Molecular Dynamic Simulation Insights into the Normal State and Restoration of p53 Function

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Advance in Molecular Simulations
2.1. Molecular Docking, MD Simulation and Binding Free Energy Prediction
2.2. Analysis of p53-Protein Interaction
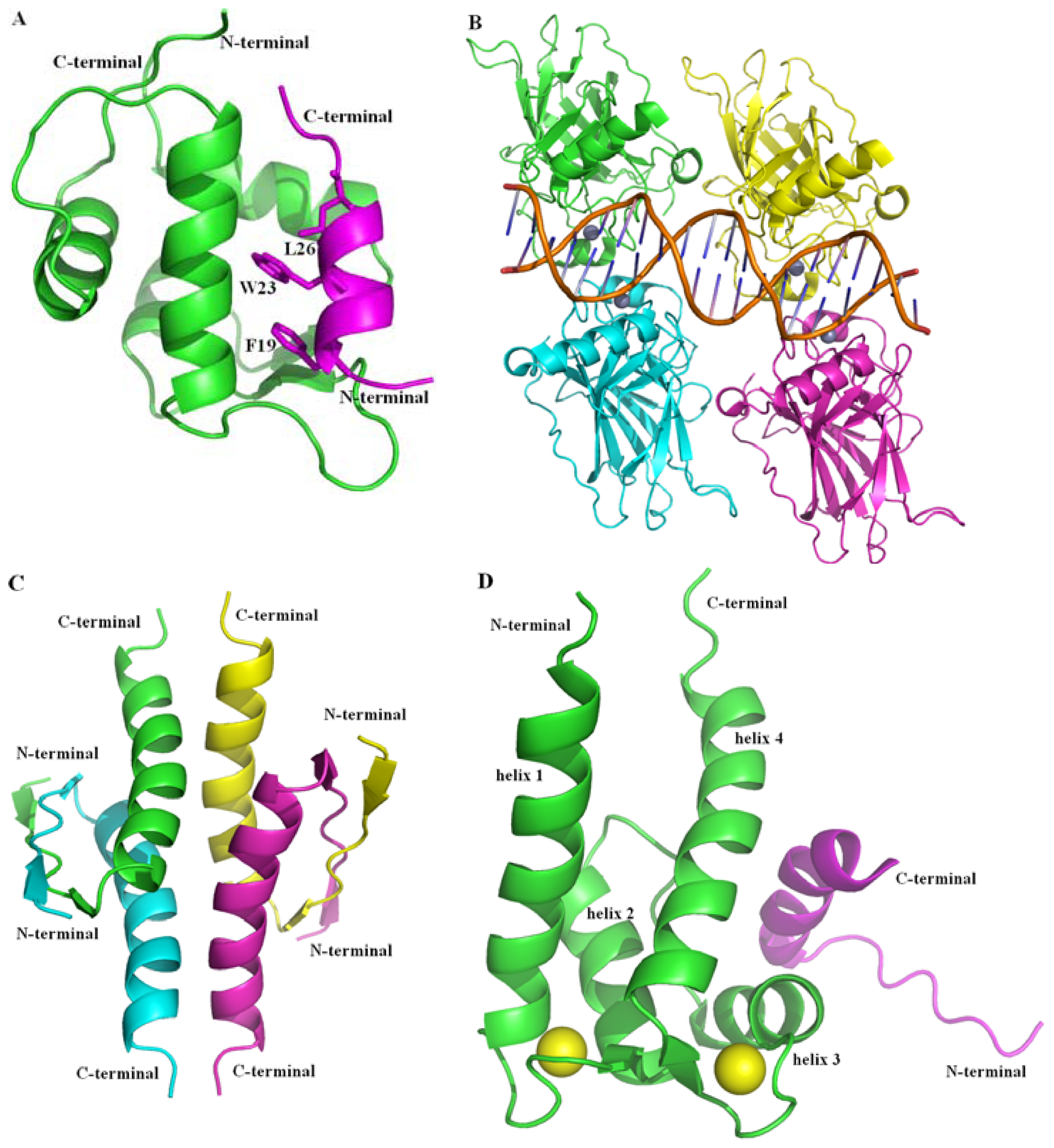
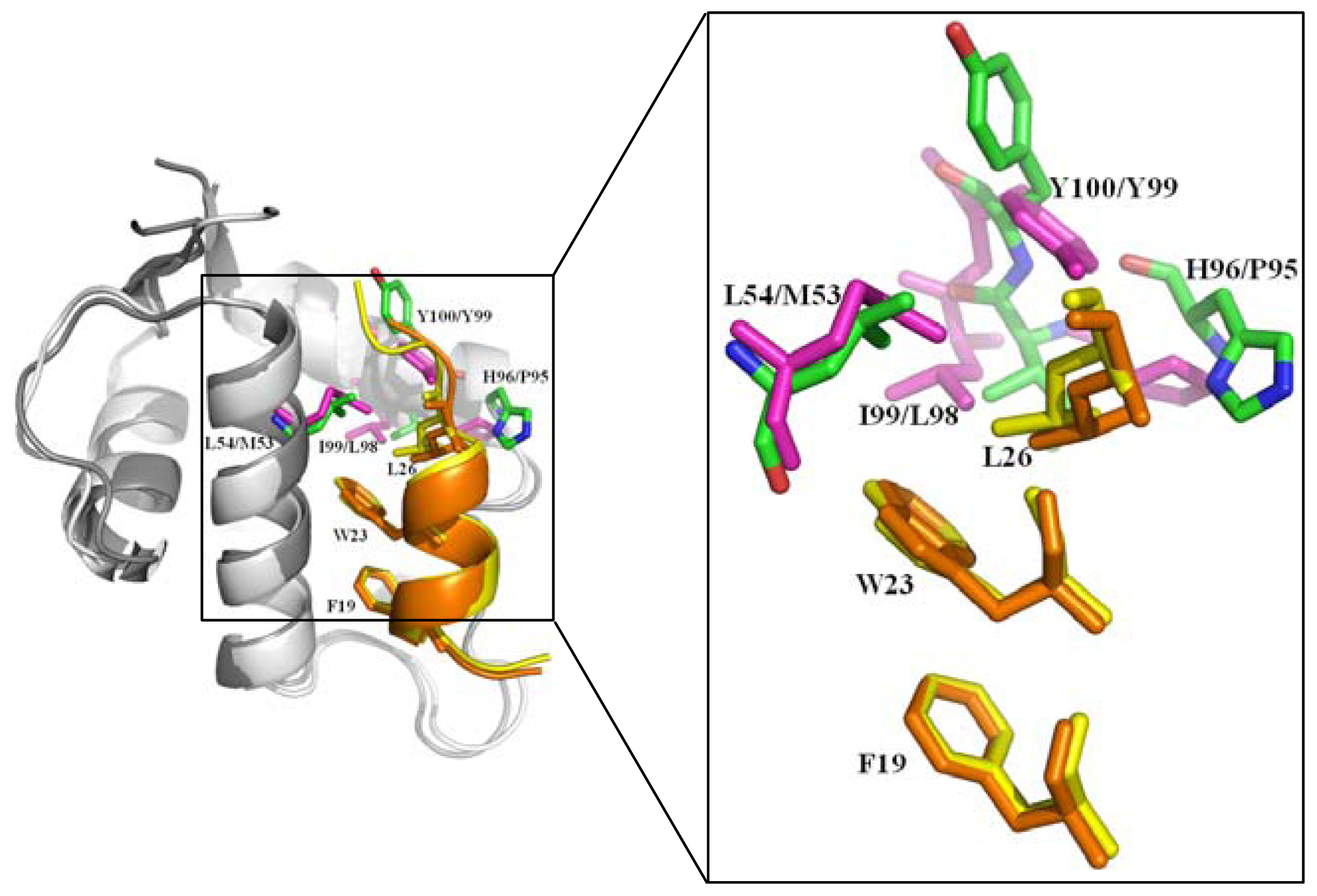
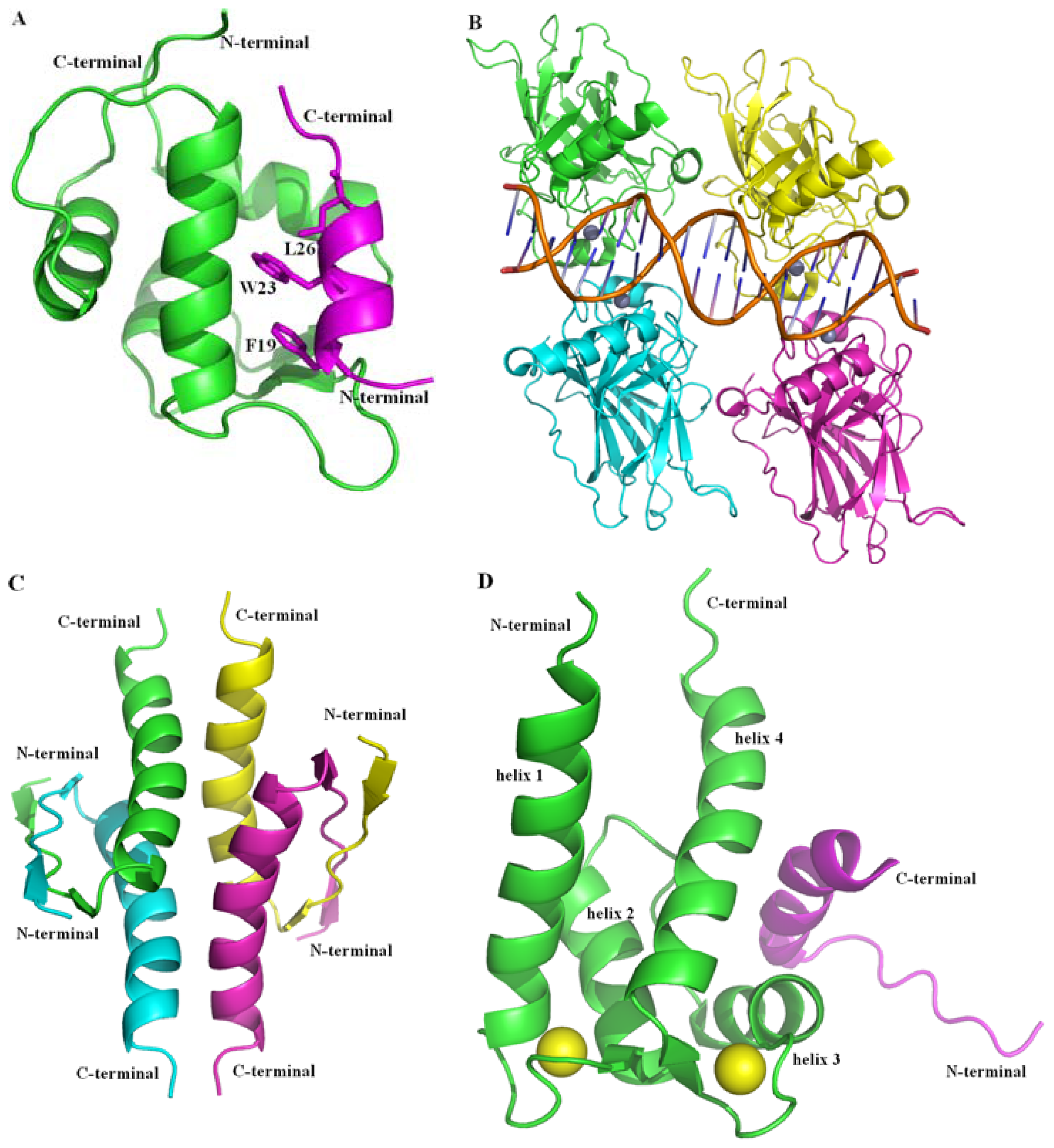
2.2.1. Protein-Protein Interaction Located in TAD of p53
2.2.2. Protein-Protein Interactions Located in DBD and OD of p53
2.2.3. Protein-Protein Interactions Located in CTD of p53
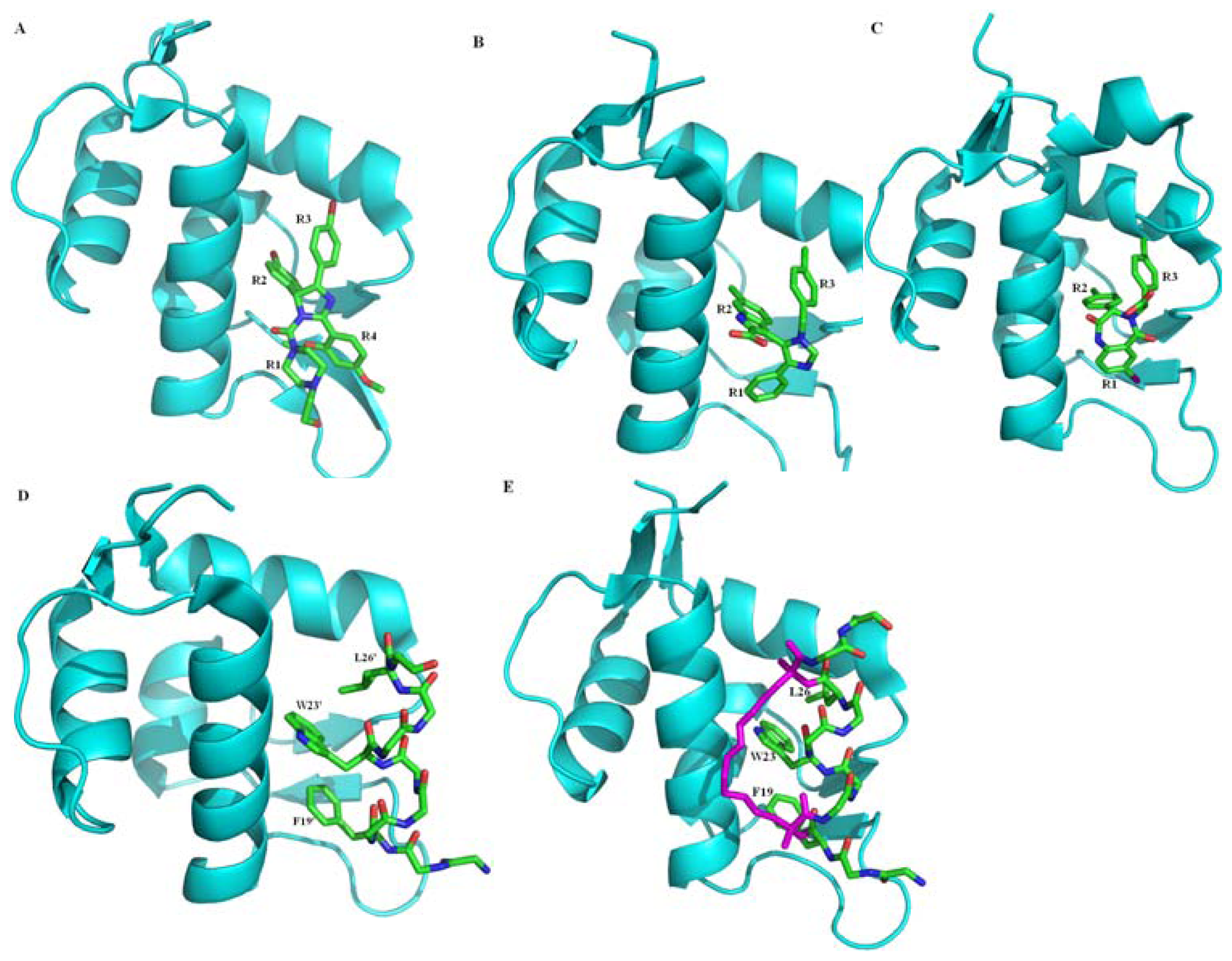
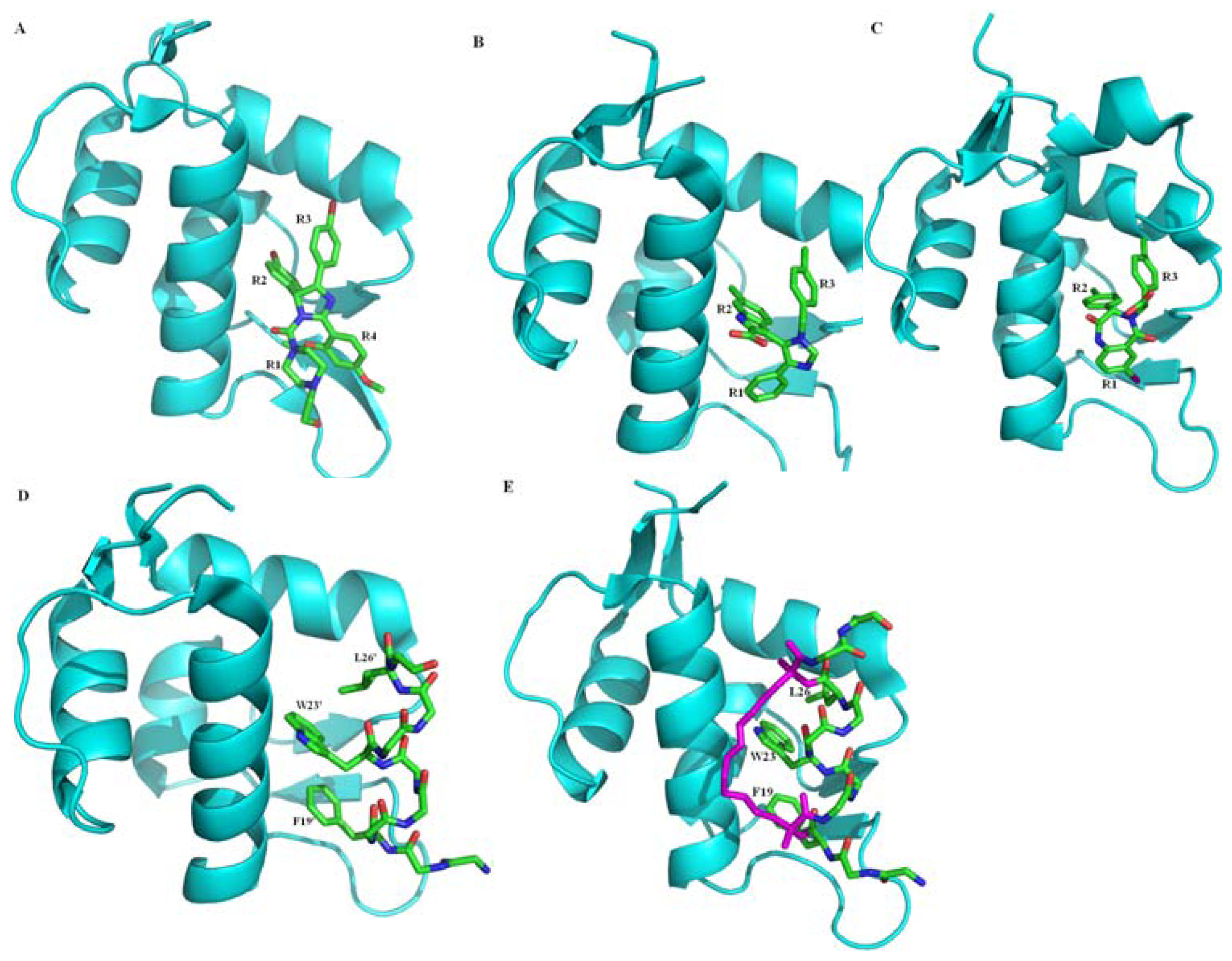
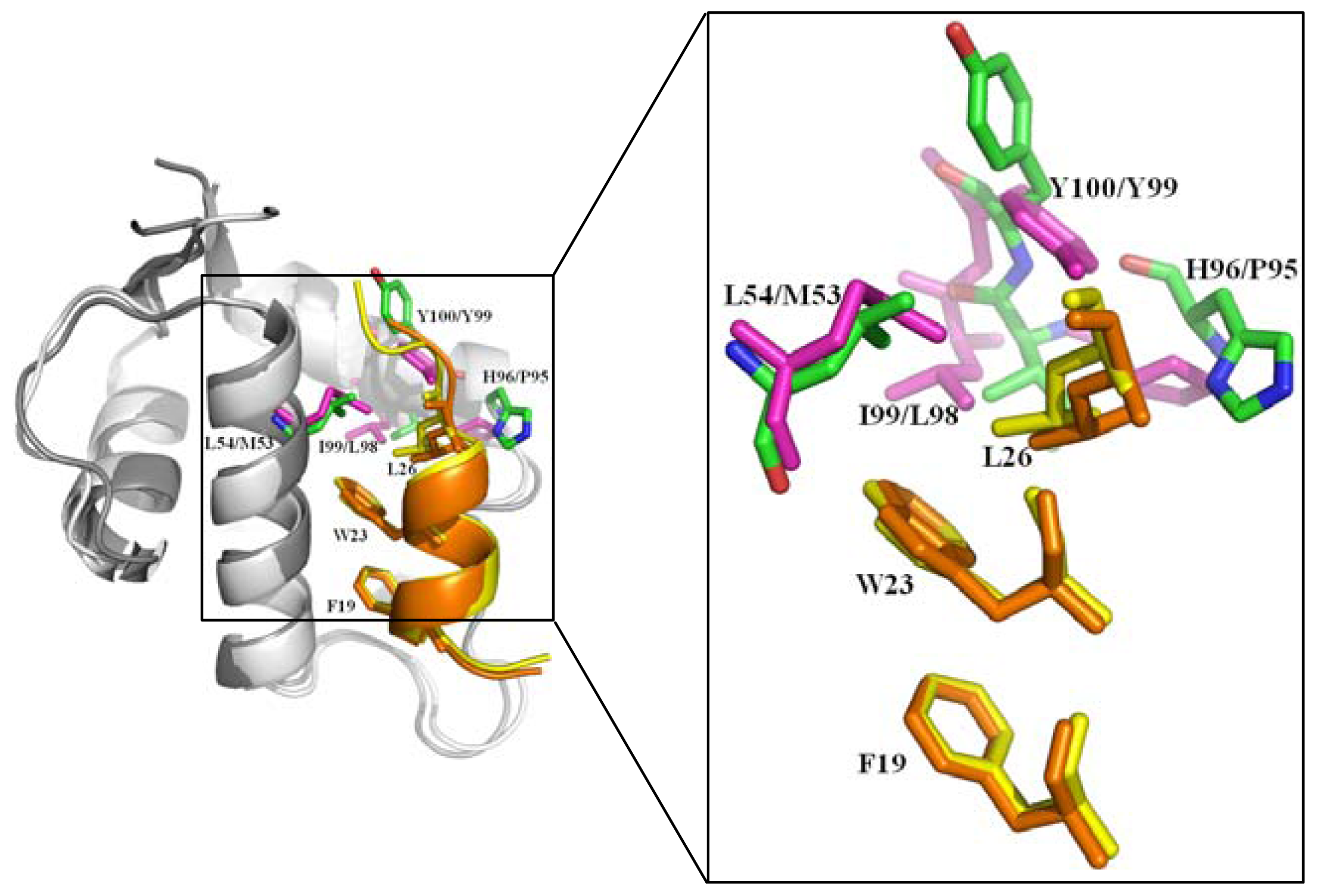
2.3. Small Molecule and Peptide Inhibitors Computational Modeling and Design
3. Outlook
Acknowledgments
References
- DeLeo, A.B.; Jay, G.; Appella, E.; Dubois, G.C.; Law, L.W.; Old, L.J. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 2420–2424. [Google Scholar]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar]
- Teodoro, J.G.; Evans, S.K.; Green, M.R. Inhibition of tumor angiogenesis by p53: A new role for the guardian of the genome. J. Mol. Med. (Berl.) 2007, 85, 1175–1186. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol 2007, 8, 275–283. [Google Scholar]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov 2008, 7, 979–987. [Google Scholar]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991, 51, 6304–6311. [Google Scholar]
- Pustisek, N.; Situm, M. UV-radiation, apoptosis and skin. Coll. Antropol 2011, 35, 339–341. [Google Scholar]
- Campbell, H.G.; Mehta, R.; Neumann, A.A.; Rubio, C.; Baird, M.; Slatter, T.L.; Braithwaite, A.W. Activation of p53 following ionizing radiation, but not other stressors, is dependent on the proline-rich domain (PRD). Oncogene 2012. [Google Scholar] [CrossRef]
- Pennica, D.; Goeddel, D.V.; Hayflick, J.S.; Reich, N.C.; Anderson, C.W.; Levine, A.J. The amino acid sequence of murine p53 determined from a c-DNA clone. Virology 1984, 134, 477–482. [Google Scholar]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev 1993, 7, 2556–2564. [Google Scholar]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol 2010, 17, 423–429. [Google Scholar]
- Chen, Y.; Dey, R.; Chen, L. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure 2010, 18, 246–256. [Google Scholar]
- Lee, W.; Harvey, T.S.; Yin, Y.; Yau, P.; Litchfield, D.; Arrowsmith, C.H. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Biol 1994, 1, 877–890. [Google Scholar]
- Bell, S.; Klein, C.; Müller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol 2002, 322, 917–927. [Google Scholar]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar]
- Rustandi, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol 2000, 7, 570–574. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.2r3pre; DeLano Scientific: San Carlos, CA, USA. Available online: http://pymol.org accessed on 1 January 2012.
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998, 12, 2831–2841. [Google Scholar]
- Warnock, L.J.; Knox, A.; Mee, T.R.; Raines, S.A.; Milner, J. Influence of tetramerisation on site-specific post-translational modifications of p53: Comparison of human and murine p53 tumor suppressor protein. Cancer Biol. Ther 2008, 7, 1481–1489. [Google Scholar]
- Scoumanne, A.; Chen, X. Protein methylation: A new mechanism of p53 tumor suppressor regulation. Histol. Histopathol 2008, 23, 1143–1149. [Google Scholar]
- Taira, N.; Yoshida, K. Post-translational modifications of p53 tumor suppressor: Determinants of its functional targets. Histol. Histopathol 2012, 27, 437–443. [Google Scholar]
- Murray, J.K.; Gellman, S.H. Targeting protein-protein interactions: Lessons from p53/MDM2. Biopolymers 2007, 88, 657–686. [Google Scholar]
- Tzakos, A.G.; Fokas, D.; Johannes, C.; Moussis, V.; Hatzimichael, E.; Briasoulis, E. Targeting oncogenic protein-protein interactions by diversity oriented synthesis and combinatorial chemistry approaches. Molecules 2011, 16, 4408–4427. [Google Scholar]
