Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
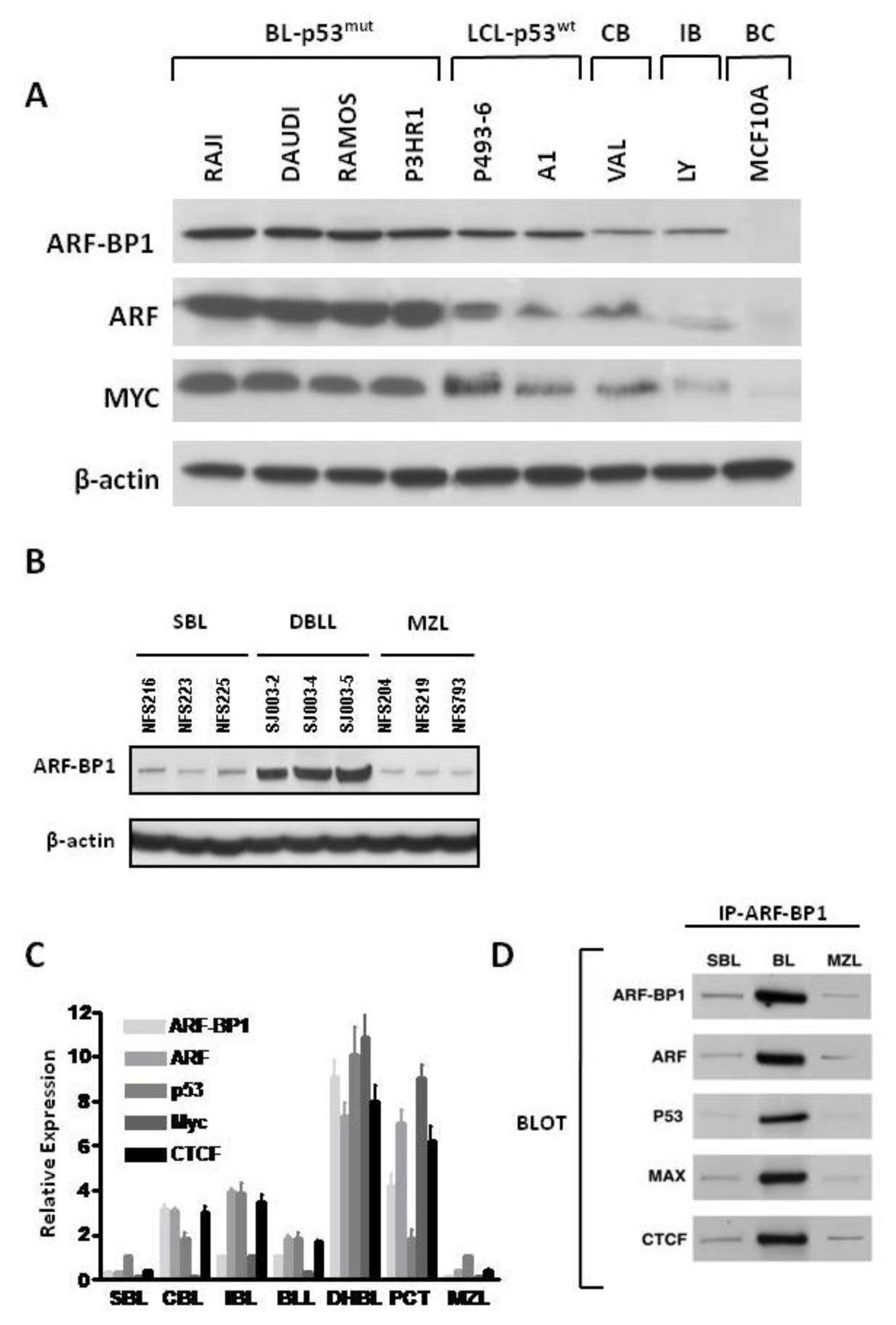
2.1. ARF-BP-1 Is Expressed at High Levels in MYC-Driven Human BL and Mouse DBLL
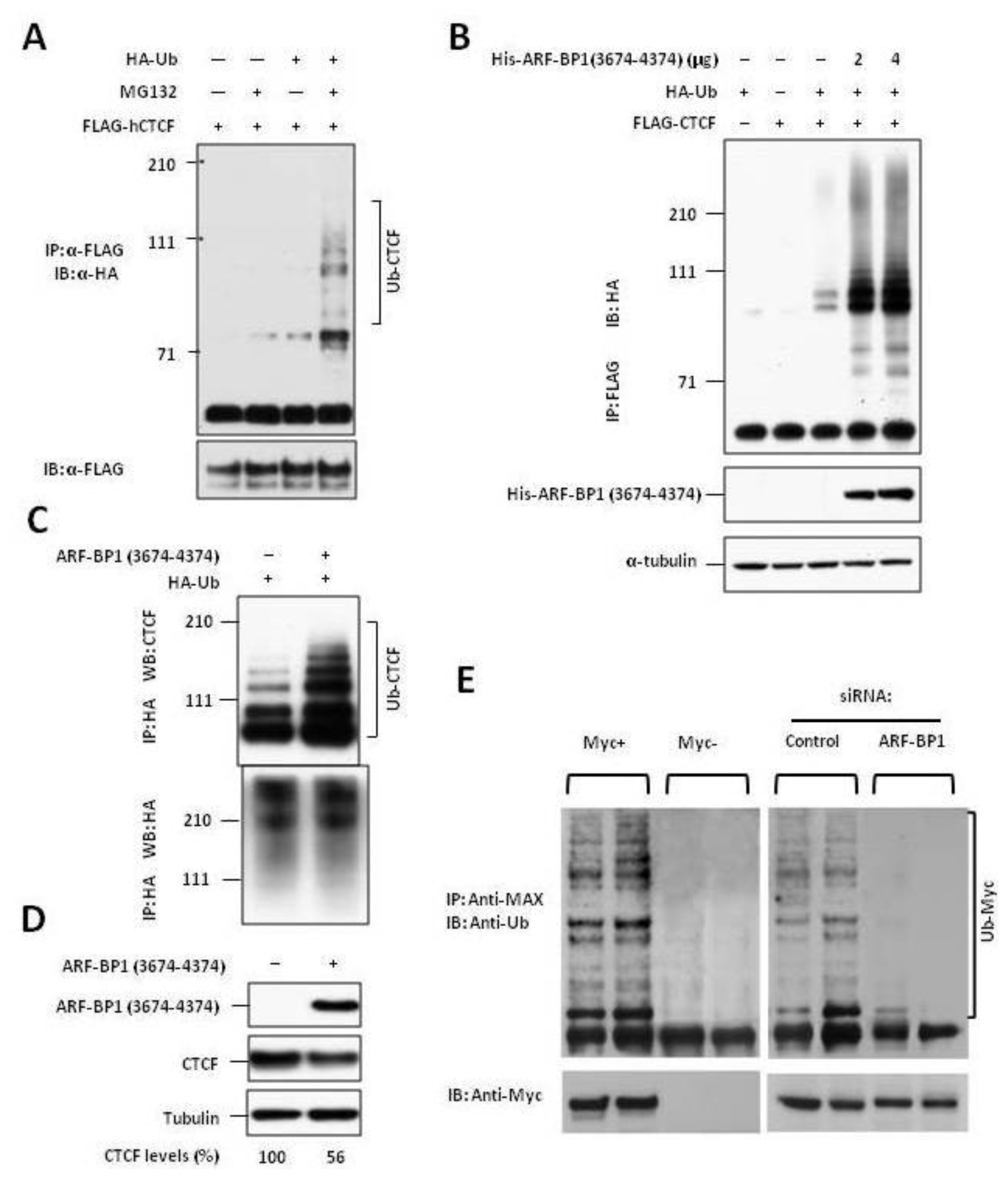
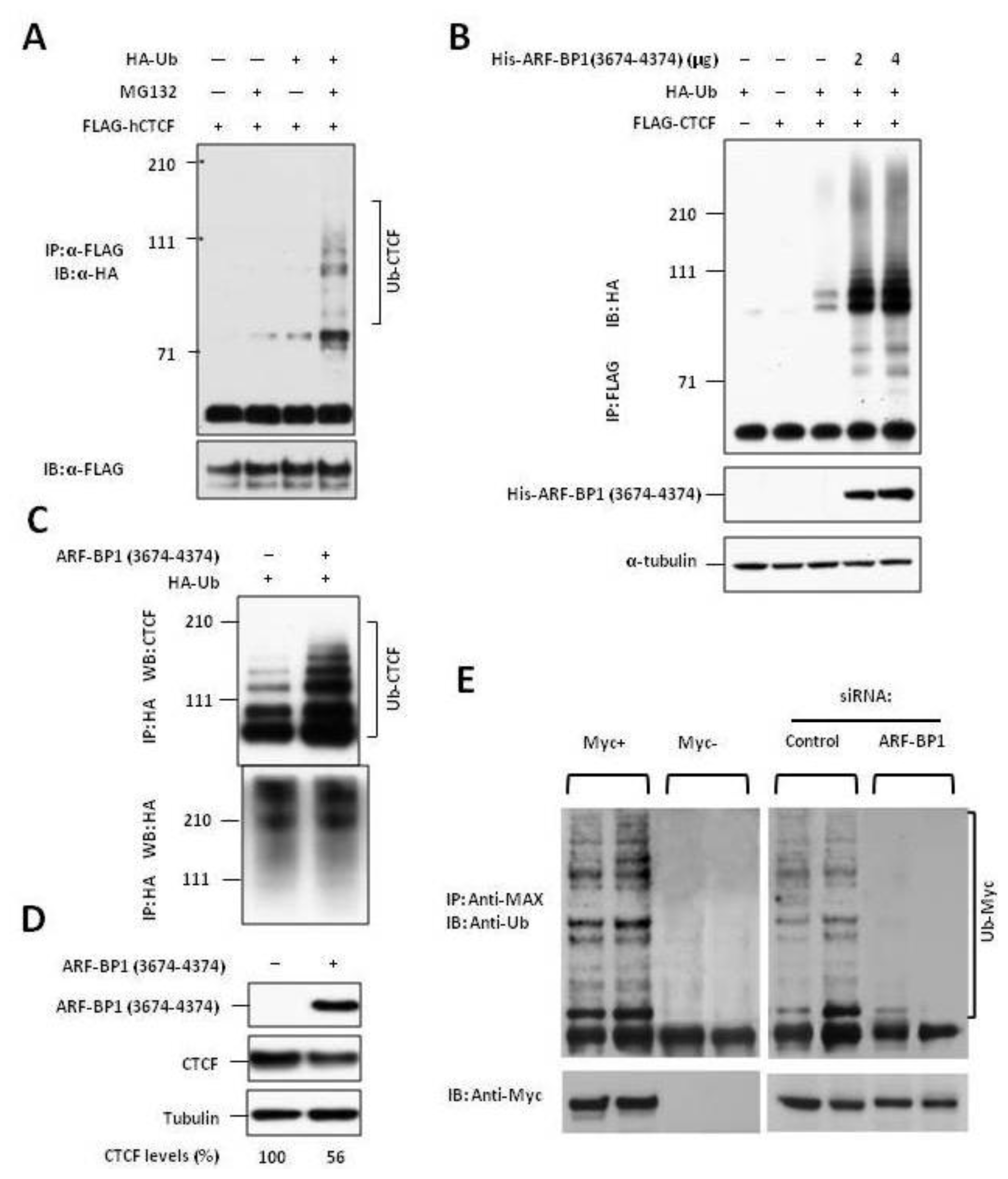
2.2. ARF-BP1 Binds to and Ubiquitylates CTCF
2.3. ARF-BP1 Ubiquitylates MYC in B Lineage Cells
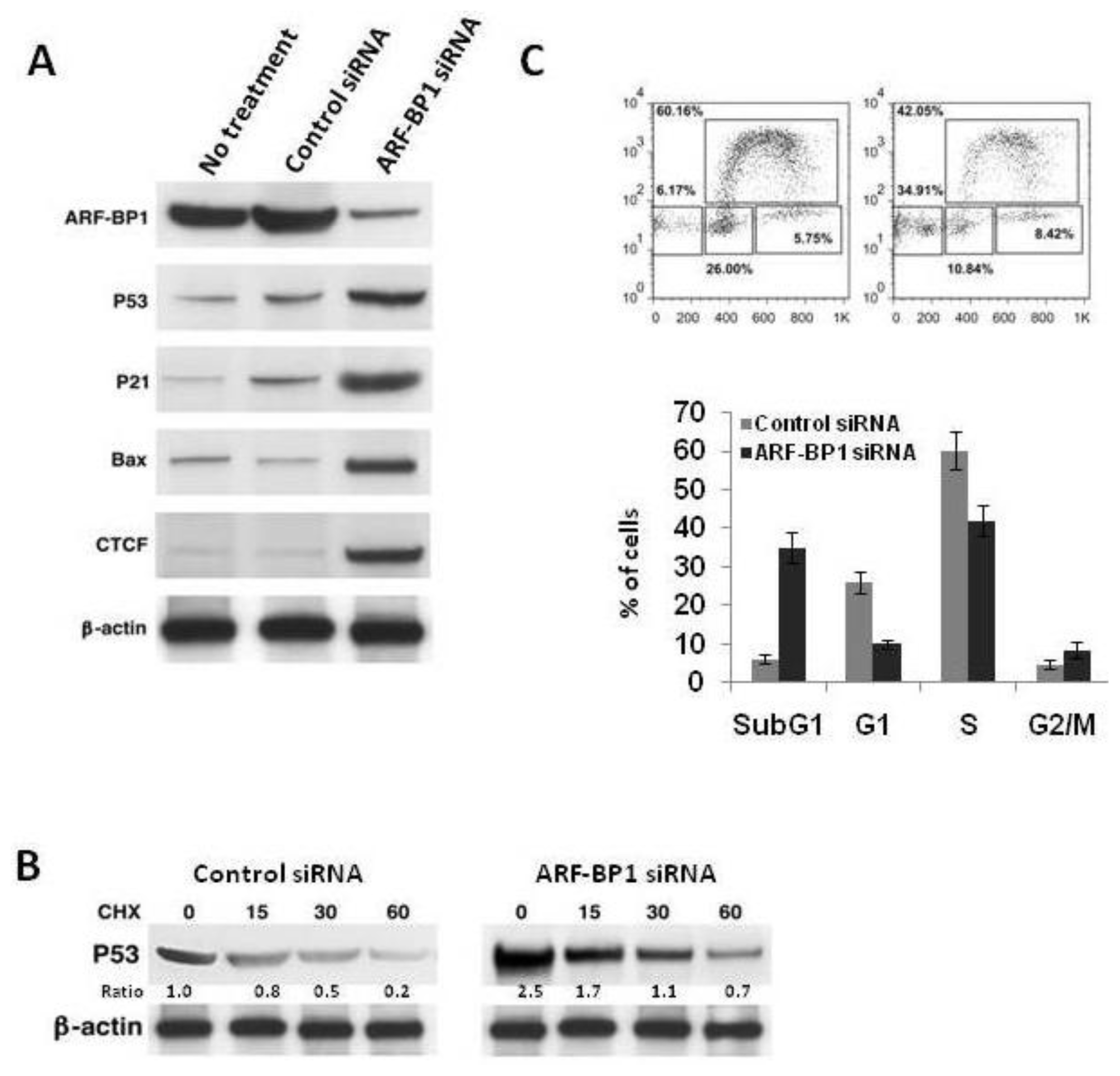
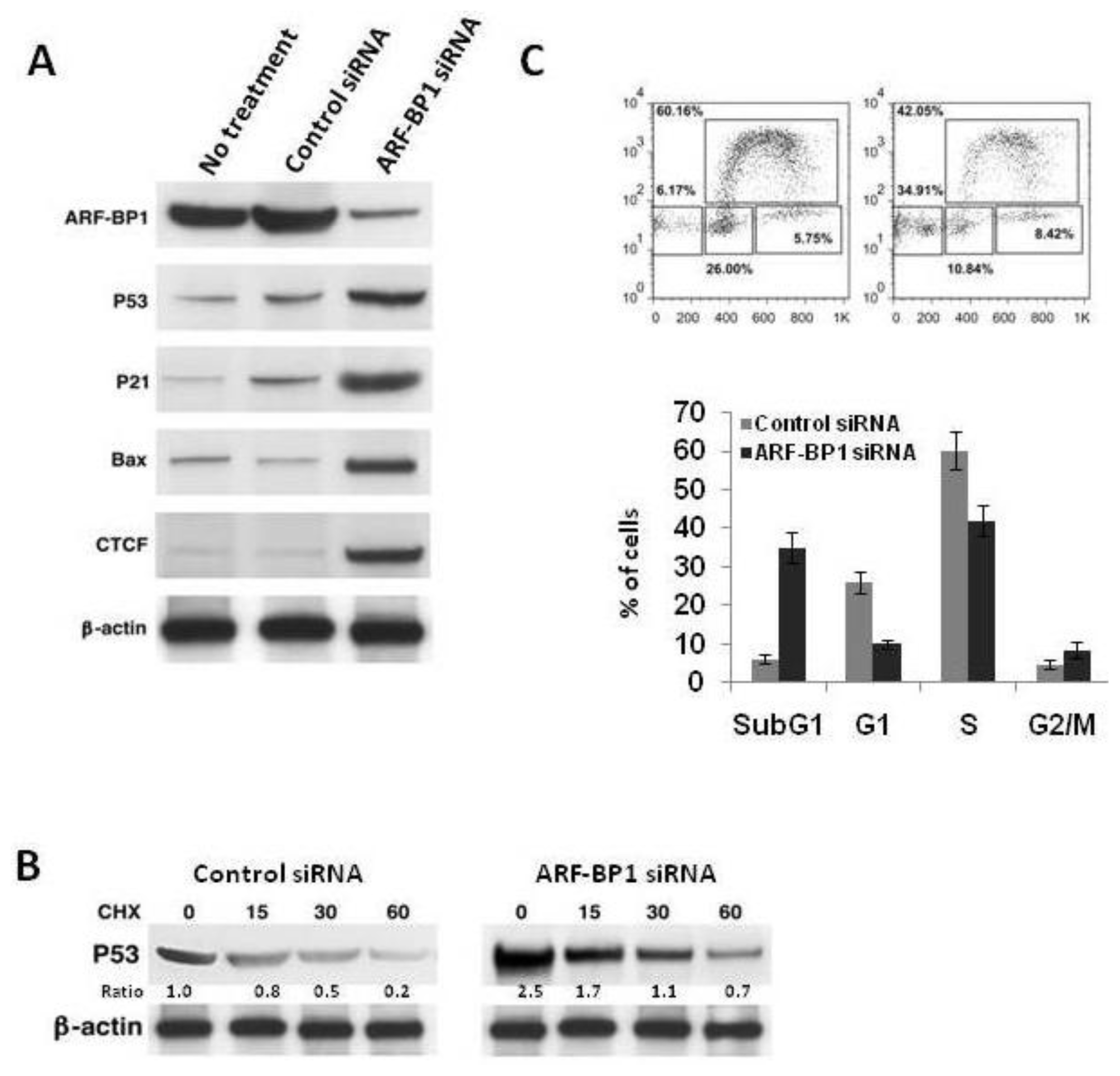
2.4. Inactivation of ARF-BP1 Stabilizes p53, Induces p53-Dependent Apoptosis, and Reduces Cell Proliferation in λ-MYC TG Cells
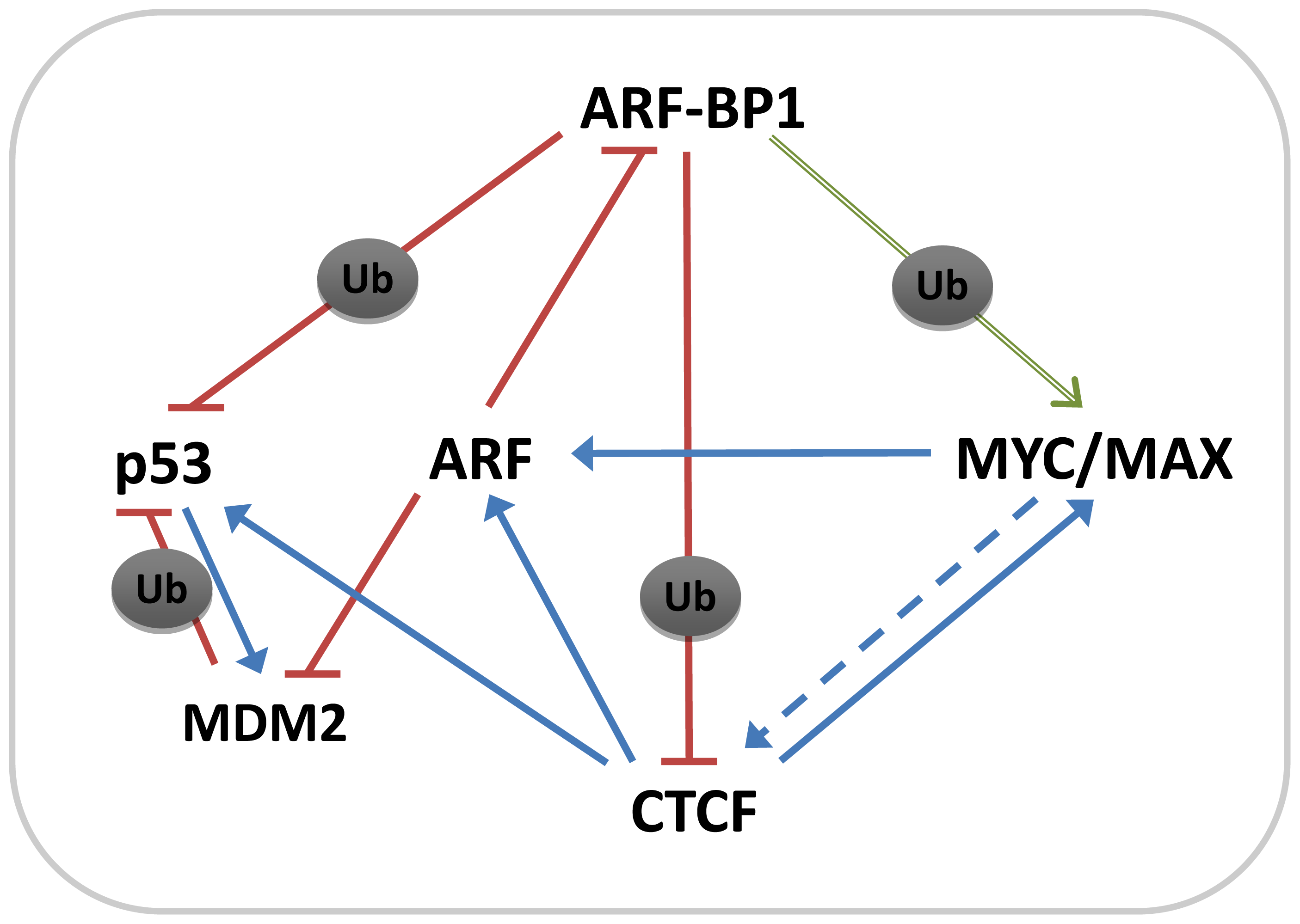
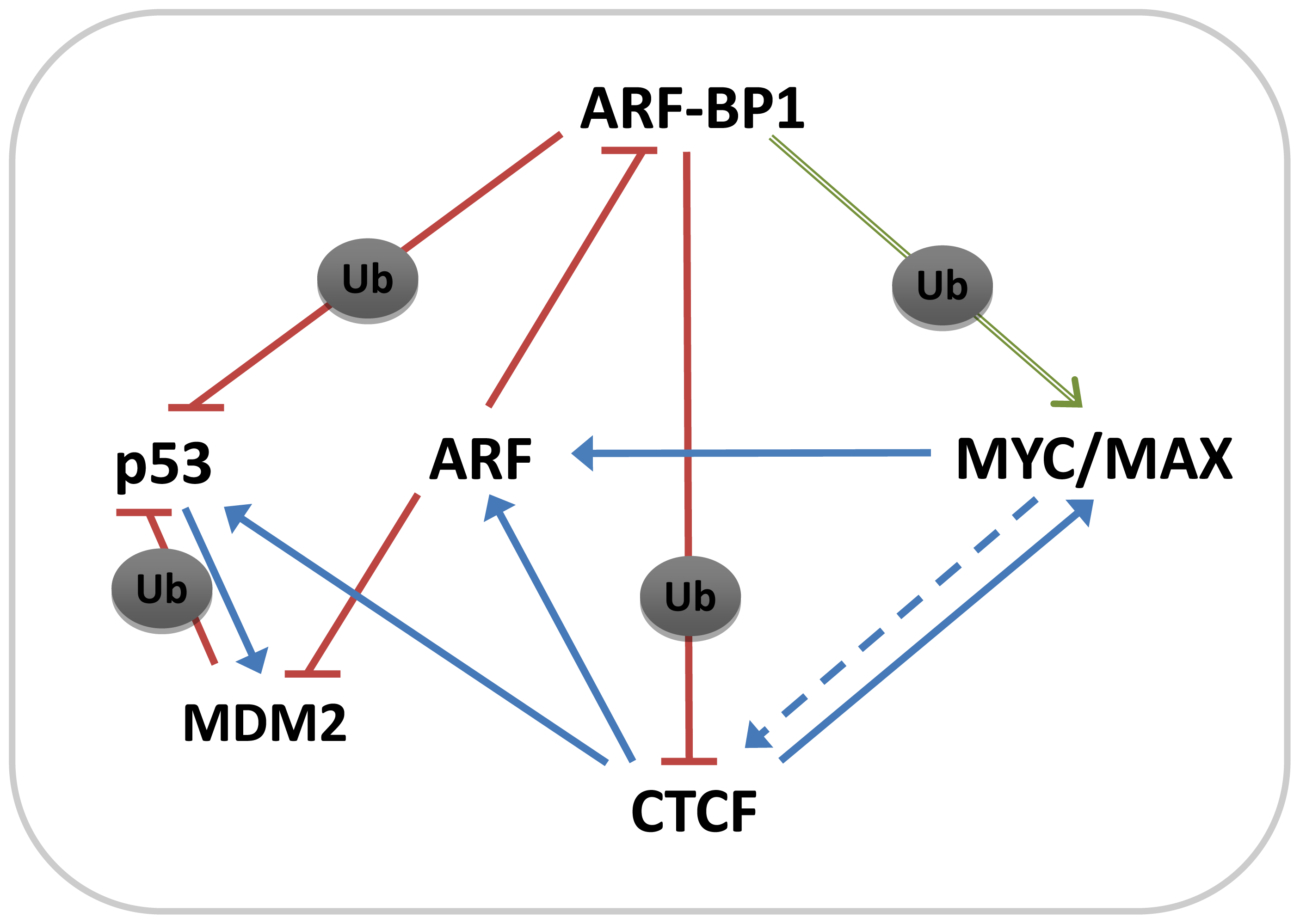
3. Discussion
4. Experimental Section
4.1. Mice and Cell Lines
4.2. Protein extraction, Western Blotting, and Immunoprecipitation
4.3. RNA Isolation and Analysis by Quantitative RT-PCR (qRT-PCR)
4.4. Ubiquitylation Assay
4.5. Cell Cycle Analysis
5. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
References
- Dave, S.S.; Fu, K.; Wright, G.W.; Lam, L.T.; Kluin, P.; Boerma, E.J.; Greiner, T.C.; Weisenburger, D.D.; Rosenwald, A.; Ott, G.; et al. Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med 2006, 354, 2431–2442. [Google Scholar]
