Deleterious Effects of High Dose Connexin 43 Mimetic Peptide Infusion After Cerebral Ischaemia in Near-Term Fetal Sheep

Abstract
:1. Introduction
2. Results and Discussion
2.1. Biochemistry and Survival
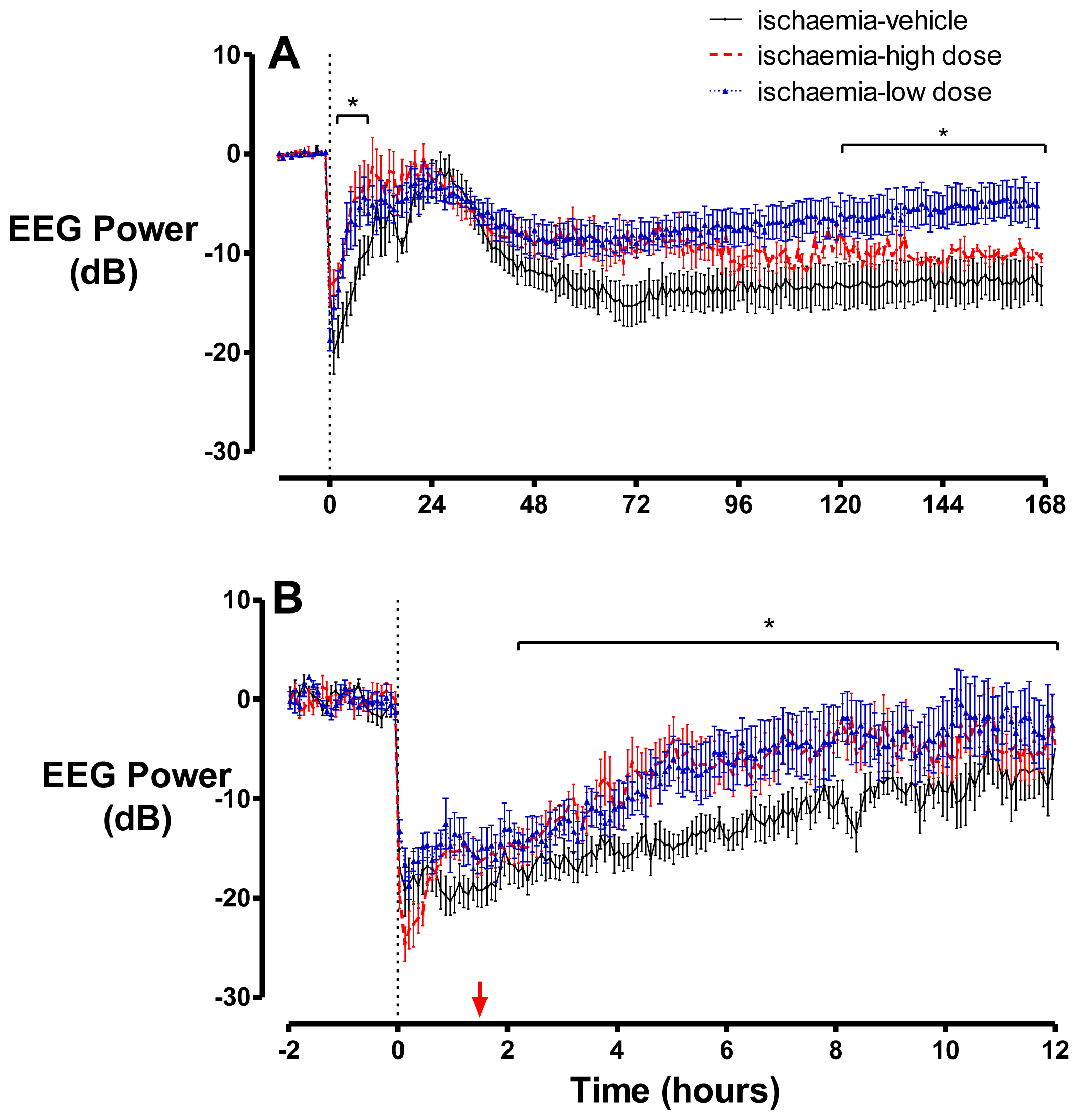
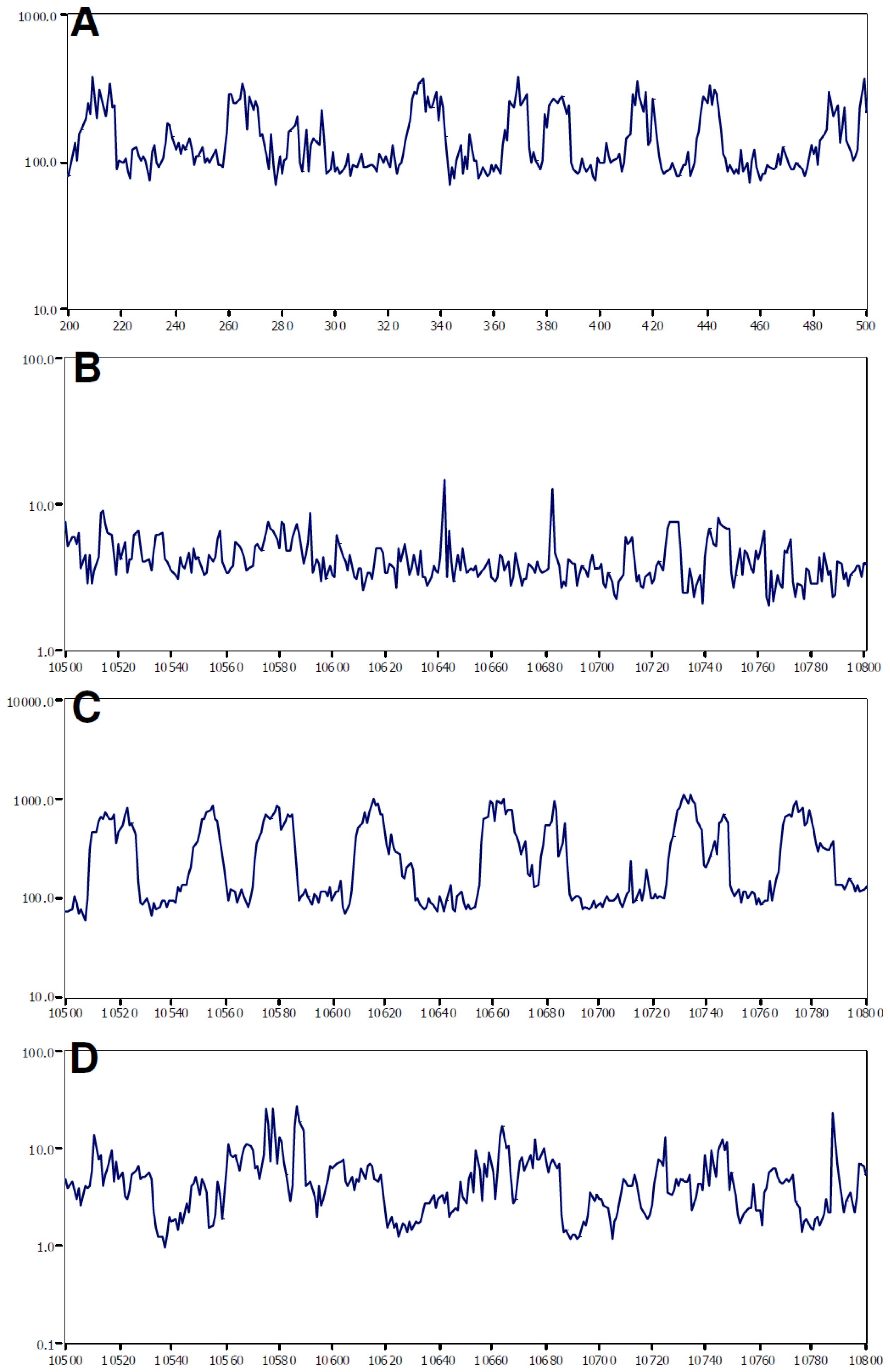
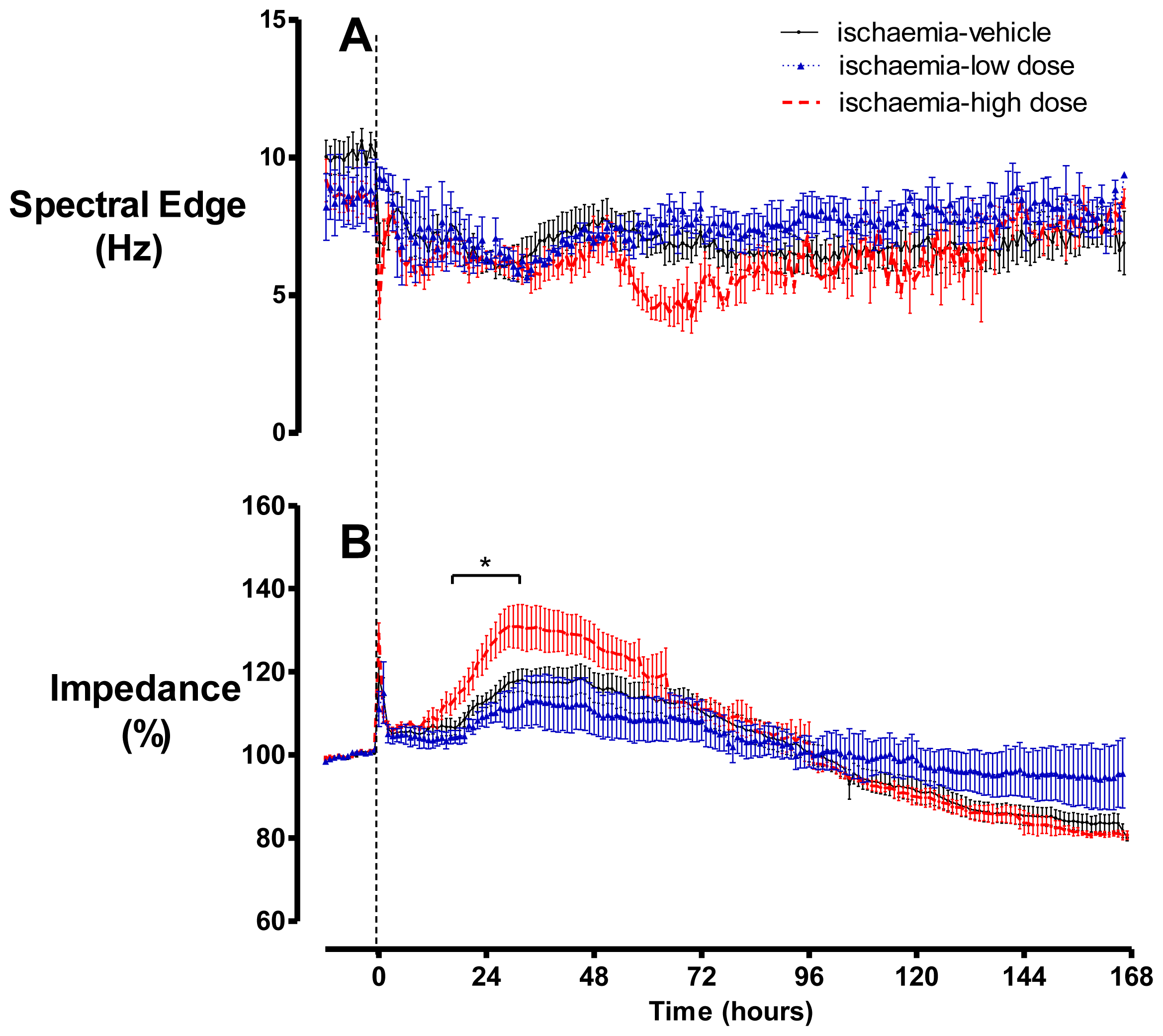
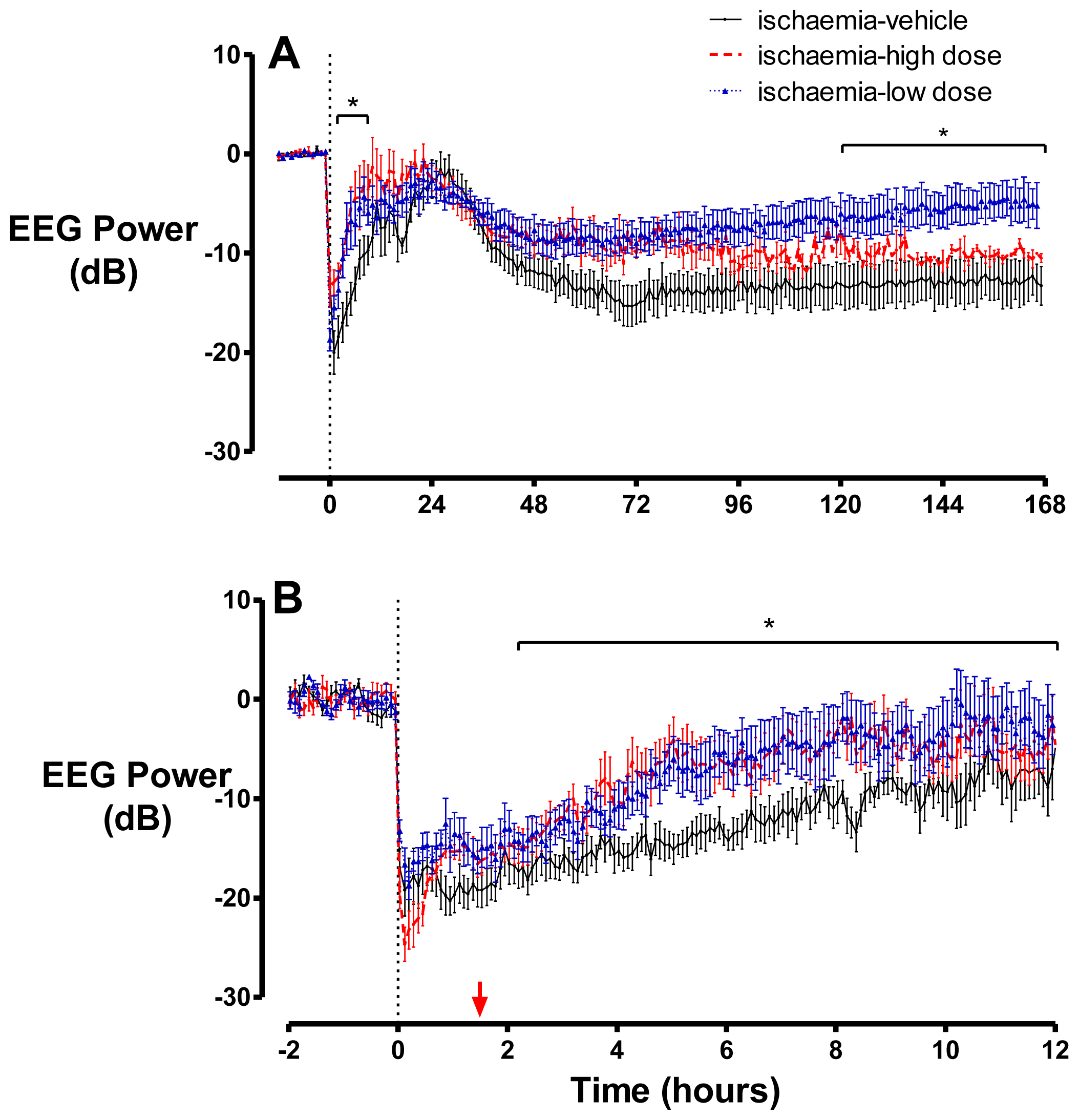
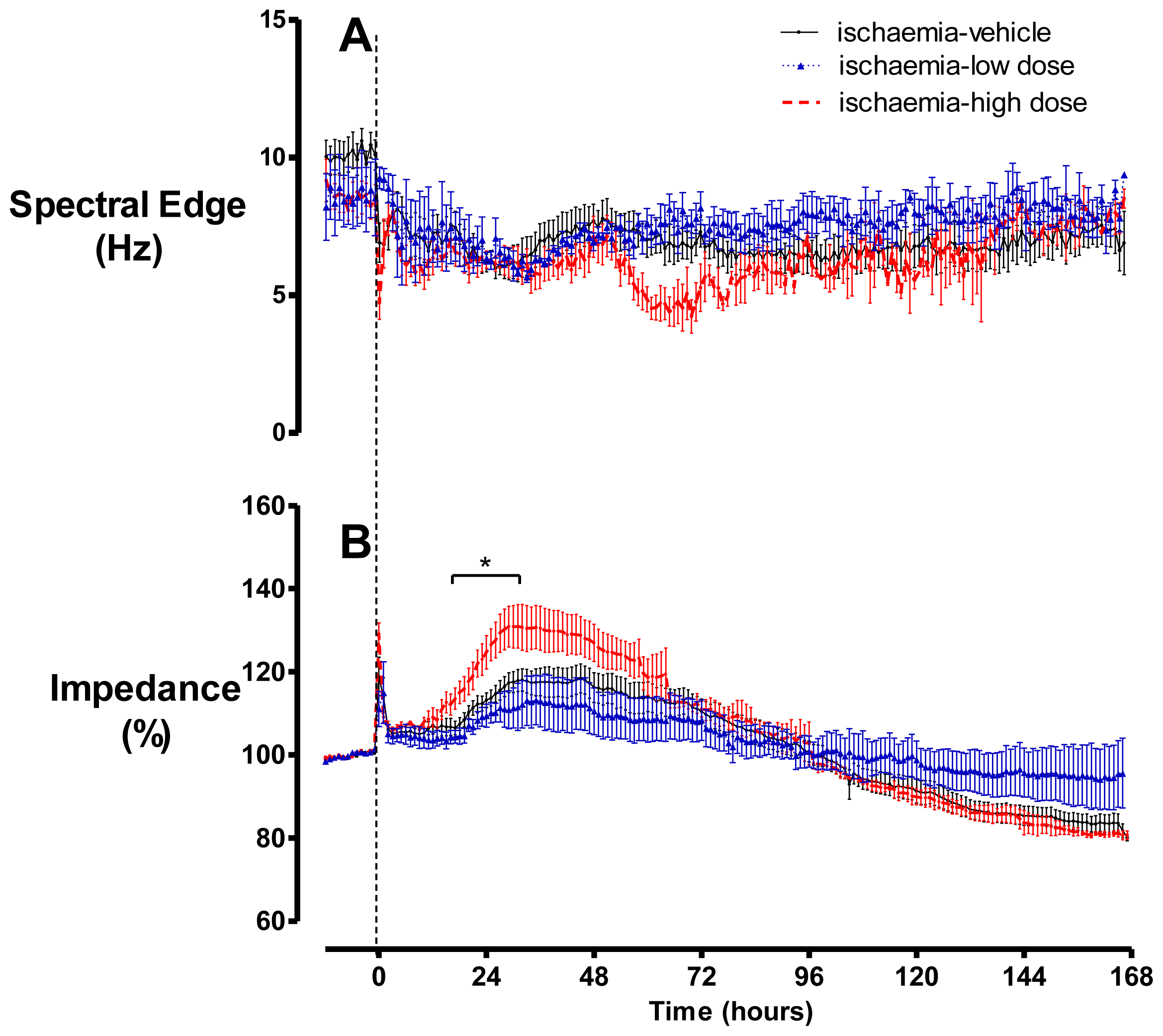
2.2. Brain Activity and Impedance
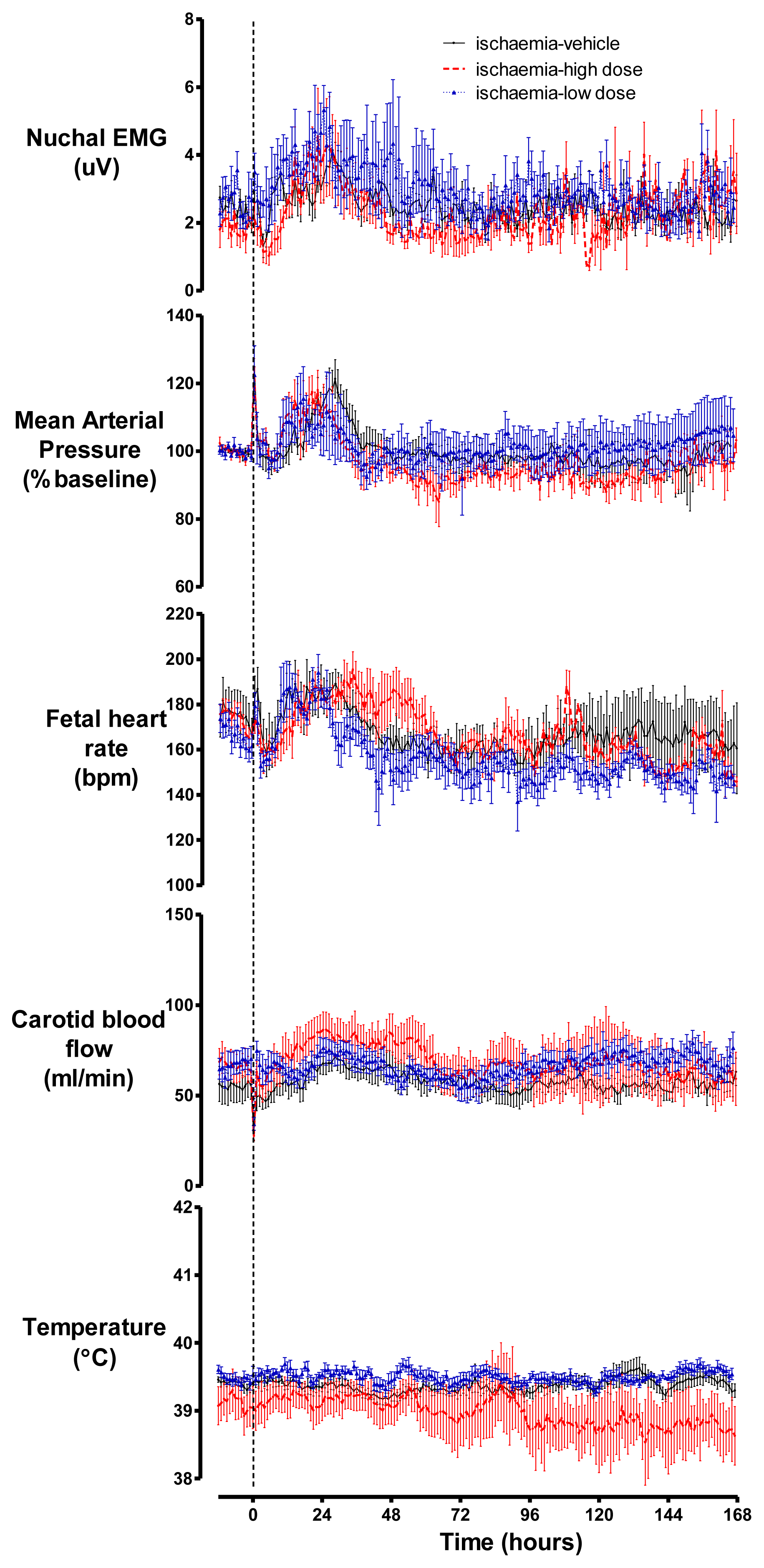
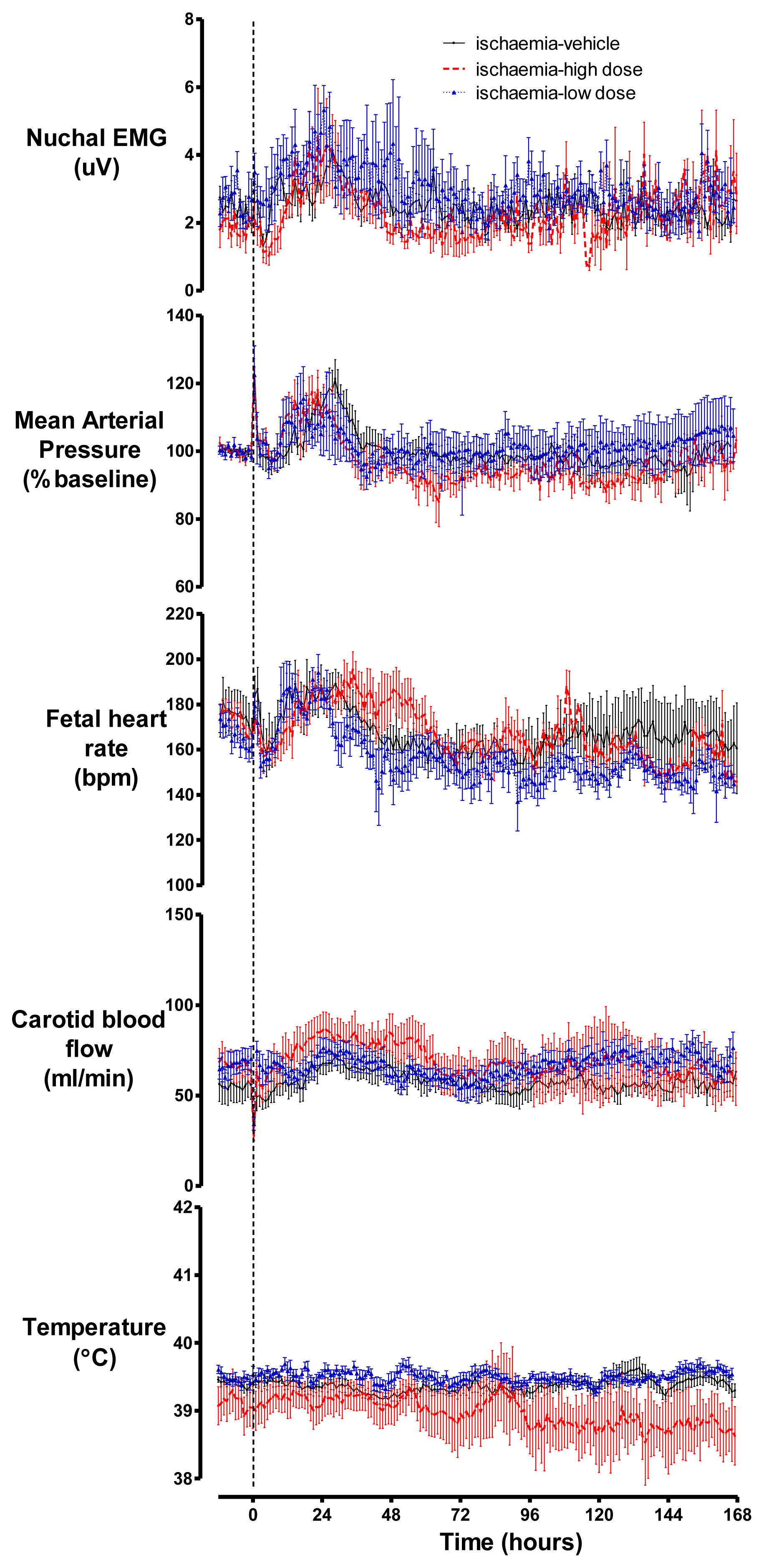
2.3. Cardiovascular Parameters, Body Movements and Temperature
2.4. Discussion
3. Experimental Section
3.1. Fetal Surgery
3.2. Post-Operative Care
3.3. Data Recording
3.4. Experimental Protocols
3.5. Data Analysis
4. Conclusions
Acknowledgments
References
- Vannucci, R.C. Hypoxic-ischemic encephalopathy. Am. J. Perinatol 2000, 17, 113–120. [Google Scholar]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. Br. Med. J 2010, 340. [Google Scholar] [CrossRef]
- Thornton, J.S.; Ordidge, R.J.; Penrice, J.; Cady, E.B.; Amess, P.N.; Punwani, S.; Clemence, M.; Wyatt, J.S. Temporal and anatomical variations of brain water apparent diffusion coefficient in perinatal cerebral hypoxic-ischemic injury: Relationships to cerebral energy metabolism. Magn. Reson. Med 1998, 39, 920–927. [Google Scholar]
- Williams, C.E.; Gunn, A.J.; Mallard, C.; Gluckman, P.D. Outcome after ischemia in the developing sheep brain: An electroencephalographic and histological study. Ann. Neurol 1992, 31, 14–21. [Google Scholar]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The “bystander effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res 1993, 53, 5274–5283. [Google Scholar]
- Lin, J.H.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.; Nedergaard, M. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci 1998, 1, 494–500. [Google Scholar]