- Popowicz, G.M.; Czarna, A.; Holak, T.A. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 2008, 7, 2441–2443. [Google Scholar]
- Chipuk, J.E.; Bouchier-Hayes, L.; Kuwana, T.; Newmeyer, D.D.; Green, D.R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005, 309, 1732–1735. [Google Scholar]
- Jiang, P.; Du, W.; Heese, K.; Wu, M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol. Cell. Biol 2006, 26, 9071–9082. [Google Scholar]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; Thompson, C.B.; Fesik, S.W. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Chen, M.L.; Kimbara, K.; Majumdar, D.; Cunningham, E.; Gupta, T.K.D.; Chakrabarty, A.M. Bacterial redox protein Azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 14098–14103. [Google Scholar]
- Punj, V.; Gupta, T.K.D.; Chakrabarty, A.M. Bacterial cupredoxin azurin and its interactions with the tumor suppressor protein p53. Biochem. Biophys. Res. Commun 2003, 312, 109–114. [Google Scholar]
- Yamada, T.; Hiraoka, Y.; Ikehata, M.; Kimbara, K.; Avner, B.S.; Gupta, T.K.D.; Chakrabarty, A.M. Apoptosis or growth arrest: Modulation of tumor suppressor p53’s specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 2004, 101, 4770–4775. [Google Scholar]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; Bochkarev, A. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar]
- Di Lello, P.; Jenkins, L.M.; Jones, T.N.; Nguyen, B.D.; Hara, T.; Yamaguchi, H.; Dikeakos, J.D.; Appella, E.; Legault, P.; Omichinski, J.G. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar]
- Lilyestrom, W.; Klein, M.G.; Zhang, R.; Joachimiak, A.; Chen, X.S. Crystal structure of SV40 large T-antigen bound to p53: Interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev 2006, 20, 2373–2382. [Google Scholar]
- Ahn, J.; Prives, C. The C-terminus of p53: The more you learn the less you know. Nat. Struct. Biol 2001, 8, 730–732. [Google Scholar]
- Avalos, J.L.; Celic, I.; Muhammad, S.; Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol. Cell 2002, 10, 523–535. [Google Scholar]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; Barlev, N.A.; Reinberg, D. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–560. [Google Scholar]
- Lowe, E.D.; Tews, I.; Cheng, K.Y.; Brown, N.R.; Gul, S.; Noble, M.E.; Gamblin, S.J.; Johnson, L.N. Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry 2002, 41, 15625–15634. [Google Scholar]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res 1994, 54, 4855–4878. [Google Scholar]
- Pfeifer, G.P.; Besaratinia, A. Mutational spectra of human cancer. Hum. Genet 2009, 125, 493–506. [Google Scholar]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol 2011, 223, 116–126. [Google Scholar]
- Popowicz, G.M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Dömling, A.; Holak, T.A. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar]
- Dickens, M.P.; Fitzgerald, R.; Fischer, P.M. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin. Cancer Biol 2010, 20, 10–18. [Google Scholar]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; Lu, W. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar]
- Phan, J.; Li, Z.; Kasprzak, A.; Li, B.; Sebti, S.; Guida, W.; Schönbrunn, E.; Chen, J. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem 2010, 285, 2174–2183. [Google Scholar]
- Li, C.; Pazgier, M.; Li, C.; Yuan, W.; Liu, M.; Wei, G.; Lu, W.Y.; Lu, W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J. Mol. Biol 2010, 398, 200–213. [Google Scholar]
- Dudkina, A.S.; Lindsley, C.W. Small molecule protein-protein inhibitors for the p53-MDM2 interaction. Curr. Top. Med. Chem 2007, 7, 952–960. [Google Scholar]
- Zhan, C.; Lu, W. Peptide activators of the p53 tumor suppressor. Curr. Pharm. Des 2011, 17, 603–609. [Google Scholar]
- Hu, C.Q.; Hu, Y.Z. Small molecule inhibitors of the p53-MDM2. Curr. Med. Chem 2008, 15, 1720–1730. [Google Scholar]