- Hann, S.R.; Eisenman, R.N. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol. Cell Biol 1984, 4, 2486–2497. [Google Scholar]
- Malempati, S.; Tibbitts, D.; Cunningham, M.; Akkari, Y.; Olson, S.; Fan, G.; Sears, R.C. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 2006, 20, 1572–1581. [Google Scholar]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 regulates Myc protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar]
- von der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; Nakayama, K.I.; et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004, 23, 2116–2125. [Google Scholar]
- Zhao, X.; Heng, J.I.; Guardavaccaro, D.; Jiang, R.; Pagano, M.; Guillemot, F.; Iavarone, A.; Lasorella, A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol 2008, 10, 643–653. [Google Scholar]
- Choi, S.H.; Wright, J.B.; Gerber, S.A.; Cole, M.D. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev 2010, 24, 1236–1241. [Google Scholar]
- Old, J.B.; Kratzat, S.; Hoellein, A.; Graf, S.; Nilsson, J.A.; Nilsson, L.; Nakayama, K.I.; Peschel, C.; Cleveland, J.L.; Keller, U.B. Skp2 directs Myc-mediated suppression of p27Kip1 yet has modest effects on Myc-driven lymphomagenesis. Mol. Cancer Res 2010, 8, 353–362. [Google Scholar]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar]