- Frantseva, M.V.; Kokarovtseva, L.; Naus, C.G.; Carlen, P.L.; MacFabe, D.; Perez Velazquez, J.L. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J. Neurosci 2002, 22, 644–653. [Google Scholar]
- Nodin, C.; Nilsson, M.; Blomstrand, F. Gap junction blockage limits intercellular spreading of astrocytic apoptosis induced by metabolic depression. J. Neurochem 2005, 94, 1111–1123. [Google Scholar]
- Rawanduzy, A.; Hansen, A.; Hansen, T.W.; Nedergaard, M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J. Neurosurg 1997, 87, 916–920. [Google Scholar]
- de Pina-Benabou, M.H.; Szostak, V.; Kyrozis, A.; Rempe, D.; Uziel, D.; Urban-Maldonado, M.; Benabou, S.; Spray, D.C.; Federoff, H.J.; Stanton, P.K.; et al. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 2005, 36, 2232–2237. [Google Scholar]
- Nakase, T.; Sohl, G.; Theis, M.; Willecke, K.; Naus, C.C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am. J. Pathol 2004, 164, 2067–2075. [Google Scholar]
- Nakase, T.; Yoshida, Y.; Nagata, K. Enhanced connexin 43 immunoreactivity in penumbral areas in the human brain following ischemia. Glia 2006, 54, 369–375. [Google Scholar]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; O’Carroll, S.J.; Fraser, M.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol 2012, 71, 121–132. [Google Scholar]
- Li, H.; Liu, T.F.; Lazrak, A.; Peracchia, C.; Goldberg, G.S.; Lampe, P.D.; Johnson, R.G. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol 1996, 134, 1019–1030. [Google Scholar]
- Kondo, R.P.; Wang, S.Y.; John, S.A.; Weiss, J.N.; Goldhaber, J.I. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J. Mol. Cell. Cardiol 2000, 32, 1859–1872. [Google Scholar]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar]