- Patel, S.; Player, M.R. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin. Investig. Drugs 2008, 17, 1865–1882. [Google Scholar]
- Riedinger, C.; McDonnell, J.M. Inhibitors of MDM2 and MDMX: A structural perspective. Future Med. Chem 2009, 1, 1075–1094. [Google Scholar]
- Millard, M.; Pathania, D.; Grande, F.; Xu, S.; Neamati, N. Small-molecule inhibitors of p53-MDM2 interaction: The 2006–2010 update. Curr. Pharm. Des 2011, 17, 536–559. [Google Scholar]
- Vu, B.T.; Vassilev, L. Small-molecule inhibitors of the p53-MDM2 interaction. Curr. Top. Microbiol. Immunol 2011, 348, 151–172. [Google Scholar]
- Popowicz, G.M.; Dömling, A.; Holak, T.A. The structure-based design of Mdm2/Mdmx-p53 inhibitors gets serious. Angew. Chem. Int. Ed. Engl 2011, 50, 2680–2688. [Google Scholar]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol 2010, 2. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; Fotouhi, N.; Liu, E.A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem 2005, 48, 909–912. [Google Scholar]
- Baek, S.; Kutchukian, P.S.; Verdine, G.L.; Huber, R.; Holak, T.A.; Lee, K.W.; Popowicz, G.M. Structure of the stapled p53 peptide bound to Mdm2. J. Am. Chem. Soc 2012, 134, 103–106. [Google Scholar]
- Rustandi, R.R.; Drohat, A.C.; Baldisseri, D.M.; Wilder, P.T.; Weber, D.J. The Ca(2+)-dependent interaction of S100B(betabeta) with a peptide derived from p53. Biochemistry 1998, 37, 1951–1960. [Google Scholar]
- Delphin, C.; Ronjat, M.; Deloulme, J.C.; Garin, G.; Debussche, L.; Higashimoto, Y.; Sakaguchi, K.; Baudier, J. Calcium-dependent interaction of S100B with the C-terminal domain of the tumor suppressor p53. J. Biol. Chem 1999, 274, 10539–10544. [Google Scholar]
- Markowitz, J.; MacKerell, A.D., Jr; Weber, D.J. A search for inhibitors of S100B, a member of the S100 family of calcium-binding proteins. Mini Rev. Med. Chem. 2007, 7, 609–616. [Google Scholar]
- Chen, J.; Zhang, S.; Liu, X.; Zhang, Q. Insights into drug resistance of mutations D30N and I50V to HIV-1 protease inhibitor TMC-114: Free energy calculation and molecular dynamic simulation. J. Mol. Model 2010, 16, 459–468. [Google Scholar]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar]
- Lane, D.P.; Hall, P.A. MDM2-arbiter of p53’s destruction. Trends Biochem. Sci 1997, 22, 372–374. [Google Scholar]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res 2003, 1, 1001–1008. [Google Scholar]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; Jochemsen, A.G. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J 1996, 15, 5349–5357. [Google Scholar]
- Mancini, F.; Moretti, F. Mitochondrial MDM4 (MDMX): An unpredicted role in the p53-mediated intrinsic apoptotic pathway. Cell Cycle 2009, 8, 3854–3859. [Google Scholar]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci 2011, 32, 53–62. [Google Scholar]
- Lee, S.H.; Park, B.J. p53 activation by blocking Snail: A novel pharmacological strategy for cancer. Curr. Pharm. Des 2011, 17, 610–617. [Google Scholar]
- Kalid, O.; Ben-Tal, N. Study of MDM2 binding to p53-analogues: Affinity, helicity, and applicability to drug design. J. Chem. Inf. Model 2009, 49, 865–876. [Google Scholar]
- Espinoza-Fonseca, L.M. Leucine-rich hydrophobic clusters promote folding of the N-terminus of the intrinsically disordered transactivation domain of p53. FEBS Lett 2009, 583, 556–560. [Google Scholar]
- Dastidar, S.G.; Lane, D.P.; Verma, C.S. Multiple peptide conformations give rise to similar binding affinities: Molecular simulations of p53-MDM2. J. Am. Chem. Soc 2008, 130, 13514–13515. [Google Scholar]
- Macchiarulo, A.; Giacchè, N.; Carotti, A.; Baroni, M.; Cruciani, G.; Pellicciari, R. Targeting the conformational transitions of MDM2 and MDMX: Insights into dissimilarities and similarities of p53 recognition. J. Chem. Inf. Model 2008, 48, 1999–2009. [Google Scholar]
