Molecular Mechanisms of Ursodeoxycholic Acid Toxicity & Side Effects: Ursodeoxycholic Acid Freezes Regeneration & Induces Hibernation Mode

{kind=link}
{kind=link}
Abstract
:1. Introduction
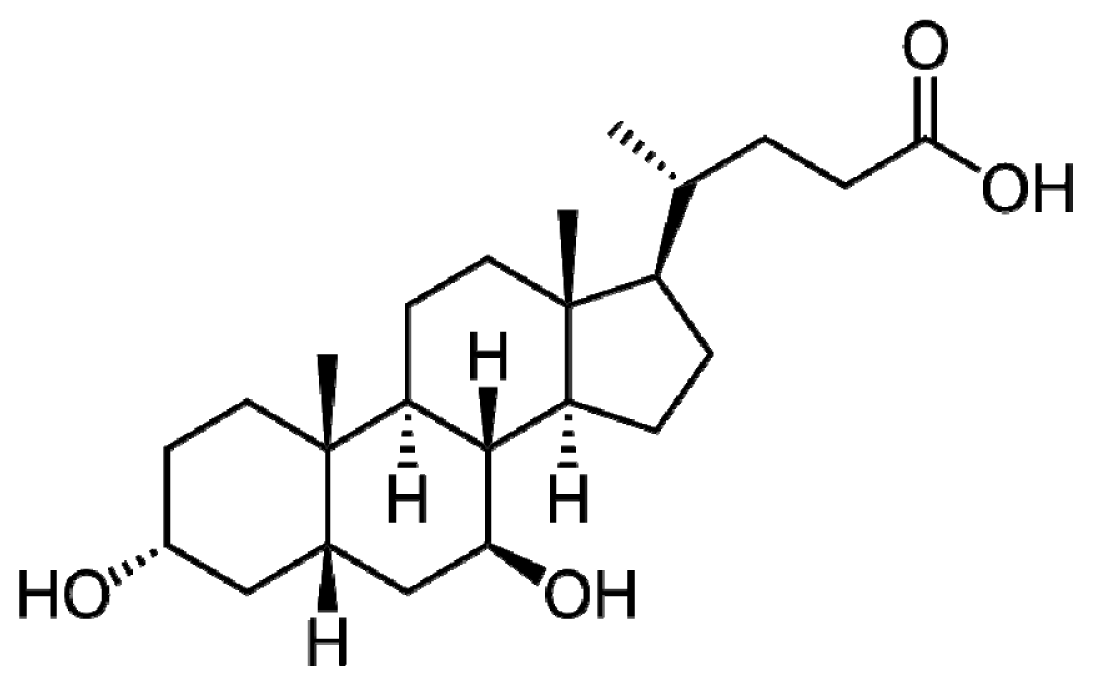
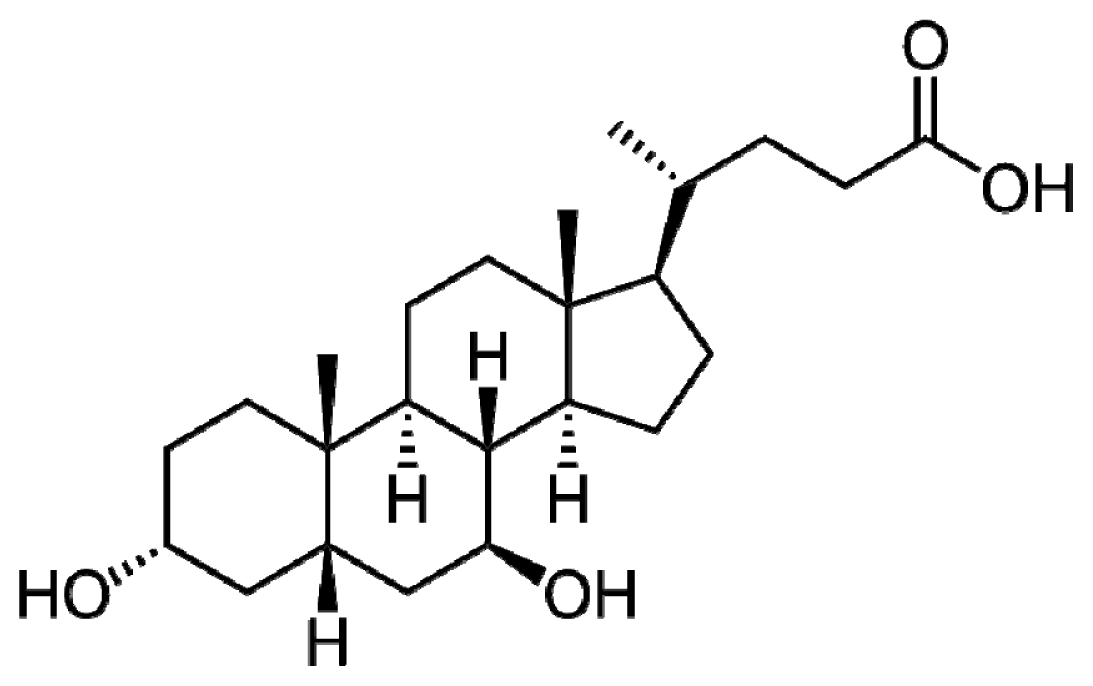
2. UDCA Structure
3. Potentially Toxic Molecular Properties of UDCA
3.1. UDCA Breaks Down into Toxic Lithocholic Acid
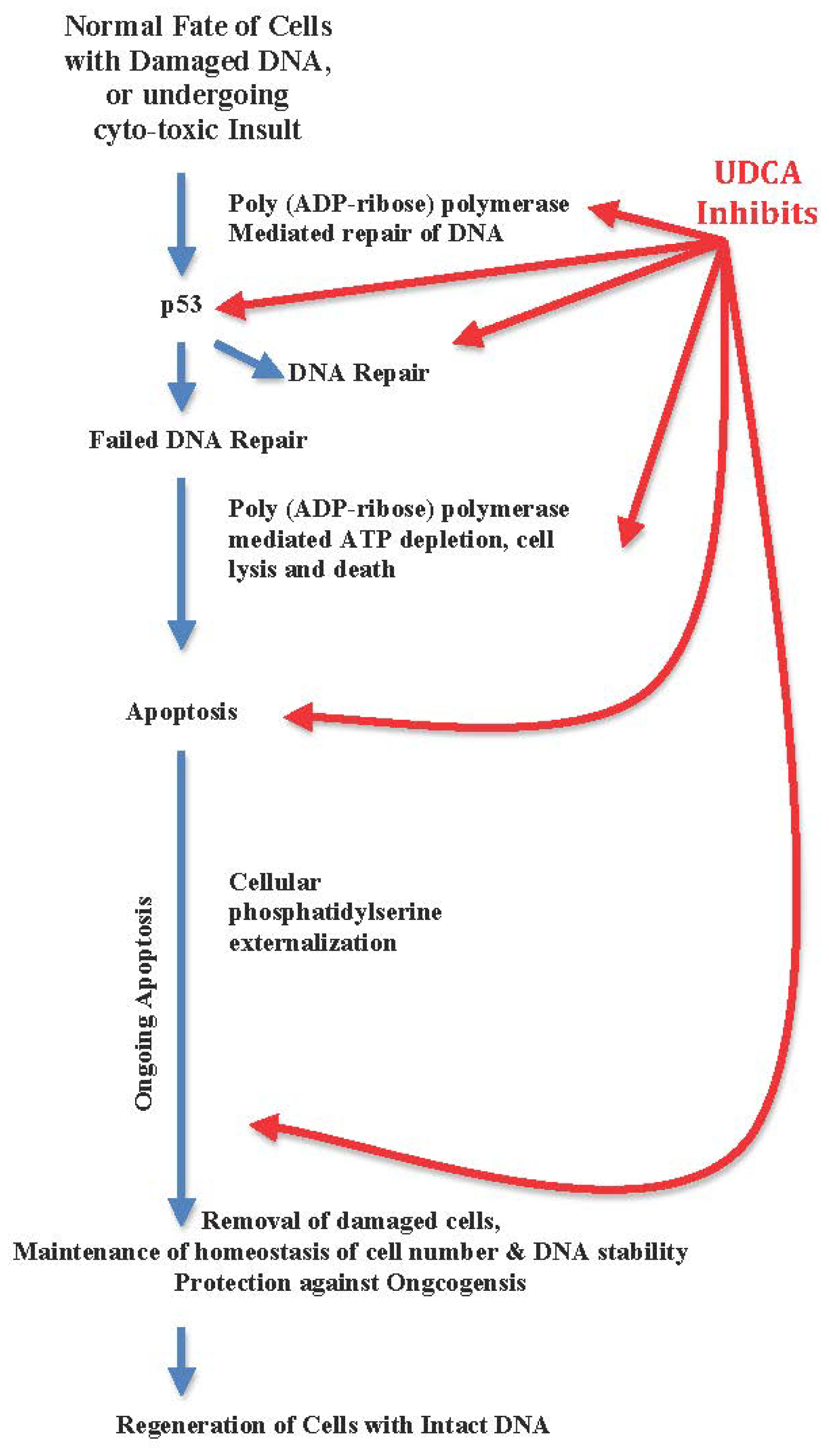
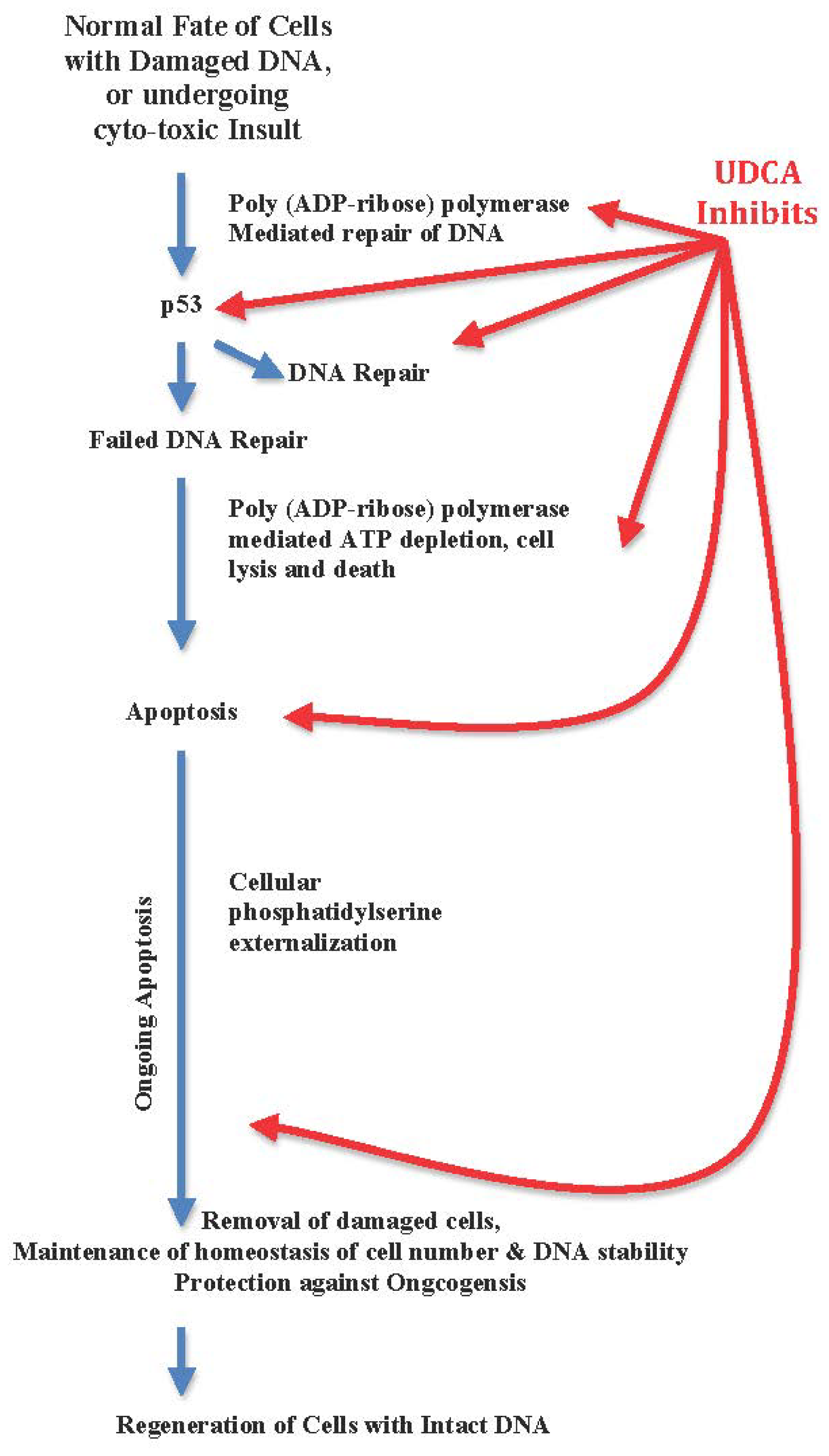
3.2. UDCA Inhibits Apoptosis, Arrests Cellular Regeneration, and Blocks DNA Repair
3.3. UDCA Inhibits Co-Enzyme A
3.4. UDCA Is a Transcriptional Factor
3.5. UDCA Is Immune-Modulatory
3.6. UDCA Interferes with Drug Metabolism and Detoxification
3.7. UDCA Is Choleretic
3.8. UDCA Is Hydrophilic
3.9. UDCA Suppresses Central Nervous System Microglia Activation
3.10. UDCA Anti-Proliferative Potential
3.11. UDCA Potentiates Cellular Cytotoxicity
3.12. UDCA Controls Bilirubin and Hepatic Transaminases
4. UDCA as a Medicine
4.1. UDCA in Primary Biliary Cirrhosis
4.2. UDCA in Primary Sclerosing Cholangitis
4.3. UDCA in Pediatric Cholestasis
4.4. UDCA in Hepatitis B, and C Virus Infection
4.5. UDCA in Cystic Fibrosis
4.6. UDCA in Non-Alcoholic Steato-Hepatitis
4.7. UDCA in Gall Stone Dissolution
4.8. UDCA in Cancer Colon
5. UDCA Associated Adverse Drug Reactions
6. Why Were Ineffectiveness and Deaths Associated with Double Dose UDCA Thought to Be “Unanticipated”?
6.1. “Hepatoprotective” as a Term Is Not Tangible, Not Objective, Is Immeasurable, Not Quantifiable, and with Unknown Sequel or Clinical Effects, or Benefits, and Is of No Prognostic Value
6.2. Relying on False Surrogate Markers
6.3. Overrating UDCA Hydrophilic Properties & Underestimating Its Tight Therapeutic Index
7. Lessons to Be Learned. What Masked UDCA Toxicity?
7.1. Clinical Trial Flaws and Bias
- UDCA results in a drop of 25% of serum bilirubin in patients compared to control groups. Though a 25% drop is of statistical significance, it is of marginal clinical and prognostic significance [8].
- UDCA clinical trials depended on hepatic transaminases and serum bilirubin control as surrogate markers of effectiveness of UDCA. Liver transaminases and serum bilirubin are not surrogate markers of liver histopathology, and do not rule out underlying fibrosis, cirrhosis, or malignancy [215].
- UDCA is an RNA transcriptional factor. Not all clinical trials of UDCA were long enough beyond 2 years to foresee the full-blown effects of UDCA [8].