- Kovalchuk, A.L.; Qi, C.F.; Torrey, T.A.; Taddesse-Heath, L.; Feigenbaum, L.; Park, S.S.; Gerbitz, A.; Klobeck, G.; Hoertnagel, K.; Polack, A.; et al. Burkitt lymphoma in the mouse. J. Exp. Med 2000, 192, 1183–1190. [Google Scholar]
- Janz, S.; Morse, H.C.; Teitell, M.A. Mouse Models of Human Mature B-Cell and Plasma Cell Neoplasms. In Mouse Models Human Blood Cancers; Li, S., Ed.; Springer: New York, NY, USA, 2008; pp. 179–225. [Google Scholar]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005, 121, 1071–1083. [Google Scholar]
- Hao, Z.; Duncan, G.S.; Su, Y.W.; Li, W.Y.; Silvester, J.; Hong, C.; You, H.; Brenner, D.; Gorrini, C.; Haight, J.; et al. The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med 2012, 209, 173–186. [Google Scholar]
- Chen, D.; Brooks, C.L.; Gu, W. ARF-BP1 as a potential therapeutic target. Br. J. Cancer 2006, 94, 1555–1558. [Google Scholar]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev 2008, 22, 2755–2766. [Google Scholar]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar]
- Smith, D.P.; Bath, M.L.; Metcalf, D.; Harris, A.W.; Cory, S. MYC levels govern hematopoietic tumor type and latency in transgenic mice. Blood 2006, 108, 653–661. [Google Scholar]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol 2005, 6, 635–645. [Google Scholar]