- Lauria, A.; Tutone, M.; Ippolito, M.; Pantano, L.; Almerico, A.M. Molecular modeling approaches in the discovery of new drugs for anti-cancer therapy: The investigation of p53-MDM2 interaction and its inhibition by small molecules. Curr. Med. Chem 2010, 17, 3142–3154. [Google Scholar]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Conformational changes of the p53-binding cleft of MDM2 revealed by molecular dynamics simulations. Biopolymers 2006, 83, 365–373. [Google Scholar]
- Terakawa, T.; Takada, S. Multiscale ensemble modeling of intrinsically disordered proteins: P53 N-terminal domain. Biophys. J 2011, 101, 1450–1458. [Google Scholar]
- Xiong, K.; Zwier, M.C.; Myshakina, N.S.; Burger, V.M.; Asher, S.A.; Chong, L.T. Direct observations of conformational distributions of intrinsically disordered p53 peptides using UV Raman and explicit solvent simulations. J. Phys. Chem. A 2011, 115, 9520–9527. [Google Scholar]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Transient stability of the helical pattern of region F19-L22 of the N-terminal domain of p53: A molecular dynamics simulation study. Biochem. Biophys. Res. Commun. 2006, 343, 110–116. [Google Scholar]
- Mavinahalli, J.N.; Madhumalar, A.; Beuerman, R.W.; Lane, D.P.; Verma, C. Differences in the transactivation domains of p53 family members: A computational study. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Lai, Z.; Auger, K.R.; Manubay, C.M.; Copeland, R.A. Thermodynamics of p53 binding to hdm2(1–126): Effects of phosphorylation and p53 peptide length. Arch. Biochem. Biophys 2000, 381, 278–284. [Google Scholar]
- Lee, H.J.; Srinivasan, D.; Coomber, D.; Lane, D.P.; Verma, C.S. Modulation of the p53-MDM2 interaction by phosphorylation of Thr18: A computational study. Cell Cycle 2007, 6, 2604–2611. [Google Scholar]
- Brown, C.J.; Srinivasan, D.; Jun, L.H.; Coomber, D.; Verma, C.S.; Lane, D.P. The electrostatic surface of MDM2 modulates the specificity of its interaction with phosphorylated and unphosphorylated p53 peptides. Cell Cycle 2008, 7, 608–610. [Google Scholar]
- Dastidar, S.G.; Lane, D.P.; Verma, C.S. Modulation of p53 binding to MDM2: Computational studies reveal important roles of Tyr100. BMC Bioinforma 2009, 10. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Madhumalar, A.; Fuentes, G.; Lane, D.P.; Verma, C.S. Forces mediating protein-protein interactions: A computational study of p53 “approaching” MDM2. Theor. Chem. Acc 2010, 125, 621–635. [Google Scholar]
- Carotti, A.; Macchiarulo, A.; Giacchè, N.; Pellicciari, R. Targeting the conformational transitions of MDM2 and MDMX: Insights into key residues affecting p53 recognition. Proteins 2009, 77, 524–535. [Google Scholar]
- Brown, C.J.; Dastidar, S.G.; Quah, S.T.; Lim, A.; Chia, B.; Verma, C.S. C-terminal substitution of MDM2 interacting peptides modulates binding affinity by distinctive mechanisms. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Raghunathan, D.; Nicholson, J.; Hupp, T.R.; Lane, D.P.; Verma, C.S. Chemical states of the N-terminal “lid” of MDM2 regulate p53 binding: Simulations reveal complexities of modulation. Cell Cycle 2011, 10, 82–89. [Google Scholar]
- Bharatham, N.; Chi, S-W.; Yoon, H.S. Molecular Basis of Bcl-XL-p53 Interaction: Insights from Molecular Dynamics Simulations. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Modeling the interaction between the N-terminal domain of the tumor suppressor p53 and azurin. J. Mol. Recognit 2009, 22, 215–222. [Google Scholar]
- Duan, J.; Nilsson, L. Effect of Zn2+ on DNA recognition and stability of the p53 DNA-binding domain. Biochemistry 2006, 45, 7483–7492. [Google Scholar]
- Wang, T.; Shao, X.; Cai, W.; Xue, Y.; Wang, S.; Feng, X. Predicting the coordination geometry for Mg2+ in the p53 DNA-binding domain: Insights from computational studies. Phys. Chem. Chem. Phys 2011, 13, 1140–1151. [Google Scholar]
- De Grandis, V.; Bizzarri, A.R.; Cannistraro, S. Docking study and free energy simulation of the complex between p53 DNA-binding domain and azurin. J. Mol. Recognit 2007, 20, 215–226. [Google Scholar]