- Decrock, E.; de Vuyst, E.; Vinken, M.; van Moorhem, M.; Vranckx, K.; Wang, N.; van Laeken, L.; de Bock, M.; D’Herde, K.; Lai, C.P.; et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ 2009, 16, 151–163. [Google Scholar]
- Orellana, J.A.; Hernandez, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.; Giaume, C.; Saez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci 2008, 28, 4702–4711. [Google Scholar]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci 2003, 23, 3588–3596. [Google Scholar]
- Rodriguez-Sinovas, A.; Cabestrero, A.; Lopez, D.; Torre, I.; Morente, M.; Abellan, A.; Miro, E.; Ruiz-Meana, M.; Garcia-Dorado, D. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol 2007, 94, 219–232. [Google Scholar]
- Quist, A.P.; Rhee, S.K.; Lin, H.; Lal, R. Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J. Cell Biol 2000, 148, 1063–1074. [Google Scholar]
- Gourine, A.V.; Dale, N.; Llaudet, E.; Poputnikov, D.M.; Spyer, K.M.; Gourine, V.N. Release of ATP in the central nervous system during systemic inflammation: Real-time measurement in the hypothalamus of conscious rabbits. J. Physiol 2007, 585, 305–316. [Google Scholar]
- Peng, W.; Cotrina, M.L.; Han, X.; Yu, H.; Bekar, L.; Blum, L.; Takano, T.; Tian, G.F.; Goldman, S.A.; Nedergaard, M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2009, 106, 12489–12493. [Google Scholar]
- Evans, C.A.; Reynolds, J.M.; Reynolds, M.L.; Saunders, N.R.; Segal, M.B. The development of a blood-brain barrier mechanism in foetal sheep. J. Physiol 1974, 238, 371–386. [Google Scholar]
- Pillai, D.R.; Dittmar, M.S.; Baldaranov, D.; Heidemann, R.M.; Henning, E.C.; Schuierer, G.; Bogdahn, U.; Schlachetzki, F. Cerebral ischemia-reperfusion injury in rats—A 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J. Cereb. Blood Flow Metab 2009, 29, 1846–1855. [Google Scholar]
- Dobbing, J.; Sands, J. Timing of neuroblast multiplication in developing human brain. Nature 1970, 226, 639–640. [Google Scholar]