7.2. Undermining Evidence
- Highlighting UDCA’s down-regulation of cellular functions of hepatocytes, phagocytes and micoglia, and anti-apoptosis as an advantage while apoptosis eliminates DNA damaged cells beyond repair. Anti-apoptosis is a virulence factor of hepatitis C virus, and the virulence factor of hepatocelluar carcinoma [31–33,219–221].
- Describing the use of UDCA as “widely used”, while UDCA approved use is strictly limited to primary biliary cirrhosis, and gall stone dissolution. UDCA is not licensed for use in children [10].
- Insinuation of a “hepato-protective” role of UDCA as an indication for its use, when evidence-based medicine proved that UDCA has no effect on histopathology or morbidity or mortality or quality of life. Moreover, in trying to force an increasing dose to allow for more “hepato-protection”, patients suffered more than double fold deaths [13,26].
- Propagation of the hydrophilic property of UDCA as an advantage. It is important to note that the more hydrophilic the compound is, the more it escapes detoxification and allows systemic dissemination and longer half-life [223–225]. Thus, UDCA half-life is 3.5–8 days, and it leads to suppression of central nervous system microglia activation [30]. It is important to highlight that micoglia injury by bilirubin intoxication in neonates is detrimental to developing central nervous system in neonates [137].
7.3. Confounders
- Under representation of review articles written by authors with no conflict of interest, and an abundance of review articles written by authors acknowledging conflicts of interest in leading journals of high impact factors [227].
- Cofounding investigational use of UDCA with UDCA approved use as a medicine encourages off-label use in un-approved indications [22].
- Use of the term “hepato-protective” that lacks objective definition. This insinuation encourages widening of scope of off-label use of UDCA to unapproved indications. This insinuated term shaped a “cultural” room for UDCA, when it failed to earn evidence based room as a curing medication [8]. The word ‘Hepatoprotective’ needs definition before being applied as an indication for drug intake. It encouraged wide UDCA off-label use, and disseminated the unforeseen hepatotoxic UDCA.
- Prescribing habits are not congruent with scientific evidence. Approximately 91% of gastroenterologists in UK prescribe UDCA to primary biliary cirrhosis patients despite conflicting evidence of UDCA effectiveness or safety [229].