- Santini, S.; Bizzarri, A.R.; Cannistraro, S. Modelling the interaction between the p53 DNA-binding domain and the p28 peptide fragment of Azurin. J. Mol. Recognit 2011, 24, 1043–1055. [Google Scholar]
- Chène, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar]
- Mittl, P.R.; Chène, P.; Grütter, M.G. Crystallization and structure solution of p53 (residues 326–356) by molecular replacement using an NMR model as template. Acta Crystallogr. D 1998, 54, 86–89. [Google Scholar]
- Chong, L.T.; Snow, C.D.; Rhee, Y.M.; Pande, V.S. Dimerization of the p53 oligomerization domain: Identification of a folding nucleus by molecular dynamics simulations. J. Mol. Biol 2005, 345, 869–878. [Google Scholar]
- Duan, J.; Nilsson, L. Thermal unfolding simulations of a multimeric protein-transition state and unfolding pathways. Proteins 2005, 59, 170–182. [Google Scholar]
- Galea, C.; Bowman, P.; Kriwacki, R.W. Disruption of an intermonomer salt bridge in the p53 tetramerization domain results in an increased propensity to form amyloid fibrils. Protein Sci 2005, 14, 2993–3003. [Google Scholar]
- Lubin, D.J.; Butler, J.S.; Loh, S.N. Folding of tetrameric p53: Oligomerization and tumorigenic mutations induce misfolding and loss of function. J. Mol. Biol 2010, 395, 705–716. [Google Scholar]
- Lwin, T.Z.; Durant, J.J.; Bashford, D. A fluid salt-bridging cluster and the stabilization of p53. J. Mol. Biol 2007, 373, 1334–1347. [Google Scholar]
- Baudier, J.; Delphin, C.; Grunwald, D.; Khochbin, S.; Lawrence, J.J. Characterization of the tumor suppressor protein p53 as a protein kinase C substrate and a S100b-binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 11627–11631. [Google Scholar]
- Fernandez-Fernandez, M.R.; Veprintsev, D.B.; Fersht, A.R. Proteins of the S100 family regulate the oligomerization of p53 tumor suppressor. Proc. Natl. Acad. Sci. USA 2005, 102, 4735–4740. [Google Scholar]
- Fernandez-Fernandez, M.R.; Rutherford, T.J.; Fersht, A.R. Members of the S100 family bind p53 in two distinct ways. Protein Sci 2008, 17, 1663–1670. [Google Scholar]
- Van Dieck, J.; Fernandez-Fernandez, M.R.; Veprintsev, D.B.; Fersht, A.R. Modulation of the oligomerization state of p53 by differential binding of proteins of the S100 family to p53 monomers and tetramers. J. Biol. Chem 2009, 284, 13804–13811. [Google Scholar]
- Rustandi, R.R.; Baldisseri, D.M.; Drohat, A.C.; Weber, D.J. Structural changes in the C-terminus of Ca2+-bound rat S100B (beta beta) upon binding to a peptide derived from the C-terminal regulatory domain of p53. Protein Sci 1999, 8, 1743–1751. [Google Scholar]
- Lin, J.; Blake, M.; Tang, C.; Zimmer, D.; Rustandi, R.R.; Weber, D.J.; Carrier, F. Inhibition of p53 transcriptional activity by the S100B calcium-binding protein. J. Biol. Chem 2001, 276, 35037–35041. [Google Scholar]
- Chen, J. Intrinsically disordered p53 extreme C-terminus binds to S100B(betabeta) through “fly-casting”. J. Am. Chem. Soc 2009, 131, 2088–2089. [Google Scholar]
- Wilder, P.T.; Lin, J.; Bair, C.L.; Charpentier, T.H.; Yang, D.; Liriano, M.; Varney, K.M.; Lee, A.; Oppenheim, A.B.; Adhya, S.; Carrier, F.; Weber, D.J. Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys. Acta 2006, 1763, 1284–1297. [Google Scholar]
- Drohat, A.C.; Baldisseri, D.M.; Rustandi, R.R.; Weber, D.J. Solution structure of calcium-bound rat S100B(betabeta) as determined by nuclear magnetic resonance spectroscopy. Biochemistry 1998, 37, 2729–2740. [Google Scholar]
- Deichmann, M.; Benner, A.; Bock, M.; Jäckel, A.; Uhl, K.; Waldmann, V.; Näher, H. S100-Beta, melanoma-inhibiting activity, and lactate dehydrogenase discriminate progressive from nonprogressive American Joint Committee on Cancer stage IV melanoma. J. Clin. Oncol 1999, 17, 1891–1896. [Google Scholar]
- Jäckel, A.; Deichmann, M.; Waldmann, V.; Bock, M.; Näher, H. S-100 beta protein in serum, a tumor marker in malignant melanoma-current state of knowledge and clinical experience. Hautarzt 1999, 50, 250–256. [Google Scholar]
