 2-negative breast cancer with acquired endocrine resistance. Clin. Cancer Res 2010, 16, 1486–1497. [Google Scholar]
- Emde, A.; Mahlknecht, G.; Maslak, K.; Ribba, B.; Sela, M.; Possinger, K.; Yarden, Y. Simultaneous inhibition of estrogen receptor and the HER2 pathway in breast cancer: Effects of HER2 abundance. Transl. Oncol 2011, 4, 293–300. [Google Scholar]
- Buzdar, A.U. Role of biologic therapy and chemotherapy in hormone receptor- and HER2-positive breast cancer. Ann. Oncol 2009, 20, 993–999. [Google Scholar]


{kind=link}
{kind=link}
| Overexpression | Underexpression | Reference |
|---|---|---|
| miR-221 | miR-342 | [176,177] |
| miR-222 | miR-489 | [176] |
| miR-181 | miR-21 | [176,178] |
| miR-375 | miR-24 | [176] |
| miR-171 | miR-27 | [176] |
| miR-213 | miR-23 | [176,177] |
| miR-203 | miR-200 | [176] |
| miR-321 | miR-451 | [176,179] |
| miR-1308 | miR-1180 | [177] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
García-Becerra, R.; Santos, N.; Díaz, L.; Camacho, J. Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance. Int. J. Mol. Sci. 2013, 14, 108-145. https://doi.org/10.3390/ijms14010108
García-Becerra R, Santos N, Díaz L, Camacho J. Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance. International Journal of Molecular Sciences. 2013; 14(1):108-145. https://doi.org/10.3390/ijms14010108
Chicago/Turabian StyleGarcía-Becerra, Rocío, Nancy Santos, Lorenza Díaz, and Javier Camacho. 2013. "Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance" International Journal of Molecular Sciences 14, no. 1: 108-145. https://doi.org/10.3390/ijms14010108