- Schuhmacher, M.; Staege, M.S.; Pajic, A.; Polack, A.; Weidle, U.H.; Bornkamm, G.W.; Eick, D.; Kohlhuber, F. Control of cell growth by c-Myc in the absence of cell division. Curr. Biol 1999, 9, 1255–1258. [Google Scholar]
- Tai, E.; Benchimol, S. TRIMming p53 for ubiquitination. Proc. Natl. Acad. Sci. USA 2009, 106, 11431–11432. [Google Scholar]
- Qi, Y.; Gregory, M.A.; Li, Z.; Brousal, J.P.; West, K.; Hann, S.R. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 2004, 431, 712–717. [Google Scholar]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar]
- Lobanenkov, V. Laboratory of Immunogenetics. Personal communication, NIAID/NIH: Rockville, MD, USA, 2012. [Google Scholar]
- El-Kady, A.; Klenova, E. Regulation of the transcription factor, CTCF, by phosphorylation with protein kinase CK2. FEBS Lett 2005, 579, 1424–1434. [Google Scholar]
- MacPherson, M.J.; Beatty, L.G.; Zhou, W.; Du, M.; Sadowski, P.D. The CTCF insulator protein is posttranslationally modified by SUMO. Mol. Cell Biol 2009, 29, 714–725. [Google Scholar]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat. Genet 2004, 36, 1105–1110. [Google Scholar]