- Harpio, R.; Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem 2004, 37, 512–518. [Google Scholar]
- Gieldon, A.; Mori, M.; Del Conte, R. Theoretical study on binding of S100B protein. J. Mol. Model 2007, 13, 1123–1131. [Google Scholar]
- Allen, W.J.; Capelluto, D.G.; Finkielstein, C.V.; Bevan, D.R. Modeling the relationship between the p53 C-terminal domain and its binding partners using molecular dynamics. J. Phys. Chem. B 2010, 114, 13201–13213. [Google Scholar]
- Pirolli, D.; Alinovi, C.C.; Capoluongo, E.; Satta, M.A.; Concolino, P.; Giardina, B.; de Rosa, M.C. Insight into a novel p53 single point mutation (G389E) by molecular dynamics simulations. Int. J. Mol. Sci 2010, 12, 128–140. [Google Scholar]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar]
- Scolnick, D.M.; Chehab, N.H.; Stavridi, E.S.; Lien, M.C.; Caruso, L.; Moran, E.; Berger, S.L.; Halazonetis, T.D. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res 1997, 57, 3693–3696. [Google Scholar]
- Livengood, J.A.; Scoggin, K.E.; Van Orden, K.; McBryant, S.J.; Edayathumangalam, R.S.; Laybourn, P.J.; Nyborg, J.K. p53 Transcriptional activity is mediated through the SRC1-interacting domain of CBP/p300. J. Biol. Chem 2002, 277, 9054–9061. [Google Scholar]
- Okoshi, R.; Ando, K.; Suenaga, Y.; Sang, M.; Kubo, N.; Kizaki, H.; Nakagawara, A.; Ozaki, T. Transcriptional regulation of tumor suppressor p53 by cAMP-responsive element-binding protein/AMP-activated protein kinase complex in response to glucose deprivation. Genes Cells 2009, 14, 1429–1440. [Google Scholar]
- Sachchidanand; Resnick-Silverman, L.; Yan, S.; Mutjaba, S.; Liu, W.J.; Zeng, L.; Manfredi, J.J.; Zhou, M.M. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006, 13, 81–90. [Google Scholar]
- Borah, J.C.; Mujtaba, S.; Karakikes, I.; Zeng, L.; Muller, M.; Patel, J.; Moshkina, N.; Morohashi, K.; Zhang, W.; Gerona-Navarro, G.; Hajjar, R.J.; Zhou, M.M. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol 2011, 18, 531–541. [Google Scholar]
- Gerona-Navarro, G.; Yoel-Rodríguez; Mujtaba, S.; Frasca, A.; Patel, J.; Zeng, L.; Plotnikov, A.N.; Osman, R.; Zhou, M.M. Rational design of cyclic peptide modulators of the transcriptional coactivator CBP: A new class of p53 inhibitors. J. Am. Chem. Soc. 2011, 133, 2040–2043. [Google Scholar]
- Eichenbaum, K.D.; Rodríguez, Y.; Mezei, M.; Osman, R. The energetics of the acetylation switch in p53-mediated transcriptional activation. Proteins 2010, 78, 447–456. [Google Scholar]
- Chandrasekaran, R.; Thompson, M. Polybromo-1-bromodomains bind histone H3 at specific acetyl-lysine positions. Biochem. Biophys. Res. Commun 2007, 355, 661–666. [Google Scholar]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins-novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov 2008, 7, 841–853. [Google Scholar]
- Milne, J.C.; Denu, J.M. The Sirtuin family: Therapeutic targets to treat diseases of aging. Curr. Opin. Chem. Biol 2008, 12, 11–17. [Google Scholar]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 2002, 21, 2383–2396. [Google Scholar]
- Smith, J. Human Sir2 and the ‘silencing’ of p53 activity. Trends Cell Biol 2002, 12, 404–406. [Google Scholar]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F.; Reinberg, D.; Barlev, N.A. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol 2007, 27, 6756–6769. [Google Scholar]
- Zhang, X.; Bruice, T.C. Mechanism of product specificity of AdoMet methylation catalyzed by lysine methyltransferases: Transcriptional factor p53 methylation by histone lysine methyltransferase SET7/9. Biochemistry 2008, 47, 2743–2748. [Google Scholar]
- Bai, Q.; Shen, Y.; Yao, X.; Wang, F.; Du, Y.; Wang, Q.; Jin, N.; Hai, J.; Hu, T.; Yang, J. Modeling a new water channel that allows SET9 to dimethylate p53. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Ding, Y.; Mei, Y.; Zhang, J.Z. Quantum mechanical studies of residue-specific hydrophobic interactions in p53-MDM2 binding. J. Phys. Chem. B 2008, 112, 11396–11401. [Google Scholar]