8. Conclusions
Acknowledgments
References
- Hagey, L.R.; Crombie, D.L.; Espinosa, E.; Carey, M.C.; Igimi, H.; Hofmann, A.F. Ursodeoxycholic acid in the Ursidae: Biliary bile acids of bears, pandas, and related carnivores. J. Lipid. Res 1993, 34, 1911–1917. [Google Scholar]
- Hofmann, A.F. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand. J. Gastroenterol 1994, 204, S1–S15. [Google Scholar]
- Bachrach, W.H.; Hofmann, A.F. Ursodeoxycholic acid in the treatment of cholesterol cholelithiasis. Part I. Dig. Dis. Sci 1982, 27, 737–761. [Google Scholar]
- Roma, M.G.; Toledo, F.D.; Boaglio, A.C.; Basiglio, C.L.; Crocenzi, F.A.; Sánchez Pozzi, E.J. Ursodeoxycholic acid in cholestasis: Linking action mechanisms to therapeutic applications. Clin. Sci. (Lond.) 2011, 121, 523–544. [Google Scholar]
- Leuschner, U.; Leuschner, M.; Sieratzki, J.; Kurtz, W.; Hübner, K. Gallstone dissolution with ursodeoxycholic acid in patients with chronic active hepatitis and two years follow-up. A pilot study. Dig. Dis. Sci 1985, 30, 642–649. [Google Scholar]
- Heathcote, E.J.; Cauch-Dudek, K.; Walker, V.; Bailey, R.J.; Blendis, L.M.; Ghent, C.N.; Michieletti, P.; Minuk, G.Y.; Pappas, S.C.; Scully, L.J.; et al. The Canadian multicentre double blind randomized controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1994, 19, 1149–1156. [Google Scholar]
- Degott, C.; Zafrani, E.S.; Callard, P.; Balkau, B.; Poupon, R.E.; Poupon, R. Histopathological study of primary biliary cirrhosis and the effect of ursodeoxycholic acid treatment on histology progression. Hepatology 1999, 29, 1007–1012. [Google Scholar]
- Gong, Y.; Huang, Z.B.; Christensen, E.; Gluud, C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst. Rev 2008, 16, CD000551. [Google Scholar]
- Reichen, J. Review: Ursodeoxycholic acid does not reduce risk for mortality or liver transplantation in primary cirrhosis. ACP J. Club 2008, 148, 17. [Google Scholar]
- Paediatric Formulary Committee, British National Formulary for Children; Pharmaceutical Press: London, UK, 2010.
- Kotb, M.A. Review of a historical cohort: Ursodeoxycholic acid in extrahepatic biliary atresia. J. Pediatr. Surg 2008, 43, 1321–1327. [Google Scholar]
- Kotb, M.A. Ursodeoxycholic acid in neonatal hepatitis and infantile paucity of intrahepatic bile ducts: Review of a historical cohort. Dig. Dis. Sci 2009, 54, 2231–2241. [Google Scholar]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar]
- Rudolph, G.; Gotthardt, D.; Kloeters-Plachky, P.; Rost, D.; Kulaksiz, H.; Stiehl, A. In PSC with dominant bile duct stenosis, IBD is associated with an increase of carcinomas and reduced survival. J. Hepatol 2010, 53, 313–317. [Google Scholar]
- Rudolph, G.; Gotthardt, D.N.; Kloeters-Plachky, P.; Kulaksiz, H.; Schirmacher, P.; Stiehl, A. In PSC with colitis treated with UDCA, most colonic carcinomas develop in the first years after the start of treatment. Dig. Dis. Sci 2011, 56, 3624–3630. [Google Scholar]
- Kuiper, E.M.; Hansen, B.E.; Adang, R.P.; van Nieuwkerk, C.M.; Timmer, R.; Drenth, J.P.; Spoelstra, P.; Brouwer, H.T.; Kuyvenhoven, J.P.; van Buuren, H.R. Dutch PBC Study Group. Relatively high risk for hepatocellular carcinoma in patients with primary biliary cirrhosis not responding to ursodeoxycholic acid. Eur. J. Gastroenterol. Hepatol 2010, 22, 1495–1502. [Google Scholar]
- Leuschner, M.; Dietrich, C.F.; You, T.; Seidl, C.; Raedle, J.; Herrmann, G.; Ackermann, H.; Leuschner, U. Characterisation of patients with primary biliary cirrhosis responding to long term ursodeoxycholic acid treatment. Gut 2000, 46, 121–126. [Google Scholar]
- Bhandari, B.M.; Bayat, H.; Rothstein, K.D. Primary biliary cirrhosis. Gastroenterol. Clin. North Am 2011, 40. [Google Scholar]
- Burnat, G.; Majka, J.; Konturek, P.C. Bile acids are multifunctional modulators of the Barrett’s carcinogenesis. J. Physiol. Pharmacol 2010, 61, 185–192. [Google Scholar]
- Material Safety Data Sheet: Ursodiol MSDS. 2010. Available online: http://www.sciencelab.com/xMSDS-Ursodiol-9925395 accessed on 13 June 2010.
- Nie, B.; Park, H.M.; Kazantzis, M.; Lin, M.; Henkin, A.; Ng, S.; Song, S.; Chen, Y.; Tran, H.; Lai, R.; et al. Specific bile acids inhibit hepatic fatty acid uptake. Hepatology 2012, 24. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol 2009, 15, 1677–1689. [Google Scholar]
- Iaizzo, P.A.; Laske, T.G.; Harlow, H.J.; McClay, C.B.; Garshelis, D.L. Wound healing during hibernation by black bears (Ursus americanus) in the wild: Elicitation of reduced scar formation. Integr. Zool 2012, 7, 48–60. [Google Scholar]
- Schiedermaier, P.; Hansen, S.; Asdonk, D.; Brensing, K.; Sauerbruch, T. Effects of ursodeoxycholic acid on splanchnic and systemic hemodynamics. A double-blind, cross-over, placebo-controlled study in healthy volunteers. Digestion 2000, 61, 107–112. [Google Scholar]
- Nair, P.; Turjman, N. Role of bile acids and neutral sterols in familial cancer syndromes of the colon. Dis. Colon Rectum 1983, 26, 629–632. [Google Scholar]
- Sinakos, E.; Marschall, H.-U.; Kowdley, K.V.; Befeler, A.; Keach, J.; Lindor, K. Bile acid changes after high-dose ursodeoxycholic acid treatment in primary sclerosing cholangitis: Relation to disease progression. Hepatology 2010, 52, 197–203. [Google Scholar]
- Fedorowski, T.; Salen, G.; Tint, G.S.; Mosbach, E. Transformation of chenodeoxycholic acid and ursodeoxycholic acid by human intestinal bacteria. Gastroenterology 1979, 77, 1068–1073. [Google Scholar]
- Bazzoli, F.; Fromm, H.; Sarva, R.P.; Sembrat, R.F.; Ceryak, S. Comparative formation of lithocholic acid from chenodeoxycholic and ursodeoxycholic acids in the colon. Gastroenterology 1982, 83, 753–760. [Google Scholar]
- Thistle, J.L.; Larusso, N.F.; Hofmann, A.F.; Turcotte, J.; Carlson, G.L.; Ott, B.J. Differing effects of ursodeoxycholic or chenodeoxycholic acid on biliary cholesterol saturation and bile acid metabolism in man. A dose-response study. Dig. Dis. Sci 1982, 27, 161–168. [Google Scholar]
- Angulo, P. Use of ursodeoxycholic acid in patients with liver disease. Curr. Gastroenterol. Rep 2002, 4, 37–44. [Google Scholar]
- Guyot, C.; Combe, C.; Balabaud, C.; Bioulac-Sage, P.; Desmoulière, A. Fibrogenic cell fate during fibrotic tissue remodeling observed in rat and human cultured liver slices. J. Hepatol 2007, 46, 142–150. [Google Scholar]
- Hansell, C.; Nibbs, R. Professional and part-time chemokine decoys in the resolution of inflammation. Sci. STKE 2007, 384, pe18. [Google Scholar]
- Brenner, D.A. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc 2009, 120, 361–368. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimmer dissociation. Nature 2003, 421, 499–506. [Google Scholar]
- Jung, D.; Alt, F.W. Unraveling V(D)J recombination; insights into gene regulation. Cell 2004, 116, 299–311. [Google Scholar]
- Braig, M.; Schmitt, C.A. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res 2006, 66, 2881–2884. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Rev. Mol. Cell. Biol 2007, 8, 729–740. [Google Scholar]
- Lynch, M.D. How does cellular senescence prevent cancer? DNA Cell Biol 2006, 25, 69–78. [Google Scholar]
- Li, M.O.; Sarkisian, M.R.; Mehal, W.Z.; Rakic, P.; Flavell, R.A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 2003, 302, 1560–1563. [Google Scholar]
- Powell, A.A.; Akare, S.; Qi, W.; Herzer, P.; Jean-Louis, S.; Feldman, R.A.; Martinez, J.D. Resistance to ursodeoxycholic acid-induced growth arrest can also result in resistance to deoxycholic acid-induced apoptosis and increased tumorgenicity. BMC Cancer 2006, 6. [Google Scholar] [CrossRef]
- Solá, S.; Aranha, M.M.; Steer, C.J.; Rodrigues, C.M. Game and players: Mitochondrial apoptosis and the therapeutic potential of ursodeoxycholic acid. Curr. Issues Mol. Biol 2007, 9, 123–138. [Google Scholar]