- McIntosh, G.H.; Baghurst, K.I.; Potter, B.J.; Hetzel, B.S. Foetal brain development in the sheep. Neuropathol. Appl. Neurobiol 1979, 5, 103–114. [Google Scholar]
- Williams, C.E.; Gunn, A.J.; Synek, B.; Gluckman, P.D. Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr. Res 1990, 27, 561–565. [Google Scholar]
- Van Rooij, L.G.; Toet, M.C.; Osredkar, D.; van Huffelen, A.C.; Groenendaal, F.; de Vries, L.S. Recovery of amplitude integrated electroencephalographic background patterns within 24 hours of perinatal asphyxia. Arch. Dis. Child. Fetal Neonatal Ed 2005, 90, F245–F251. [Google Scholar]
- Murray, D.M.; Boylan, G.B.; Ryan, C.A.; Connolly, S. Early EEG findings in hypoxic-ischemic encephalopathy predict outcomes at 2 years. Pediatrics 2009, 124, e459–e467. [Google Scholar]
- Williams, C.E.; Gunn, A.; Gluckman, P.D. Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 1991, 22, 516–521. [Google Scholar]
- Araque, A.; Sanzgiri, R.P.; Parpura, V.; Haydon, P.G. Astrocyte-induced modulation of synaptic transmission. Can. J. Physiol. Pharmacol 1999, 77, 699–706. [Google Scholar]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFα. Science 2002, 295, 2282–2285. [Google Scholar]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–1086. [Google Scholar]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar]
- Cotrina, M.L.; Lin, J.H.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar]
- Fiacco, T.A.; McCarthy, K.D. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci 2004, 24, 722–732. [Google Scholar]
- Parri, H.R.; Gould, T.M.; Crunelli, V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat. Neurosci 2001, 4, 803–812. [Google Scholar]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzan, T.; Volterra, A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998, 391, 281–285. [Google Scholar]