- Hu, G.; Wang, D.; Liu, X.; Zhang, Q. A computational analysis of the binding model of MDM2 with inhibitors. J. Comput. Aided Mol. Des 2010, 24, 687–697. [Google Scholar]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Wang, S. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem 2006, 49, 3432–3435. [Google Scholar]
- Chen, J.; Wang, J.; Xu, B.; Zhu, W.; Li, G. Insight into mechanism of small molecule inhibitors of the MDM2-p53 interaction: Molecular dynamics simulation and free energy analysis. J. Mol. Graph. Model 2011, 30, 46–53. [Google Scholar]
- Wang, F.; Li, Y.; Ma, Z.; Wang, X.; Wang, Y. Structural determinants of benzodiazepinedione/peptide-based p53-HDM2 inhibitors using 3D-QSAR, docking and molecular dynamics. J. Mol. Model 2012, 18, 295–306. [Google Scholar]
- Liu, Y.; Lane, D.P.; Verma, C.S. Systematic mutational analysis of an ubiquitin ligase (MDM2)-binding peptide: Computational studies. Theor. Chem. Acc 2011, 130, 1145–1154. [Google Scholar]
- Cheng, W.; Chen, J.; Liang, Z.; Li, G.; Yi, C.; Wang, W.; Wang, K. A computational analysis of interaction mechanisms of peptide and non-peptide inhibitors with MDMX based on molecular dynamics simulation. Comp. Theor. Chem 2012, 984, 43–50. [Google Scholar]
- Barakat, K.; Mane, J.; Friesen, D.; Tuszynski, J. Ensemble-based virtual screening reveals dual-inhibitors for the p53-MDM2/MDMX interactions. J. Mol. Graph. Model 2010, 28, 555–568. [Google Scholar]
- Joseph, T.L.; Madhumalar, A.; Brown, C.J.; Lane, D.P.; Verma, C.S. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle 2010, 9, 1167–1181. [Google Scholar]
- Lu, S.Y.; Jiang, Y.J.; Zou, J.W.; Wu, T.X. Molecular modeling and molecular dynamics simulation studies on pyrrolopyrimidine-based α-helix mimetic as dual inhibitors of MDM2 and MDMX. J. Mol. Graph. Model 2011, 30, 167–178. [Google Scholar]
- Chen, J.; Zhang, D.; Zhang, Y.; Li, G. Computational studies of difference in binding modes of peptide and non-peptide inhibitors to MDM2/MDMX based on molecular dynamics simulations. Int. J. Mol. Sci 2012, 13, 2176–2195. [Google Scholar]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc 2007, 129, 2456–2457. [Google Scholar]
- Madden, M.M.; Muppidi, A.; Li, Z.; Li, X.; Chen, J.; Lin, Q. Synthesis of cell-permeable stapled peptide dual inhibitors of the p53-Mdm2/Mdmx interactions via photoinduced cycloaddition. Bioorg Med. Chem. Lett 2011, 21, 1472–1475. [Google Scholar]
- Muppidi, A.; Li, X.; Chen, J.; Lin, Q. Conjugation of spermine enhances cellular uptake of the stapled peptide-based inhibitors of p53-Mdm2 interaction. Bioorg. Med. Chem. Lett 2011, 21, 7412–7415. [Google Scholar]
- Crunkhorn, S. Anticancer drugs: Stapled peptide rescues p53. Nat. Rev. Drug Discov 2011, 10. [Google Scholar] [CrossRef]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the alpha-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des 2010, 75, 348–359. [Google Scholar]
- Joseph, T.L.; Lane, D.; Verma, C.S. Stapled peptides in the p53 pathway: Computer simulations reveal novel interactions of the staples with the target protein. Cell Cycle 2010, 9, 4560–4568. [Google Scholar]
- Lin, J.; Yang, Q.; Yan, Z.; Markowitz, J.; Wilder, P.T.; Carrier, F.; Weber, D.J. Inhibiting S100B restores p53 levels in primary malignant melanoma cancer cells. J. Biol. Chem 2004, 279, 34071–34077. [Google Scholar]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The calcium-binding protein S100B down-regulates p53 and apoptosis in malignant melanoma. J. Biol. Chem 2010, 285, 27487–27498. [Google Scholar]
- Markowitz, J.; Chen, I.; Gitti, R.; Baldisseri, D.M.; Pan, Y.; Udan, R.; Carrier, F.; MacKerell, A.D., Jr; Weber, D.J. Identification and characterization of small molecule inhibitors of the calcium-dependent S100B-p53 tumor suppressor interaction. J. Med. Chem. 2004, 47, 5085–5093. [Google Scholar]