- Castro, R.E.; Amaral, J.D.; Solá, S.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Differential regulation of cyclin D1 and cell death by bile acids in primary rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol 2007, 293, G327–G334. [Google Scholar]
- Sola, S.; Ma, X.; Castro, R.E.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Ursodeoxycholic acid modulates E2F-1 and p53 expression through a caspase-independent mechanism in transforming growth factor beta1-induced apoptosis of rat hepatocytes. J. Biol. Chem 2003, 278, 48831–48838. [Google Scholar]
- Im, E.; Martinez, J.D. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J. Nutr 2004, 134, 483–486. [Google Scholar]
- Rodrigues, C.M.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med 1998, 4, 165–178. [Google Scholar]
- Koh, H.; Lee, K.H.; Kim, D.; Kim, S.; Kim, J.W.; Chung, J. Inhibition of Akt and its anti-apoptotic activities by tumor necrosis factor-induced protein kinase C-related kinase 2 (PRK2) cleavage. J. Biol. Chem 2000, 275, 34451–34458. [Google Scholar]
- Akare, S.; Jean-Louis, S.; Chen, W.; Wood, D.J.; Powell, A.A.; Martinez, J.D. Ursodeoxycholic acid modulates histone acetylation and induces differentiation and senescence. Int. J. Cancer 2006, 119, 2958–2969. [Google Scholar]
- Rodrigues, C.M.; Fan, G.; Ma, X.; Kren, B.T.; Steer, C.J. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Invest 1998, 101, 2790–2799. [Google Scholar]
- Trauner, M.; Graziadei, I.W. Review article: Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment. Pharmacol. Ther 1998, 13, 979–996. [Google Scholar]
- Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Ursodeoxycholic acid mechanisms of action and clinical use in hepatobiliary disorders. J. Hepatol 2010, 35, 134–146. [Google Scholar]
- Fickert, P.; Trauner, M.; Fuchsbichler, A.; Stumptner, C.; Zatloukal, K.; Denk, H. Cytokeratins as targets for bile acid-induced toxicity. Am. J. Pathol 2002, 160, 491–599. [Google Scholar]
- Martinez-Diez, M.C.; Serrano, M.A.; Monte, M.J.; Marin, J.J. Comparison of the effects of bile acids on cell viability and DNA synthesis by rat hepatocytes in primary culture. Biochim. Biophys. Acta 2000, 1500, 153–160. [Google Scholar]
- Popov, Y.; Sverdlov, D.Y.; Bhaskar, K.R.; Sharma, A.K.; Millonig, G.; Patsenker, E.; Krahenbuhl, S.; Krahenbuhl, L.; Schuppan, D. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol 2010, 298, G323–G334. [Google Scholar]
- Garewal, H.; Bernstein, H.; Bernstein, C.; Sampliner, R.; Payne, C. Reduced bile acid-induced apoptosis in “normal” colorectal mucosa: A potential biological marker for cancer risk. Cancer Res 1996, 56, 1480–1483. [Google Scholar]
- Abate, N.; Carubbi, F.; Bozzoli, M.; Bertolotti, M.; Farah, I.; Rosi, A.; Carulli, N. Effect of chenodeoxycholic acid and ursodeoxycholic acid administration on acyl-CoA: Cholesterol acyltransferase activity in human liver. Ital. J. Gastroenterol 1994, 26, 287–293. [Google Scholar]
- Das, S.; Chakraborty, S.; Basu, A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem 2010, 115, 537–549. [Google Scholar]
- Pontes Soares, C.; Portilho, D.M.; da Silva Sampaio, L.; Einicker-Lamas, M.; Morales, M.M.; Costa, M.L.; Dos Santos Mermelstein, C. Membrane cholesterol depletion by methyl-beta-cyclodextrin enhances the expression of cardiac differentiation markers. Cells Tissues Organs 2010, 192, 187–199. [Google Scholar]
- Weber, P.; Wagner, M.; Schneckenburger, H. Fluorescence imaging of membrane dynamics in living cells. J. Biomed. Opt 2010, 15, 046017. [Google Scholar]
- Smith, L.L. Another cholesterol hypothesis: Cholesterol as antioxidant. Free Radic. Biol. Med 1991, 11, 47–61. [Google Scholar]
- DuSell, C.D.; Nelson, E.R.; Wang, X.; Abdo, J.; Mödder, U.I.; Umetani, M.; Gesty-Palmer, D.; Javitt, N.B.; Khosla, S.; McDonnell, D.P. The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis. Endocrinology 2010, 151, 3675–3685. [Google Scholar]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med 2010, 31, 1–20. [Google Scholar]
- Bouscarel, B.; Gettys, T.W.; Fromm, H.; Dubner, H. Ursodeoxycholic acid inhibits glucagon-induced cAMP formation in hamster hepatocytes: A role for PKC. Am. J. Physiol. Gastrointest. Liver Physiol 1995, 268, G300–G310. [Google Scholar]
- Bouscarel, B.; Matsuzaki, Y.; Le, M.; Gettys, T.W.; Fromm, H. Changes in G protein expression account for impaired modulation of hepatic cAMP formation after BDL. Am. J. Physiol 1998, 274, G1151–G1159. [Google Scholar]
- Peterson, T.C.; Slysz, G.; Isbrucker, R. The inhibitory effect of ursodeoxycholic acid and pentoxifylline on platelet derived growth factor-stimulated proliferation is distinct from an effect by cyclic AMP. Immunopharmacology 1998, 39, 181–191. [Google Scholar]
- Amaral, J.D.; Castro, R.E.; Solá, S.; Steer, C.J.; Rodrigues, C.M. p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J. Biol. Chem 2007, 282, 34250–34259. [Google Scholar]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. Targeting the p53 Pathway of Apoptosis. Curr. Pharm. Des 2010, 16, 2493–2503. [Google Scholar]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27, S71–S83. [Google Scholar]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem 2002, 277, 3247–3257. [Google Scholar]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117–129. [Google Scholar]
- Georgiev, P.; Dahm, F.; Graf, R.; Clavien, P.A. Blocking the path to death: Anti-apoptotic molecules in ischemia/reperfusion injury of the liver. Curr. Pharm. Des 2006, 12, 2911–2921. [Google Scholar]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem 2008, 77, 557–582. [Google Scholar]
- Park, I.H.; Kim, M.K.; Kim, S.U. Ursodeoxycholic acid prevents apoptosis of mouse sensory neurons induced by cisplatin by reducing P53 accumulation. Biochem. Biophys. Res. Commun 2008, 377, 1025–1030. [Google Scholar]
- Ji, W.J.; Qu, Q.; Jin, Y.; Zhao, L.; He, X.D. Ursodeoxycholic acid inhibits hepatocyte-like cell apoptosis by down-regulating the expressions of Bax and Caspase-3. Zhonghua Yi Xue Za Zhi 2009, 89, 2997–3001. [Google Scholar]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell. Biol 2007, 8, 49–62. [Google Scholar]
- Miura, T.; Ouchida, R.; Yoshikawa, N.; Okamoto, K.; Makino, Y.; Nakamura, T.; Morimoto, C.; Makino, I.; Tanaka, H. Functional modulation of the glucocorticoid receptor and suppression of NF-kappaB-dependent transcription by ursodeoxycholic acid. J. Biol. Chem 2001, 276, 47371–47378. [Google Scholar]
- Garg, A.; Aggarwal, B.B. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia 2002, 16, 1053–1068. [Google Scholar]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-kappaB activation: From bench to bedside. Exp. Biol. Med 2008, 233, 21–31. [Google Scholar]
- Schuster, A.; Schilling, T.; De Laurenzi, V.; Koch, A.F.; Seitz, S.; Staib, F.; Teufel, A.; Thorgeirsson, S.S.; Galle, P.R.; Melino, G.; et al. DeltaNp73beta is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle 2010, 9, 2629–2639. [Google Scholar]
- Higuchi, H.; Grambihler, A.; Canbay, A.; Bronk, S.F.; Gores, G.J. Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J. Biol. Chem 2004, 279, 51–60. [Google Scholar]
- Choi, Y.H.; Im, E.O.; Suh, H.; Jin, Y.; Yoo, Y.H.; Kim, N.D. Apoptosis and modulation of cell cycle control by synthetic derivatives of ursodeoxycholic acid and chenodeoxycholic acid in human prostate cancer cells. Cancer Lett 2003, 199, 157–167. [Google Scholar]
- Im, E.; Akare, S.; Powell, A.; Martinez, J.D. Ursodeoxycholic acid can suppress deoxycholic acid-induced apoptosis by stimulating Akt/PKB-dependent survival signaling. Nutr. Cancer 2005, 51, 110–116. [Google Scholar]
- Horowitz, N.S.; Hua, J.; Powell, M.A.; Gibb, R.K.; Mutch, D.G.; Herzog, T.J. Novel cytotoxic agents from an unexpected source: Bile acids and ovarian tumor apoptosis. Gynecol. Oncol 2007, 107, 344–349. [Google Scholar]