- van de Nobelen, S.; Rosa-Garrido, M.; Leers, J.; Heath, H.; Soochit, W.; Joosen, L.; Jonkers, I.; Demmers, J.; van der Reijden, M.; Torrano, V.; et al. CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 2010, 3. [Google Scholar] [CrossRef]
- Soto-Reyes, E.; Recillas-Targa, F. Epigenetic regulation of the human p53 gene promoter by the CTCF transcription factor in transformed cell lines. Oncogene 2010, 29, 2217–2227. [Google Scholar]
- Rodriguez, C.; Borgel, J.; Court, F.; Cathala, G.; Forne, T.; Piette, J. CTCF is a DNA methylation-sensitive positive regulator of the INK/ARF locus. Biochem. Biophys. Res. Commun 2010, 392, 129–134. [Google Scholar]
- Gombert, M.W.; Krumm, A. Targeted deletion of multiple CTCF-binding elements in the human C-MYC gene reveals a requirement for CTCF in C-MYC expression. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Rasko, J.E.; Klenova, E.M.; Leon, J.; Filippova, G.N.; Loukinov, D.I.; Vatolin, S.; Robinson, A.F.; Hu, Y.J.; Ulmer, J.; Ward, M.D.; et al. Cell growth inhibition by the multifunctional multivalent zinc-finger factor CTCF. Cancer Res 2001, 61, 6002–6007. [Google Scholar]
- Qi, C.F.; Martensson, A.; Mattioli, M.; Dalla-Favera, R.; Lobanenkov, V.V.; Morse, H.C., III. CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc. Natl. Acad. Sci. USA 2003, 100, 633–638. [Google Scholar]