- Kang, J.; Jiang, L.; Goldman, S.A.; Nedergaard, M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci 1998, 1, 683–692. [Google Scholar]
- Gordon, G.R.; Baimoukhametova, D.V.; Hewitt, S.A.; Rajapaksha, W.R.; Fisher, T.E.; Bains, J.S. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci 2005, 8, 1078–1086. [Google Scholar]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar]
- Sarkar, S.; Barks, J.D.; Donn, S.M. Should amplitude-integrated electroencephalography be used to identify infants suitable for hypothermic neuroprotection? J. Perinatol 2008, 28, 117–122. [Google Scholar]
- Keogh, M.J.; Drury, P.P.; Bennet, L.; Davidson, J.O.; Mathai, S.; Gunn, E.R.; Booth, L.C.; Gunn, A.J. Limited predictive value of early changes in EEG spectral power for neural injury after asphyxia in preterm fetal sheep. Pediatr. Res 2012, 71, 345–353. [Google Scholar]
- Cotrina, M.L.; Kang, J.; Lin, J.H.; Bueno, E.; Hansen, T.W.; He, L.; Liu, Y.; Nedergaard, M. Astrocytic gap junctions remain open during ischemic conditions. J. Neurosci 1998, 18, 2520–2537. [Google Scholar]
- Lurtz, M.M.; Louis, C.F. Intracellular calcium regulation of connexin43. Am. J. Physiol. Cell physiol 2007, 293, C1806–C1813. [Google Scholar]
- Kristian, T.; Katsura, K.; Siesjo, B.K. The influence of moderate hypothermia on cellular calcium uptake in complete ischaemia: Implications for the excitotoxic hypothesis. Acta Physiol. Scand 1992, 146, 531–532. [Google Scholar]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes 2008, 15, 27–42. [Google Scholar]
- Ment, L.R.; Peterson, B.S.; Vohr, B.; Allan, W.; Schneider, K.C.; Lacadie, C.; Katz, K.H.; Maller-Kesselman, J.; Pugh, K.; Duncan, C.C.; et al. Cortical recruitment patterns in children born prematurely compared with control subjects during a passive listening functional magnetic resonance imaging task. J. Pediatr 2006, 149, 490–498. [Google Scholar]