- Khare, S.; Mustafi, R.; Cerda, S.; Yuan, W.; Jagadeeswaran, S.; Dougherty, U.; Tretiakova, M.; Samarel, A.; Cohen, G.; Wang, J.; et al. Ursodeoxycholic acid suppresses Cox-2 expression in colon cancer: Roles of Ras, p38, and CCAAT/enhancer-binding protein. Nutr. Cancer 2008, 60, 389–400. [Google Scholar]
- Su, J.G.; Liao, P.J.; Huang, M.C.; Chu, W.C.; Lin, S.C.; Chang, Y.J. Aldo-keto reductase 1C is essential for 1-nitropyrene’s but not for benzo[a]pyrene’s induction of p53 phosphorylation and apoptosis. Toxicology 2008, 244, 257–270. [Google Scholar]
- Wimmer, R.; Hohenester, S.; Pusl, T.; Denk, G.U.; Rust, C.; Beuers, U. Tauroursodeoxycholic acid exerts anticholestatic effects by a cooperative cPKC alpha-/PKA-dependent mechanism in rat liver. Gut 2008, 57, 1448–1454. [Google Scholar]
- Andersson, Y.; Juell, S.; Fodstad, Ø. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin a immunotoxin. Int. J. Cancer 2004, 112, 475–483. [Google Scholar]
- Rolo, A.P.; Palmeira, C.M.; Holy, J.M.; Wallace, K.B. Role of mitochondrial dysfunction in combined bile acid-induced cytotoxicity: The switch between apoptosis and necrosis. Toxicol. Sci 2004, 79, 196–204. [Google Scholar]
- Zollner, G.; Trauner, M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br. J. Pharmacol 2009, 156, 7–27. [Google Scholar]
- Huang, J.; Plass, C.; Gerhauser, C. Cancer chemoprevention by targeting the epigenome. Curr. Drug Targets 2011, 12, 1925–1956. [Google Scholar]
- Oike, T.; Ogiwara, H.; Torikai, K.; Nakano, T.; Yokota, J.; Kohno, T. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int. J. Radiat. Oncol. Biol. Phys 2012, in press. [Google Scholar]
- Calmus, Y.; Gane, P.; Rouger, P.; Poupon, R. Hepatic expression of class I and class II histocompatibility complex molecules in primary biliary cirrhosis: Effect of ursodeoxycholic acid. Hepatology 1990, 11, 12–15. [Google Scholar]
- Yoshikawa, M.; Tsujii, T.; Matsumura, K.; Yamao, J.; Matsumura, Y.; Kubo, R.; Fukui, H.; Ishizaka, S. Immunomodulatory effects of ursodeoxycholic acid on immune responses. Hepatology 1992, 16, 358–364. [Google Scholar]
- Quist, R.G.; Ton-Nu, H.T.; Lillienau, J.; Hofmann, A.F.; Barrett, K.E. Activation of mast cells by bile acids. Gastroenterology 1991, 101, 446–456. [Google Scholar]
- Solá, S.; Amaral, J.D.; Aranha, M.M.; Steer, C.J.; Rodrigues, C.M. Modulation of hepatocyte apoptosis: Cross-talk between bile acids and nuclear steroid receptors. Curr. Med. Chem 2006, 13, 3039–3051. [Google Scholar]
- Solá, S.; Amaral, J.D.; Castro, R.E.; Ramalho, R.M.; Borralho, P.M.; Kren, B.T.; Tanaka, H.; Steer, C.J.; Rodrigues, C.M. Nuclear translocation of UDCA by the glucocorticoid receptor is required to reduce TGF-beta1-induced apoptosis in rat hepatocytes. Hepatology 2005, 42, 925–934. [Google Scholar]
- Weitzel, C.; Stark, D.; Kullmann, F.; Schölmerich, J.; Holstege, A.; Falk, W. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur. J. Gastroenterol. Hepatol 2005, 17, 169–177. [Google Scholar]
- Amaral, J.D.; Solá, S.; Steer, C.J.; Rodrigues, C.M. Role of nuclear steroid receptors in apoptosis. Curr. Med. Chem 2006, 16, 3886–3902. [Google Scholar]
- Jacquemin, E.; Hermans, D.; Myara, A.; Habes, D.; Debray, D.; Hadchouel, M.; Sokal, E.M.; Bernard, O. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology 1997, 25, 519–523. [Google Scholar]
- Masferrer, J.L.; Seibert, K. Regulation of prostaglandin synthesis by glucocorticoids. Receptor 1994, 4, 25–30. [Google Scholar]
- Goppelt-Struebe, M. Molecular mechanisms involved in the regulation of prostaglandin biosynthesis by glucocorticoids. Biochem. Pharmacol 1997, 53, 1389–1395. [Google Scholar]
- Ikegami, T.; Matsuzaki, Y.; Fukushima, S.; Shoda, J.; Olivier, J.L.; Bouscarel, B.; Tanaka, N. Suppressive effect of ursodeoxycholic acid on type IIA phospholipase A2 expression in HepG2 cells. Hepatology 2005, 41, 896–905. [Google Scholar]
- Mitsuyoshi, H.; Nakashima, T.; Inaba, K.; Ishikawa, H.; Nakajima, Y.; Sakamoto, Y.; Matsumoto, M.; Okanoue, T.; Kashima, K. Ursodeoxycholic acid enhances glucocorticoid-induced tyrosine aminotransferase-gene expression in cultured rat hepatocytes. Biochem. Biophys. Res. Commun 1997, 240, 732–736. [Google Scholar]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar]
- Savory, J.G.; Préfontaine, G.G.; Lamprecht, C.; Liao, M.; Walther, R.F.; Lefebvre, Y.A.; Haché, R.J. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol. Cell. Biol 2001, 21, 781–793. [Google Scholar]
- Solá, S.; Castro, R.E.; Kren, B.T.; Steer, C.J.; Rodrigues, C.M. Modulation of nuclear steroid receptors by ursodeoxycholic acid inhibits TGF-beta1-induced E2F-1/p53-mediated apoptosis of rat hepatocytes. Biochemistry 2004, 43, 8429–8438. [Google Scholar]
- Shi, Q.Y.; Kong, B.H.; Ma, K.D.; Zhang, X.L.; Jiang, S. Effects of ursodeoxycholic acid on the liver plasma membrane fluidity, hepatic glutathione concentration, hepatic estrogen receptors and progesterone receptors in pregnant rats with ethinylestradiol and progesterone induced intrahepatic cholestasis. Zhonghua Fu Chan Ke Za Zhi 2003, 38, 680–682, Abstract. [Google Scholar]
- Rodrigues, C.M.; Ma, X.; Linehan-Stieers, C.; Fan, G.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ 1999, 6, 842–854. [Google Scholar]
- Nishigaki, Y.; Ohnishi, H.; Moriwaki, H.; Muto, Y. Ursodeoxycholic acid corrects defective natural killer activity by inhibiting prostaglandin E2 production in primary biliary cirrhosis. Dig. Dis. Sci 1996, 41, 1487–1493. [Google Scholar]
- Guarino, M.P.; Carotti, S.; Morini, S.; Perrone, G.; Behar, J.; Altomare, A.; Alloni, R.; Caviglia, R.; Emerenziani, S.; Rabitti, C.; Cicala, M. Decreased number of activated macrophages in gallbladder muscle layer of cholesterol gallstone patients following ursodeoxycholic acid. Gut 2008, 57, 1740–1741. [Google Scholar]
- Paolini, M.; Pozzetti, L.; Montagnani, M.; Potenza, G.; Sabatini, L.; Antelli, A.; Cantelli-Forti, G.; Roda, A. Ursodeoxycholic acid (UDCA) prevents DCA effects on male mouse liver via up-regulation of CYP [correction of CXP] and preservation of BSEP activities. Hepatology 2002, 36, 305–314. [Google Scholar]
- Pemberton, P.W.; Aboutwerat, A.; Smith, A.; Warnes, T.W. Ursodeoxycholic acid in primary biliary cirrhosis improves glutathione status but fails to reduce lipid peroxidation. Redox. Rep 2006, 11, 117–123. [Google Scholar]
- Zollner, G.; Wagner, M.; Moustafa, T.; Fickert, P.; Silbert, D.; Gumhold, J.; Fuchsbichler, A.; Halilbasic, E.; Denk, H.; Marschall, H.U.; et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: Role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, G923–G932. [Google Scholar]
- Boyland, E.; Chasseaud, L.F. Enzymes catalysing conjugations of glutathione with alpha-beta-unsaturated carbonyl compounds. Biochem. J 1968, 109, 651–661. [Google Scholar]
- Boyland, E.; Chasseaud, L.F. Glutathione S-aralkyltransferase. Biochem. J 1969, 115, 985–991. [Google Scholar]
- Boyland, E.; Chasseaud, L.F. The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis. Adv. Enzymol. Relat. Areas. Mol. Biol 1969, 32, 173–219. [Google Scholar]
- Jakoby, W.B.; Ziegler, D.M. The enzymes of detoxication. J. Biol. Chem 1990, 265, 20715–20718. [Google Scholar]
- Ziegler, D.M.; Ansher, S.S.; Nagata, T.; Kadlubar, F.F.; Jakoby, W.B. N-methylation: Potential mechanism for metabolic activation of carcinogenic primary arylamines. Proc. Natl. Acad. Sci. USA 1988, 85, 2514–2517. [Google Scholar]
- Ujhelly, O.; Ozvegy, C.; Várady, G.; Cervenak, J.; Homolya, L.; Grez, M.; Scheffer, G.; Roos, D.; Bates, S.E.; Váradi, A.; et al. Application of a human multidrug transporter (ABCG2) variant as selectable marker in gene transfer to progenitor cells. Hum. Gene Ther 2003, 14, 403–412. [Google Scholar]
- Homolya, L.; Váradi, A.; Sarkadi, B. Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. Biofactors 2003, 17, 103–114. [Google Scholar]