- Zlatanova, J.; Caiafa, P. CTCF and its protein partners: Divide and rule? J. Cell Sci 2009, 122, 1275–1284. [Google Scholar]
- Klenova, E.M.; Chernukhin, I.V.; El-Kady, A.; Lee, R.E.; Pugacheva, E.M.; Loukinov, D.I.; Goodwin, G.H.; Delgado, D.; Filippova, G.N.; Leon, J.; et al. Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol. Cell Biol 2001, 21, 2221–2234. [Google Scholar]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar]
- Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Destruction of Myc by ubiquitin-mediated proteolysis: Cancer-associated and transforming mutations stabilize Myc. EMBO J 1999, 18, 717–726. [Google Scholar]
- Welcker, M.; Orian, A.; Grim, J.E.; Eisenman, R.N.; Clurman, B.E. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol 2004, 14, 1852–1857. [Google Scholar]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol 2004, 6, 308–318. [Google Scholar]
- Amati, B. Myc degradation: Dancing with ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2004, 101, 8843–8844. [Google Scholar]
- Kon, N.; Zhong, J.; Qiang, L.; Accili, D.; Gu, W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J. Biol. Chem 2012, 287, 5102–5111. [Google Scholar]
- Hartley, J.W.; Chattopadhyay, S.K.; Lander, M.R.; Taddesse-Heath, L.; Naghashfar, Z.; Morse, H.C., III; Fredrickson, T.N. Accelerated appearance of multiple B cell lymphoma types in NFS/N mice congenic for ecotropic murine leukemia viruses. Lab Invest 2000, 80, 159–169. [Google Scholar]
- Morse, H.C., III; Anver, M.R.; Fredrickson, T.N.; Haines, D.C.; Harris, A.W.; Harris, N.L.; Jaffe, E.S.; Kogan, S.C.; MacLennan, I.C.; Pattengale, P.K.; et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood 2002, 100, 246–258. [Google Scholar]
- Pasqualucci, L.; Bhagat, G.; Jankovic, M.; Compagno, M.; Smith, P.; Muramatsu, M.; Honjo, T.; Morse, H.C., III; Nussenzweig, M.C.; Dalla-Favera, R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet 2008, 40, 108–112. [Google Scholar]
- Bornkamm, G.W. Epstein-Barr virus and the pathogenesis of Burkitt's lymphoma: More questions than answers. Int. J. Cancer 2009, 124, 1745–1755. [Google Scholar]
- Qi, C.F.; Xiang, S.; Shin, M.S.; Hao, X.; Lee, C.H.; Zhou, J.X.; Torrey, T.A.; Hartley, J.W.; Fredrickson, T.N.; Morse, H.C., III. Expression of the cyclin-dependent kinase inhibitor p27 and its deregulation in mouse B cell lymphomas. Leukemia Res 2006, 30, 153–163. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qi, C.-F.; Kim, Y.-S.; Xiang, S.; Abdullaev, Z.; Torrey, T.A.; Janz, S.; Kovalchuk, A.L.; Sun, J.; Chen, D.; Cho, W.C.; et al. Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms. Int. J. Mol. Sci. 2012, 13, 6204-6219. https://doi.org/10.3390/ijms13056204
Qi C-F, Kim Y-S, Xiang S, Abdullaev Z, Torrey TA, Janz S, Kovalchuk AL, Sun J, Chen D, Cho WC, et al. Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms. International Journal of Molecular Sciences. 2012; 13(5):6204-6219. https://doi.org/10.3390/ijms13056204
Chicago/Turabian StyleQi, Chen-Feng, Yong-Soo Kim, Shao Xiang, Ziedulla Abdullaev, Ted A. Torrey, Siegfried Janz, Alexander L. Kovalchuk, Jiafang Sun, Delin Chen, William C. Cho, and et al. 2012. "Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms" International Journal of Molecular Sciences 13, no. 5: 6204-6219. https://doi.org/10.3390/ijms13056204
APA StyleQi, C.-F., Kim, Y.-S., Xiang, S., Abdullaev, Z., Torrey, T. A., Janz, S., Kovalchuk, A. L., Sun, J., Chen, D., Cho, W. C., Gu, W., & Morse III, H. C. (2012). Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms. International Journal of Molecular Sciences, 13(5), 6204-6219. https://doi.org/10.3390/ijms13056204