- Desplantez, T.; Verma, V.; Leybaert, L.; Evans, W.H.; Weingart, R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol. Res 2012, 65, 546–552. [Google Scholar]
- Counsell, S.J.; Edwards, A.D.; Chew, A.T.; Anjari, M.; Dyet, L.E.; Srinivasan, L.; Boardman, J.P.; Allsop, J.M.; Hajnal, J.V.; Rutherford, M.A.; et al. Specific relations between neurodevelopmental abilities and white matter microstructure in children born preterm. Brain 2008, 131, 3201–3208. [Google Scholar]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflugers Archiv. Eur. J. Physiol 2010, 460, 583–592. [Google Scholar]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol 2003, 13, 1697–1703. [Google Scholar]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar]
- Wallraff, A.; Kohling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhauser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci 2006, 26, 5438–5447. [Google Scholar]
- Takahashi, Y.; de Vroomen, M.; Gournay, V.; Roman, C.; Rudolph, A.M.; Heymann, M.A. Mechanisms of adrenomedullin-induced increase of pulmonary blood flow in fetal sheep. Pediatr. Res 1999, 45, 276–281. [Google Scholar]
- Vyskocil, F.; Kritz, N.; Bures, J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res 1972, 39, 255–259. [Google Scholar]
- Tsacopoulos, M.; Magistretti, P.J. Metabolic coupling between glia and neurons. J. Neurosci 1996, 16, 877–885. [Google Scholar]
- Siesjo, B.K. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab 1981, 1, 155–185. [Google Scholar]
- Katzman, R.; Clasen, R.; Klatzo, I.; Meyer, J.S.; Pappius, H.M.; Waltz, A.G. Report of Joint Committee for Stroke Resources. IV. Brain edema in stroke. Stroke 1977, 8, 512–540. [Google Scholar]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Invest 1997, 99, 248–256. [Google Scholar]