- Commandeur, J.N.; Stijntjes, G.J.; Vermeulen, N.P. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev 1995, 47, 271–330. [Google Scholar]
- Fickert, P.; Zollner, G.; Fuchsbichler, A.; Stumptner, C.; Weiglein, A.H.; Lammert, F.; Marschall, H.U.; Tsybrovskyy, O.; Zatloukal, K.; Denk, H.; et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology 2002, 123, 1238–1251. [Google Scholar]
- Zollner, G.; Marschall, H.U.; Wagner, M.; Trauner, M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: Pathogenetic and therapeutic considerations. Mol. Pharm 2006, 3, 231–251. [Google Scholar]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev 2003, 55, 425–461. [Google Scholar]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J 1990, 269, 1–11. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol 1997, 82, 291–295. [Google Scholar]
- Joo, S.S.; Kang, H.C.; Won, T.J.; Lee, D.I. Ursodeoxycholic acid inhibits pro-inflammatory repertoires., IL-1 beta and nitric oxide in rat microglia. Arch. Pharm. Res 2003, 26, 1067–1073. [Google Scholar]
- Joo, S.S.; Won, T.J.; Hwang, K.W.; Lee, D.I. Inhibition of iNOS expression via Ursodeoxycholic acid in murine microglial cell; BV-2 Cell Line. Immune Netw 2005, 5, 45–49. [Google Scholar]
- Joo, S.S.; Won, T.J.; Lee, D.I. Potential role of ursodeoxycholic acid in suppression of nuclear factor kappa B in microglial cell line (BV-2). Arch. Pharm. Res 2004, 27, 954–960. [Google Scholar]
- Parry, G.J.; Rodrigues, C.M.; Aranha, M.M.; Hilbert, S.J.; Davey, C.; Kelkar, P.; Low, W.C.; Steer, C.J. Safety., tolerability., and cerebrospinal fluid penetration of ursodeoxycholic Acid in patients with amyotrophic lateral sclerosis. Clin. Neuropharmacol 2010, 33, 17–21. [Google Scholar]
- Ramalho, R.M.; Viana, R.J.; Low, W.C.; Steer, C.J.; Rodrigues, C.M. Bile acids and apoptosis modulation: An emerging role in experimental Alzheimer’s disease. Trends Mol. Med 2008, 14, 54–62. [Google Scholar]
- Ritter, M.R.; Banin, E.; Moreno, S.K.; Aguilar, E.; Dorrel, M.I.; Friedlander, M. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J. Clin. Invest 2006, 116, 3266–3276. [Google Scholar]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci 2006, 29, 506–510. [Google Scholar]
- Wee Yong, V. Inflammation in neurological disorders: A help or a hindrance? Neuroscientist 2010, 16, 408–420. [Google Scholar]
- Gordo, A.C.; Falcão, A.S.; Fernandes, A.; Brito, M.A.; Silva, R.F.M.; Brites, D. Unconjugated bilirubin activates and damages microglia. J. Neurosci. Res 2006, 84, 194–201. [Google Scholar]
- Jantaratnotai, N.; Schwab, C.; Ryu, J.K.; McGeer, P.L.; McLarnon, J.G. Converging perturbed microvasculature and microglial clusters characterize alzheimer disease brain. Curr. Alzheimer Res 2010, 7, 625–636. [Google Scholar]
- Im, E.O.; Choi, Y.H.; Paik, K.J.; Suh, H.; Jin, Y.; Kim, KW.; Yoo, Y.H.; Kim, N.D. Novel bile acid derivatives induce apoptosis via a p53-independent pathway in human breast carcinoma cells. Cancer Lett 2001, 163, 83–93. [Google Scholar]
- Im, E.; Choi, S.H.; Suh, H.; Choi, Y.H.; Yoo, Y.H.; Kim, N.D. Synthetic bile acid derivatives induce apoptosis through a c-Jun N-terminal kinase and NF-kappaB-dependent process in human cervical carcinoma cells. Cancer Lett 2005, 229, 49–57. [Google Scholar]
- Pavek, P.; Merino, G.; Wagenaar, E.; Bolscher, E.; Novotna, M.; Jonker, J.W.; Schinkel, A.H. Human breast cancer resistance protein: Interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J. Pharmacol. Exp. Ther 2005, 312, 144–152. [Google Scholar]
- Fimognari, C.; Lenzi, M.; Cantelli-Forti, G.; Hrelia, P. Apoptosis and modulation of cell cycle control by bile acids in human leukemia T cells. Ann. N. Y. Acad. Sci 2009, 1171, 264–269. [Google Scholar]
- Hiramatsu, K.; Matsumoto, Y.; Miyazaki, M.; Tsubouchi, H.; Yamamoto, I.; Gohda, E. Inhibition of hepatocyte growth factor production in human fibroblasts by ursodeoxycholic acid. Biol. Pharm. Bull 2005, 28, 619–624. [Google Scholar]
- Zarnegar, R.; Michalopoulos, G.K.J. The many faces of hepatocyte growth factor: From hepatopoiesis to hematopoiesis. Cell Biol 1995, 129, 1177–1180. [Google Scholar]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor (HGF) as a tissue organizer for organogenesis and regeneration. Biochem. Biophys. Res. Commun 1997, 239, 639–644. [Google Scholar]
- Yasuda, H.; Imai, E.; Shiota, A.; Fujise, N.; Morinaga, T.; Higashio, K. Antifibrogenic effect of a deletion variant of hepatocyte growth factor on liver fibrosis in rats. Hepatology 1996, 24, 636–642. [Google Scholar]
- Mizuno, S.; Kurosawa, T.; Matsumoto, K.; Mizuno-Horikawa, Y.; Okamoto, M.; Nakamura, T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J. Clin. Invest 1998, 101, 1827–1834. [Google Scholar]
- Tsagarakis, N.J.; Drygiannakis, I.; Batistakis, A.G.; Kolios, G.; Kouroumalis, E.A. A concentration-dependent effect of ursodeoxycholate on apoptosis and caspases activities of HepG2 hepatocellular carcinoma cells. Eur. J. Pharmacol 2010, 640, 1–7. [Google Scholar]
- Ikegami, T.; Matsuzaki, Y.; Al Rashid, M.; Ceryak, S.; Zhang, Y.; Bouscarel, B. Enhancement of DNA topoisomerase I inhibitor-induced apoptosis by ursodeoxycholic acid. Mol. Cancer. Ther 2006, 5, 68–79. [Google Scholar]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res 1991, 51, 4187–4191. [Google Scholar]
- Fimognari, C.; Nüsse, M.; Cesari, R.; Cantelli-Forti, G.; Hrelia, P. Micronuclei induction, cell cycle delay and apoptosis as markers of cellular stress caused by ursodeoxycholic acid in human lymphocytes. Mutat. Res 2001, 495, 1–9. [Google Scholar]
- Norppa, H.; Falck, G.C. What do human micronuclei contain? Mutagenesis 2003, 18, 221–233. [Google Scholar]
- Irifune, H.; Tsukazaki, N.; Watanabe, M.; Nonaka, S. The influence of ursodesoxycholic acid (URSO) on griseofulvin (GF)-induced protoporphyria. Nihon Hifuka Gakkai Zasshi 1991, 101, 813–817. [Google Scholar]
- Kessel, D.; Caruso, J.A.; Reiners, J.J., Jr. Potentiation of photodynamic therapy by ursodeoxycholic acid. Cancer Res 2000, 60, 6985–6988. [Google Scholar]
- Rolo, A.P.; Palmeira, C.M.; Holy, J.M.; Wallace, K.B. Role of mitochondrial dysfunction in combined bile acid-induced cytotoxicity: The switch between apoptosis and necrosis. Toxicol. Sci 2004, 79, 196–204. [Google Scholar]
- Poropat, G.; Giljaca, V.; Stimac, D.; Gluud, C. Bile acids for liver-transplanted patients. Cochrane Database Syst. Rev 2010, 3, CD005442. [Google Scholar]
- Scharschmidt, B.F.; Lake, J.R. Hepatocellular bile acid transport and ursodeoxycholic acid hypercholeresis. Dig. Dis. Sci 1989, 34, 5S–15S. [Google Scholar]
- Katz, R.; Ducci, H.; Alessandri, H. Influence of cortisone and prednisolone on hyperbilirubinemia. J. Clin. Invest 1957, 36, 1370–1374. [Google Scholar]
- Ohkubo, H.; Okuda, K. The nicotinic acid test in constitutional conjugated hyperbilirubinemias and effects of corticosteroid. Hepatology 1984, 4, 1206–1208. [Google Scholar]
- Giljaca, V.; Poropat, G.; Stimac, D.; Gluud, C. Glucocorticosteroids for primary sclerosing cholangitis. Cochrane Database Syst. Rev 2010, 1, CD004036. [Google Scholar]
- Propst, A.; Propst, T.; Zangerl, G.; Ofner, D.; Judmaier, G.; Vogel, W. Prognosis and life expectancy in chronic liver disease. Dig. Dis. Sci 1995, 40, 1805–1815. [Google Scholar]
- Wong, G.L.; Chan, H.L. Predictors of treatment response in chronic hepatitis B. Drugs 2009, 69, 2167–2177. [Google Scholar]
- Sangfelt, P.; von Sydow, M.; Uhnoo, I.; Weiland, O.; Lindh, G.; Fischler, B.; Lindgren, S.; Reichard, O. Serum ALT levels as a surrogate marker for serum HBV DNA levels in HBeAg-negative pregnant women. Scand. J. Infect. Dis 2004, 36, 182–185. [Google Scholar]