- Markowitz, J.; Mackerell, A.D., Jr; Carrier, F.; Charpentier, T.H.; Weber, D.J. Design of inhibitors for S100B. Curr. Top. Med. Chem. 2005, 5, 1093–1108. [Google Scholar]
- Wilder, P.T.; Charpentier, T.H.; Liriano, M.A.; Gianni, K.; Varney, K.M.; Pozharski, E.; Coop, A.; Toth, E.A.; Mackerell, A.D.; Weber, D.J. In vitro screening and structural characterization of inhibitors of the S100B-p53 interaction. Int. J. High Throughput Screen 2010, 2010, 109–126. [Google Scholar]
- Charpentier, T.H.; Wilder, P.T.; Liriano, M.A.; Varney, K.M.; Zhong, S.; Coop, A.; Pozharski, E.; MacKerell, A.D., Jr; Toth, E.A.; Weber, D.J. Small molecules bound to unique sites in the target protein binding cleft of calcium-bound S100B as characterized by nuclear magnetic resonance and X-ray crystallography. Biochemistry 2009, 48, 6202–6212. [Google Scholar]
- Whitlow, J.L.; Varughese, J.F.; Zhou, Z.; Bartolotti, L.J.; Li, Y. Computational screening and design of S100B ligand to block S100B-p53 interaction. J. Mol. Graph. Model 2009, 27, 969–977. [Google Scholar]
- Zhou, Z.; Li, Y. Molecular dynamics simulation of S100B protein to explore ligand blockage of the interaction with p53 protein. J. Comput. Aided Mol. Des 2009, 23, 705–714. [Google Scholar]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 1999, 447, 5–9. [Google Scholar]
- Wang, L.; He, G.; Zhang, P.; Wang, X.; Jiang, M.; Yu, L. Interplay between MDM2, MDMX, Pirh2 and COP1: The negative regulators of p53. Mol. Biol. Rep 2011, 38, 229–236. [Google Scholar]
- Van Dieck, J.; Lum, J.K.; Teufel, D.P.; Fersht, A.R. S100 proteins interact with the N-terminal domain of MDM2. FEBS Lett 2010, 584, 3269–3274. [Google Scholar]
- Wang, M.; Lei, Y. Delaying vascular aging with Chinese medicine: Implications from an overview of the p53 and miR-34s family. Chin. J. Integr. Med 2011, 17, 635–639. [Google Scholar]
- Zhang, X.; Zhao, M.; Chen, L.; Jiao, H.; Liu, H.; Wang, L.; Ma, S. A triterpenoid from Thalictrum fortunei induces apoptosis in BEL-7402 cells through the P53-induced apoptosis pathway. Molecules 2011, 16, 9505–9519. [Google Scholar]
- Zhang, F.; Xu, L.; Qu, X.; Zhao, M.; Jin, B.; Kang, J.; Liu, Y.; Hu, X. Synergistic antitumor effect of β-elemene and etoposide is mediated via induction of cell apoptosis and cell cycle arrest in non-small cell lung carcinoma cells. Mol. Med. Rep 2011, 4, 1189–1193. [Google Scholar]
- Wang, X.Y.; Wang, Y.G.; Wang, Y.F. Ginsenoside Rb1, Rg1 and three extracts of traditional Chinese medicine attenuate ultraviolet B-induced G1 growth arrest in HaCaT cells and dermal fibroblasts involve down-regulating the expression of p16, p21 and p53. Photodermatol. Photoimmunol. Photomed 2011, 27, 203–212. [Google Scholar]
- Liu, C.H.; Chen, C.Y.; Huang, A.M.; Li, J.H. Subamolide A, a component isolated from Cinnamomum subavenium, induces apoptosis mediated by mitochondria-dependent, p53 and ERK1/2 pathways in human urothelial carcinoma cell line NTUB1. J. Ethnopharmacol 2011, 137, 503–511. [Google Scholar]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure-Activity Relationships, and Molecular Mechanisms of Action. Front. Pharmacol 2012, 3. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fu, T.; Min, H.; Xu, Y.; Chen, J.; Li, G. Molecular Dynamic Simulation Insights into the Normal State and Restoration of p53 Function. Int. J. Mol. Sci. 2012, 13, 9709-9740. https://doi.org/10.3390/ijms13089709
Fu T, Min H, Xu Y, Chen J, Li G. Molecular Dynamic Simulation Insights into the Normal State and Restoration of p53 Function. International Journal of Molecular Sciences. 2012; 13(8):9709-9740. https://doi.org/10.3390/ijms13089709
Chicago/Turabian StyleFu, Ting, Hanyi Min, Yong Xu, Jianzhong Chen, and Guohui Li. 2012. "Molecular Dynamic Simulation Insights into the Normal State and Restoration of p53 Function" International Journal of Molecular Sciences 13, no. 8: 9709-9740. https://doi.org/10.3390/ijms13089709