- Quaedackers, J.S.; Roelfsema, V.; Hunter, C.J.; Heineman, E.; Gunn, A.J.; Bennet, L. Polyuria and impaired renal blood flow after asphyxia in preterm fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol 2004, 286, R576–R583. [Google Scholar]
- Wibbens, B.; Westgate, J.A.; Bennet, L.; Roelfsema, V.; de Haan, H.H.; Hunter, C.J.; Gunn, A.J. Profound hypotension and associated ECG changes during prolonged cord occlusion in the near term fetal sheep. Am. J. Obstet. Gynecol 2005, 193, 803–810. [Google Scholar]
- Scher, M.S.; Hamid, M.Y.; Steppe, D.A.; Beggarly, M.E.; Painter, M.J. Ictal and interictal electrographic seizure durations in preterm and term neonates. Epilepsia 1993, 34, 284–288. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Baseline | 2 h | 4 h | 6 h | 1 day | 2 day | 7day |
|---|---|---|---|---|---|---|---|
| Ischaemiavehicle | 7.40 ± 0.01 | 7.39 ± 0.01 | 7.41 ± 0.01 | 7.40 ± 0.01 | 7.375 ± 0.02 | 7.39 ± 0.01 | 7.36 ± 0.01 |
| Ischaemia-low | 7.40 ± 0.01 | 7.40 ± 0.01 | 7.41 ± 0.01 | 7.41 ± 0.01 | 7.38 ± 0.02 | 7.38 ± 0.01 | 7.35 ± 0.01 * |
| Ischaemia-high | 7.40 ± 0.01 | 7.40 ± 0.01 | 7.42 ± 0.01 | 7.41 ± 0.01 | 7.31 ± 0.05 † | 7.39 ± 0.01 | 7.35 ± 0.00 |
| PCO2 (mmHg) | Baseline | 2 h | 4 h | 6 h | 1 day | 2 day | 7 day |
| Ischaemiavehicle | 44.6 ± 1.4 | 42.6 ± 1.6 | 43.4 ± 1.4 | 44.9 ± 1.9 | 44.4 ± 2.7 | 44.13 ± 1.3 | 49.4 ± 1.9 † |
| Ischaemia-low | 45.1 ± 1.7 | 44.8 ± 1.2 | 44.8 ± 1.6 | 46.6 ± 1.4 | 44.3 ± 1.4 | 42.73 ± 1.7 | 46.2 ± 1.4 |
| Ischaemia-high | 46.6 ± 0.9 | 45.2 ± 1.1 | 45.5 ± 0.7 | 46.3 ± 0.9 | 45.3 ± 0.6 | 47.98 ± 1.6 | 48.7 ± 2.1 |
| PO2 (mmHg) | Baseline | 2 h | 4 h | 6 h | 1 day * | 2 day | 7 day |
| Ischaemiavehicle | 26.0 ± 1.1 | 25.8 ± 1.2 | 25.9 ± 1.25 | 25.4 ± 1.9 | 25.0 ± 2.0 | 24.6 ± 1.5† | 23.0 ± 1.1 † |
| Ischaemia-low | 23.5 ± 0.9 | 22.9 ± 0.9 | 25.2 ± 1.48 | 24.6 ± 0.5 | 22.1 ± 1.5 | 23.4 ± 0.9 | 22.7 ± 1.0 |