- Prestileo, T.; Mazzola, G.; di Lorenzo, F.; Colletti, P.; Vitale, F.; Ferraro, D.; di Stefano, R.; Cammà, C.; Craxì, A. Response-adjusted alpha-interferon therapy for chronic hepatitis C in HIV-infected patients. Int. J. Antimicrob. Agents 2000, 16, 373–378. [Google Scholar]
- Thompson, P.A.; Wertheim, B.C.; Roe, D.J.; Ashbeck, E.L.; Jacobs, E.T.; Lance, P.; Martinez, M.E.; Alberts, D.A. Gender modifies the effect of ursodeoxycholic acid in a randomized controlled trial in colorectal adenoma patients. Cancer Prev. Res. (Phila.) 2009, 2, 1023–1030. [Google Scholar]
- Sørensen, H.T.; Thulstrup, A.M.; Mellemkjar, L.; Jepsen, P.; Christensen, E.; Olsen, J.H.; Vilstrup, H. Long-term survival and cause-specific mortality in patients with cirrhosis of the liver: A nationwide cohort study in Denmark. J. Clin. Epidemiol 2003, 56, 88–93. [Google Scholar]
- Wachtel, M.S.; Zhang, Y.; Kaye, K.E.; Chiriva-Internati, M.; Frezza, E.E. Increased age, male gender, and cirrhosis, but not steatosis or a positive viral serology, negatively impact the life expectancy of patients who undergo liver biopsy. Dig. Dis. Sci 2007, 52, 2276–2281. [Google Scholar]
- Normann, A.; Badur, S.; Onel, D.; Kilic, A.; Sidal, M.; Larouzé, B.; Massari, V.; Müller, J.; Flehmig, B. Acute hepatitis A virus infection in Turkey. J. Med. Virol 2008, 80, 785–790. [Google Scholar]
- Migita, K.; Watanabe, Y.; Jiuchi, Y.; Nakamura, Y.; Saito, A.; Yagura, M.; Morimoto, H.; Shimada, M.; Mita, E.; Hijioka, T.; et al. Evaluation of risk factors for the development of cirrhosis in autoimmune hepatitis: Japanese NHO-AIH prospective study. J. Gastroenterol 2011, 46, S56–S62. [Google Scholar]
- Sanai, F.M.; Benmousa, A.; Al-Hussaini, H.; Ashraf, S.; Alhafi, O.; Abdo, A.A.; Alameri, H.F.; Akbar, H.O.; Bzeizi, K.I. Is serum alanine transaminase level a reliable marker of histological disease in chronic hepatitis C infection? Liver Int 2008, 28, 1011–1018. [Google Scholar]
- Ali, N.; Moiz, B.; Moatter, T.; Ahmed, S.; Adil, S.N.; Khurshid, M. Correlation of hepatitis C RNA and serum alanine aminotransferase in hepatitis B and C seronegative healthy blood donors. Indian J. Pathol. Microbiol 2010, 53, 480–485. [Google Scholar]
- Thomson, E.C.; Fleming, V.M.; Main, J.; Klenerman, P.; Weber, J.; Eliahoo, J.; Smith, J.; McClure, M.O.; Karayiannis, P. Predicting spontaneous clearance of acute hepatitis C virus in a large cohort of HIV-1-infected men. Gut 2010, 60, 837–845. [Google Scholar]
- Chen, W.; Gluud, C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst. Rev 2003, 2, CD003626. [Google Scholar]
- Cheng, K.; Ashby, D.; Smyth, R. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst. Rev 2000, 2, CD000222. [Google Scholar]
- Goulis, J.; Leandro, G.; Burroughs, A.K. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: A meta-analysis. Lancet 1999, 354, 1053–1060. [Google Scholar]
- Combes, B.; Luketic, V.A.; Peters, M.G.; Zetterman, R.K.; Garcia-Tsao, G.; Munoz, S.J.; Lin, D.; Flye, N.; Carithers, R.L., Jr. Prolonged follow-up of patients in the U.S. multicenter trial of ursodeoxycholic acid for primary biliary cirrhosis. Am. J. Gastroenterol 2004, 99, 264–268. [Google Scholar]
- Combes, B.; Carithers, R.L., Jr; Maddrey, W.C.; Lin, D.; McDonald, M.F.; Wheeler, D.E.; Eigenbrodt, E.H.; Muñoz, S.J.; Rubin, R.; Garcia-Tsao, G.; et al. A randomized, double-blind, placebo-controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1995, 22, 759–766. [Google Scholar]
- Corpechot, C.; Carrat, F.; Bonnand, A.M.; Poupon, R.E.; Poupon, R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology 2000, 32, 1196–9. [Google Scholar]
- Charatcharoenwitthaya, P.; Pimentel, S.; Talwalkar, J.A.; Enders, F.T.; Lindor, K.D.; Krom, R.A.; Wiesner, R.H. Long-term survival and impact of ursodeoxycholic acid treatmentfor recurrent primary biliary cirrhosis after liver transplantation. Liver Transpl 2007, 13, 1236–1245. [Google Scholar]
- Parés, A.; Caballería, L.; Rodés,, J.; Bruguera, M.; Rodrigo, L.; García-Plaza, A.; Berenguer, J.; Rodríguez-Martínez, D.; Mercader, J.; Velicia, R. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: Results of a double-blind controlled multicentric trial. UDCA-Cooperative Group from the Spanish Association for the Study of the Liver. J. Hepatol 2000, 32, 561–566. [Google Scholar]
- Shi, J.; Wu, C.; Lin, Y.; Chen, Y.X.; Zhu, L.; Xie, W.F. Long-term effects of mid-dose ursodeoxycholic acid in primary biliary cirrhosis: A meta-analysis of randomized controlled trials. Am. J. Gastroenterol 2006, 101, 1529–1538. [Google Scholar]
- Hohenester, S.; Oude-Elferink, R.P.; Beuers, U. Primary biliary cirrhosis. Semin. Immunopathol 2009, 31, 283–307. [Google Scholar]
- Roll, J.; Boyer, J.L.; Barry, D.; Klatskin, G. The prognostic importance of clinical and histological features in asymptomatic and symptomatic primary biliary cirrhosis. N. Engl. J. Med 1983, 308, 1–7. [Google Scholar]
- Springer, J.; Cauch-Dudek, K.; O’Rourke, K.; Wanless, I.R.; Heathcote, E.J. Asymptomatic primary biliary cirrhosis: A study of its natural history and prognosis. Am. J. Gstroenterol 1999, 94, 47–53. [Google Scholar]
- Kim, W.R.; Lindor, K.D.; Locke, G.R.; Thermeau, T.M.; Homburger, H.A.; Batts, K.P.; Yawn, B.P.; Petz, J.L.; Melton, L.J., 3rd; Dickson, E.R. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology 2000, 119, 1631–1636. [Google Scholar]
- Balasubramaniam, K.; Grambsch, P.M.; Wiesner, R.H.; Lindor, K.D.; Dickson, E.R. Diminished survival in asymptomatic primary biliary cirrhosis: A prospective study. Gastroenterology 1990, 98, 1567–1571. [Google Scholar]
- Mahl, T.C.; Shockcor, W.; Boyer, J.L. Primary biliary cirrhosis: Survival of a large cohort of symptomatic and asymptomatic patients followed for 24 years. J. Hepatol 1994, 20, 707–713. [Google Scholar]
- Dickson, E.R.; Fleming, T.R.; Wiesner, R.H.; Baldus, W.P.; Fleming, C.R.; Ludwig, J.; McCall, J.T. Trial of penicillamine in advanced primary biliary cirrhosis. N. Engl. J. Med 1985, 312, 1011–1015. [Google Scholar]
- Feldstein, A.E.; Perrault, J.; El-Youssif, M.; Lindor, K.D.; Freese, D.K.; Angulo, P. Primary sclerosing cholangitis in children: A long-term follow-up study. Hepatology 2003, 38, 210–217. [Google Scholar]
- Prince, M.; Christensen, E.; Gluud, C. Glucocorticosteroids for primary biliary cirrhosis. Cochrane Database Syst. Rev 2005, 2, CD003778. [Google Scholar]
- Van Hoogstraten, H.J.; Wolfhagen, F.H.; van de Meeberg, P.C.; Kuiper, H.; Nix, G.A.; Becx, M.C.; Hoek, A.C.; van Houte, D.P.; Rijk, M.C.; Salemans, J.M.; et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: Results of a 2-year randomized controlled trial to evaluate single versus multiple daily doses. J. Hepatol 1998, 29, 417–423. [Google Scholar]
- Lindor, K.D. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis Ursodeoxycholic Acid Study Group. N. Engl. J. Med 1997, 336, 691–695. [Google Scholar]
- Kader, H.H.; Balistreri, W.F. Cholestasis. In Nelson Textbook of Pediatrics, 17th ed.; Beherman, R.E., Kleigmen, R.M., Jenson, H.B., Eds.; WB Saunders: Philadelphia, PA, USA, 2004; pp. 1314–1319. [Google Scholar]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol 2006, 168, 410–422. [Google Scholar]
- Takikawa, H.; Beppu, T.; Seyama, Y.; Obinata, K.; Nittono, H. Serum concentration of glucuronidated and sulfated bile acids in children with cholestasis. Biochem. Med 1985, 33, 381–386. [Google Scholar]
- Shoda, J.; Mahara, R.; Osuga, T.; Mahara, R.; Osuga, T.; Ohnishi, S.; Miyazaki, H.; Tanaka, N.; Matsuzaki, Y. Similarity of unusual bile acids in human umbilical cord blood and amniotic fluid from newborns and in sera and urine from adult patients with cholestatic liver diseases. J. Lipid Res 1988, 29, 847–858. [Google Scholar]
- Hofmann, A.F. Detoxification of lithocholic acid, a toxic bile acid relevance to drug hepatotoxicity. Drug Metab. Rev 2004, 36, 703–722. [Google Scholar]
- Chen, W.; Liu, J.P.; Gluud, C. Bile acids for viral hepatitis. Cochrane Database Syst. Rev 2007, 4, CD003181. [Google Scholar]
- Desmond, C.P.; Wilson, J.; Bailey, M.; Clark, D.; Roberts, S.K. The benign course of liver disease in adults with cystic fibrosis and the effect of ursodeoxycholic acid. Liver Int 2007, 27, 1402–1408. [Google Scholar]
- Feigelson, J.; Anagnostopoulos, C.; Poquet, M.; Pecau, Y.; Munck, A.; Navarro, J. Liver cirrhosis in cystic fibrosis--therapeutic implications and long term follow up. Arch. Dis. Child 1993, 68, 653–657. [Google Scholar]
- Curry, M.P.; Hegarty, J.E. The gallbladder and biliary tract in cystic fibrosis. Curr. Gastroenterol. Rep 2005, 7, 147–153. [Google Scholar]
- Colombo, C.; Battezzati, P.M.; Crosignani, A.; Assaisso, M.; Ronchi, M.; Giunta, A. Effects of taurine and ursodeoxycholic acid on liver function tests in patients with cystic fibrosis. Acta Univ. Carol. Med. (Praha) 1990, 36, 148–151. [Google Scholar]
- Cotting, J.; Lentze, M.J.; Reichen, J. Effects of ursodeoxycholic acid treatment on nutrition and liver function in patients with cystic fibrosis and longstanding cholestasis. Gut 1990, 31, 918–921. [Google Scholar]
- Merli, M.; Bertasi, S.; Servi, R.; Diamanti, S.; Martino, F.; de Santis, A.; Goffredo, F.; Quattrucci, S.; Antonelli, M.; Angelico, M. Effect of a medium dose of ursodeoxycholic acid with or without taurine supplementation on the nutritional status of patients with cystic fibrosis: A randomized, placebo-controlled, crossover trial. J. Pediatr. Gastroenterol. Nutr 1994, 19, 198–203. [Google Scholar]
- Brigman, C.; Feranchak, A. Liver involvement in cystic fibrosis. Curr. Treat. Options Gastroenterol 2006, 9, 484–496. [Google Scholar]
- Colombo, C.; Bertolini, E.; Assaisso, M.L.; Bettinardi, N.; Giunta, A.; Podda, M. Failure of ursodeoxycholic acid to dissolve radiolucent gallstones in patients with cystic fibrosis. Acta Paediatr 1993, 82, 562–565. [Google Scholar]
- Trauner, M.; Fickert, P.; Halilbasic, E.; Moustafa, T. Lessons from the toxic bile concept for the pathogenesis and treatment of cholestatic liver diseases. Wien. Med. Wochenschr 2008, 158, 542–548. [Google Scholar]
- Orlando, R.; Azzalini, L.; Orando, S.; Lirussi, F. Bile acids for non-alcoholic fatty liver disease and/or steatohepatitis. Cochrane Database Syst. Rev 2007, 1, CD005160. [Google Scholar]
- Leuschner, U.F.; Lindenthal, B.; Herrmann, G.; Arnold, J.C.; Rössle, M.; Cordes, H.J.; Zeuzem, S.; Hein, J.; Berg, T. NASH Study Group. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: A double-blind, randomized, placebo-controlled trial. Hepatology 2010, 52, 472–479. [Google Scholar]
- Di Ciaula, A.; Wang, D.Q.; Wang, H.H.; Bonfrate, L.; Portincasa, P. Targets for current pharmacologic therapy in cholesterol gallstone disease. Gastroenterol. Clin. North Am 2010, 39. [Google Scholar]
- Alberts, D.S.; Martínez, M.E.; Hess, L.M.; Einspahr, J.G.; Green, S.B.; Bhattacharyya, A.K.; Guillen, J.; Krutzsch, M.; Batta, A.K.; Salen, G.; et al. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J. Natl. Cancer Inst 2005, 97, 846–853. [Google Scholar]
- Quesada, B.M.; Kohan, G.; Roff, H.E.; Canullán, C.M.; Chiappetta Porras, L.T. Management of gallstones and gallbladder disease in patients undergoing gastric bypass. World J. Gastroenterol 2010, 16, 2075–2079. [Google Scholar]
- Ministry of Health and Welfare (MHW), Pharmaceuticals and Medical Devices Safety Information (Summary) No. 162; MHW: Tokyo, Japan, 27 September 2000.
- Mayo, M.J. Patients and patience: The pitfalls of primary biliary cirrhosis trials. Nat. Clin. Pract. Gastroenterol. Hepatol 2005, 2, 552–553. [Google Scholar]
- Lo, S.K.; Fleming, K.A.; Chapman, R.W. A 2-year follow-up study of anti-neutrophil antibody in primary sclerosing cholangitis: Relationship to clinical activity, liver biochemistry and ursodeoxycholic acid treatment. J. Hepatol 1994, 21, 974–978. [Google Scholar]
- Floreani, A.; Caroli, D.; Variola, A.; Rizzotto, E.R.; Antoniazzi, S.; Chiaramonte, M.; Cazzagon, N.; Brombin, C.; Salmaso, L.; Baldo, V. A 35-year follow-up of a large cohort of patients with primary biliary cirrhosis seen at a single centre. Liver Int 2011, 31, 361–368. [Google Scholar]
- Nash, K.L.; Allison, M.E.; McKeon, D.; Lomas, D.J.; Haworth, C.S.; Bilton, D.; Alexander, G.J. A single centre experience of liver disease in adults with cystic fibrosis 1995–2006. J. Cyst. Fibros 2008, 7, 252–257. [Google Scholar]
- Siano, M.; de Gregorio, F.; Boggia, B.; Sepe, A.; Ferri, P.; Buonpensiero, P.; di Pasqua, A.; Raia, V. Ursodeoxycholic acid treatment in patients with cystic fibrosis at risk for liver disease. Dig. Liver Dis 2010, 42, 428–431. [Google Scholar]
- Lamontagne, J.; Pinkerton, M.; Block, T.M.; Lu, X. Hepatitis B and hepatitis C virus replication upregulates serine protease inhibitor Kazal, resulting in cellular resistance to serine protease-dependent apoptosis. J. Virol 2010, 84, 907–917. [Google Scholar]
- Li, J.C.; Yang, X.R.; Sun, H.X.; Xu, Y.; Zhou, J.; Qiu, S.J.; Ke, A.W.; Cui, Y.H.; Wang, Z.J.; Wang, W.M.; et al. Up-regulation of Krüppel-Like factor 8 promotes tumor invasion and indicates poor prognosis for hepatocellular carcinoma. Gastroenterology 2010, 139, 2146–2157. [Google Scholar]
- Wang, H.C.; Chung, P.J.; Wu, C.H.; Lan, K.P.; Yang, M.Y.; Wang, C.J. Solanum nigrum L. polyphenolic extract inhibits hepatocarcinoma cell growth by inducing G2/M phase arrest and apoptosis. J. Sci. Food Agric 2011, 91, 178–185. [Google Scholar]
- De Marco, G.; Sordino, D.; Bruzzese, E.; di Caro, S.; Mambretti, D.; Tramontano, A.; Colombo, C.; Simoni, P.; Guarino, A. Early treatment with ursodeoxycholic acid for cholestasis in children on parenteral nutrition because of primary intestinal failure. Aliment. Pharmacol. Ther 2006, 24, 387–394. [Google Scholar]
- Grattagliano, I.; Bonfrate, L.; Diogo, C.V.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Biochemical mechanisms in drug-induced liver injury: Certainties and doubts. World J. Gastroenterol 2009, 15, 4865–4876. [Google Scholar]
- Barouki, R. Linking long-term toxicity of xeno-chemicals with short-term biological adaptation. Biochimie 2010, 92, 1222–1226. [Google Scholar]
- Sainis, I.; Fokas, D.; Vareli, K.; Tzakos, A.G.; Kounnis, V.; Briasoulis, E. Cyanobacterial cyclopeptides as lead compounds to novel targeted cancer drugs. Mar. Drugs 2010, 8, 629–657. [Google Scholar]
- Chapman, R.W. Primary sclerosing cholangitis: What is the role of ursodeoxycholic acid in therapy for PSC? Nat. Rev. Gastroenterol. Hepatol 2010, 7, 74–75. [Google Scholar]
- Lindor, K.; Rakela, J.; Fung, J. Conflict of interest policy. Hepatology 2008, 47, 1. [Google Scholar]
- Makino, I.; Nakagawa, S. Changes in biliary lipid and biliary bile acid composition in patients after administration of ursodeoxycholic acid. J. Lipid Res 1978, 19, 723–728. [Google Scholar]
- Verma, A.; Jazrawi, R.P.; Ahmed, H.A.; Northfield, T.C. Prescribing habits in primary biliary cirrhosis: A national survey. Eur. J. Gastroenterol. Hepatol 1999, 11, 817–820. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kotb, M.A. Molecular Mechanisms of Ursodeoxycholic Acid Toxicity & Side Effects: Ursodeoxycholic Acid Freezes Regeneration & Induces Hibernation Mode. Int. J. Mol. Sci. 2012, 13, 8882-8914. https://doi.org/10.3390/ijms13078882
Kotb MA. Molecular Mechanisms of Ursodeoxycholic Acid Toxicity & Side Effects: Ursodeoxycholic Acid Freezes Regeneration & Induces Hibernation Mode. International Journal of Molecular Sciences. 2012; 13(7):8882-8914. https://doi.org/10.3390/ijms13078882
Chicago/Turabian StyleKotb, Magd A. 2012. "Molecular Mechanisms of Ursodeoxycholic Acid Toxicity & Side Effects: Ursodeoxycholic Acid Freezes Regeneration & Induces Hibernation Mode" International Journal of Molecular Sciences 13, no. 7: 8882-8914. https://doi.org/10.3390/ijms13078882