| Ischaemia-high | 20.7 ± 1.0 | 21.4 ± 1.0 | 22.9 ± 1.45 | 21.2 ± 1.5 | 18.0 ± 1.2 | 18.8 ± 2.6 | 21.6 ± 2.5 |
| Lactate (mmol/L) | Baseline | 2 h | 4 h | 6 h | 1 day | 2 day | 7 day |
| Ischaemiavehicle | 1.1 ± 0.1 | 2.4 ± 0.5 | 2.3 ± 0.5 | 2.0 ± 0.4 | 4.7 ± 1.1 † | 0.94 ± 0.1 | 1.2 ± 0.1 |
| Ischaemia-low | 1.1 ± 0.1 | 2.1 ± 0.3 | 2.0 ± 0.2 | 1.6 ± 0.2 | 2.3 ± 0.5 † | 1.46 ± 0.2 | 1.1 ± 0.0 |
| Ischaemia-high | 1.2 ± 0.1 | 2.4 ± 0.3 | 2.1 ± 0.2 | 1.9 ± 0.2 | 10.2 ± 3.3 † | 4.21 ± 1.9 | 1.1 ± 0.1 |
| Glucose (mmol/L) | Baseline | 2 h | 4 h | 6 h | 1 day | 2 day | 7 day |
| Ischaemiavehicle | 0.9 ± 0.1 | 1.3 ± 0.1 † | 1.3 ± 0.1 † | 1.3 ± 0.1† | 1.5 ± 0.1 † | 0.9 ± 0.1 | 1.0 ± 0.1 |
| Ischaemia-low | 0.8 ± 0.1 | 1.3 ± 0.1 † | 1.2 ± 0.1 † | 1.2 ± 0.1† | 1.1 ± 0.1 † | 1.0 ± 0.1 | 0.9 ± 0.1 |
| Ischaemia-high | 0.8 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.4 ± 0.2 † | 0.9 ± 0.1 | 0.8 ± 0.2 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; Bennet, L.; Gunn, A.J. Deleterious Effects of High Dose Connexin 43 Mimetic Peptide Infusion After Cerebral Ischaemia in Near-Term Fetal Sheep. Int. J. Mol. Sci. 2012, 13, 6303-6319. https://doi.org/10.3390/ijms13056303
Davidson JO, Green CR, Nicholson LFB, Bennet L, Gunn AJ. Deleterious Effects of High Dose Connexin 43 Mimetic Peptide Infusion After Cerebral Ischaemia in Near-Term Fetal Sheep. International Journal of Molecular Sciences. 2012; 13(5):6303-6319. https://doi.org/10.3390/ijms13056303
Chicago/Turabian StyleDavidson, Joanne O., Colin R. Green, Louise F. B. Nicholson, Laura Bennet, and Alistair J. Gunn. 2012. "Deleterious Effects of High Dose Connexin 43 Mimetic Peptide Infusion After Cerebral Ischaemia in Near-Term Fetal Sheep" International Journal of Molecular Sciences 13, no. 5: 6303-6319. https://doi.org/10.3390/ijms13056303