Loss of p16Ink4a Function Rescues Cellular Senescence Induced by Telomere Dysfunction


Abstract
:1. Introduction
2. Results and Discussion
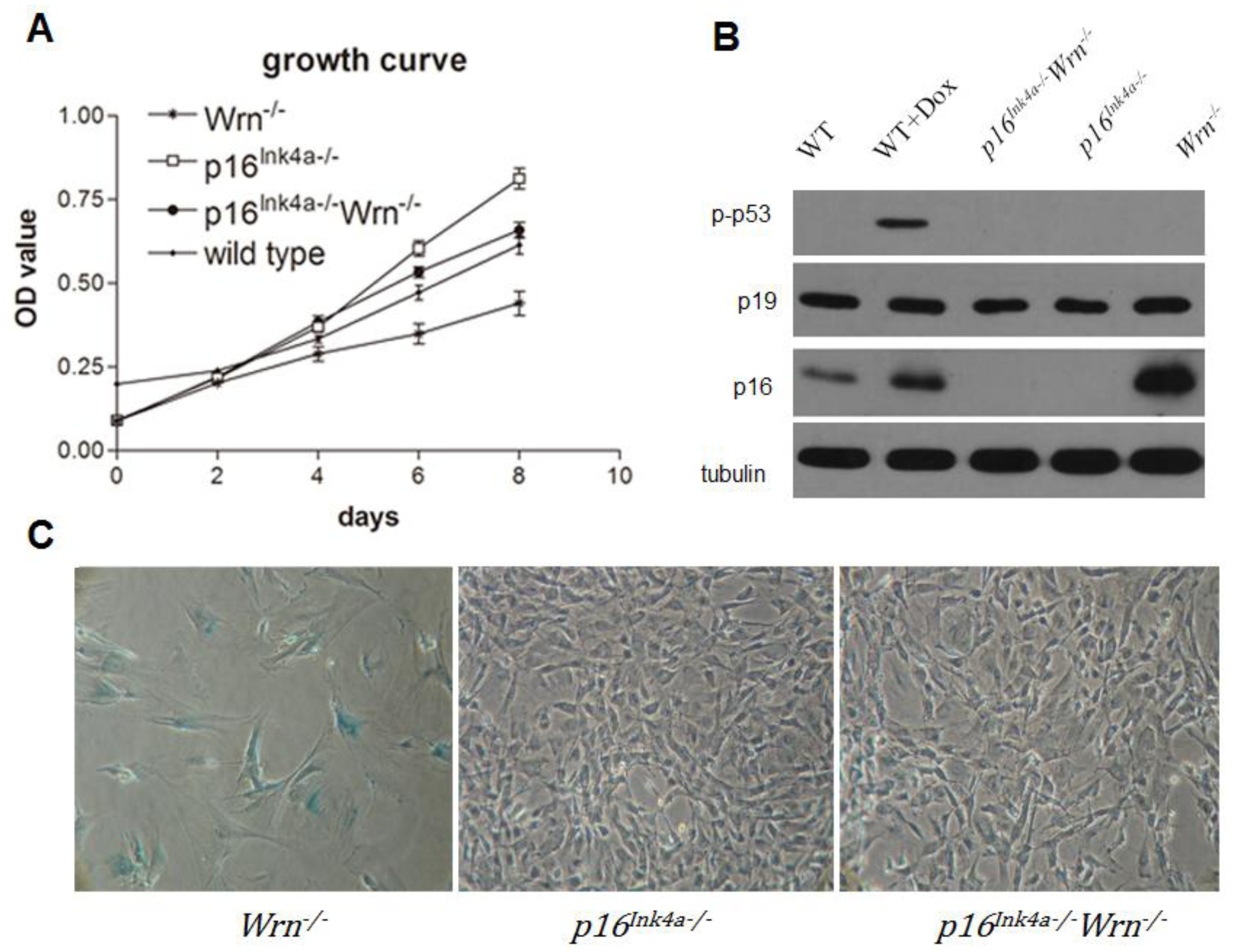
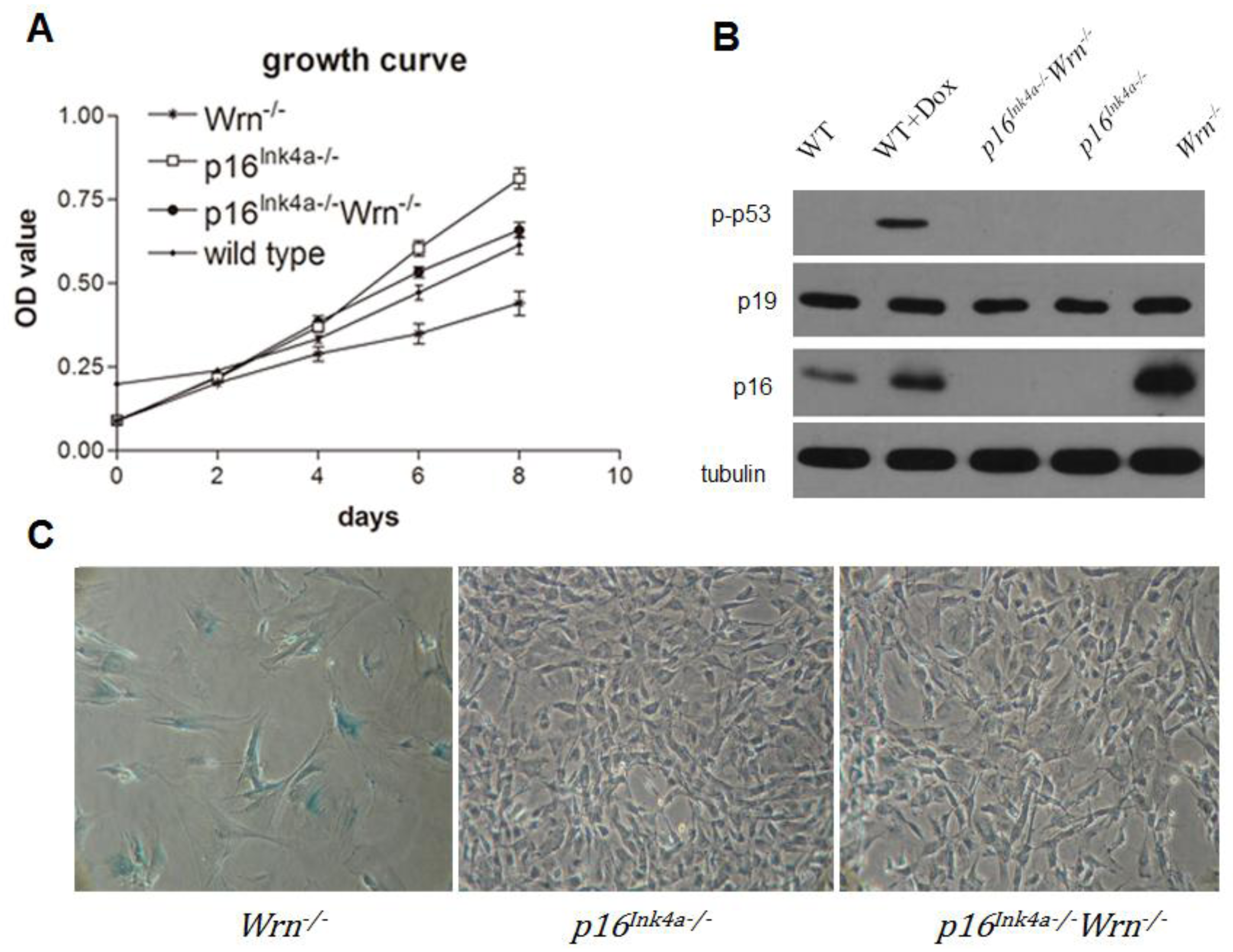
2.1. Loss of p16Ink4a Function Overcame the Growth Barrier Caused by Dysfunction of Wrn
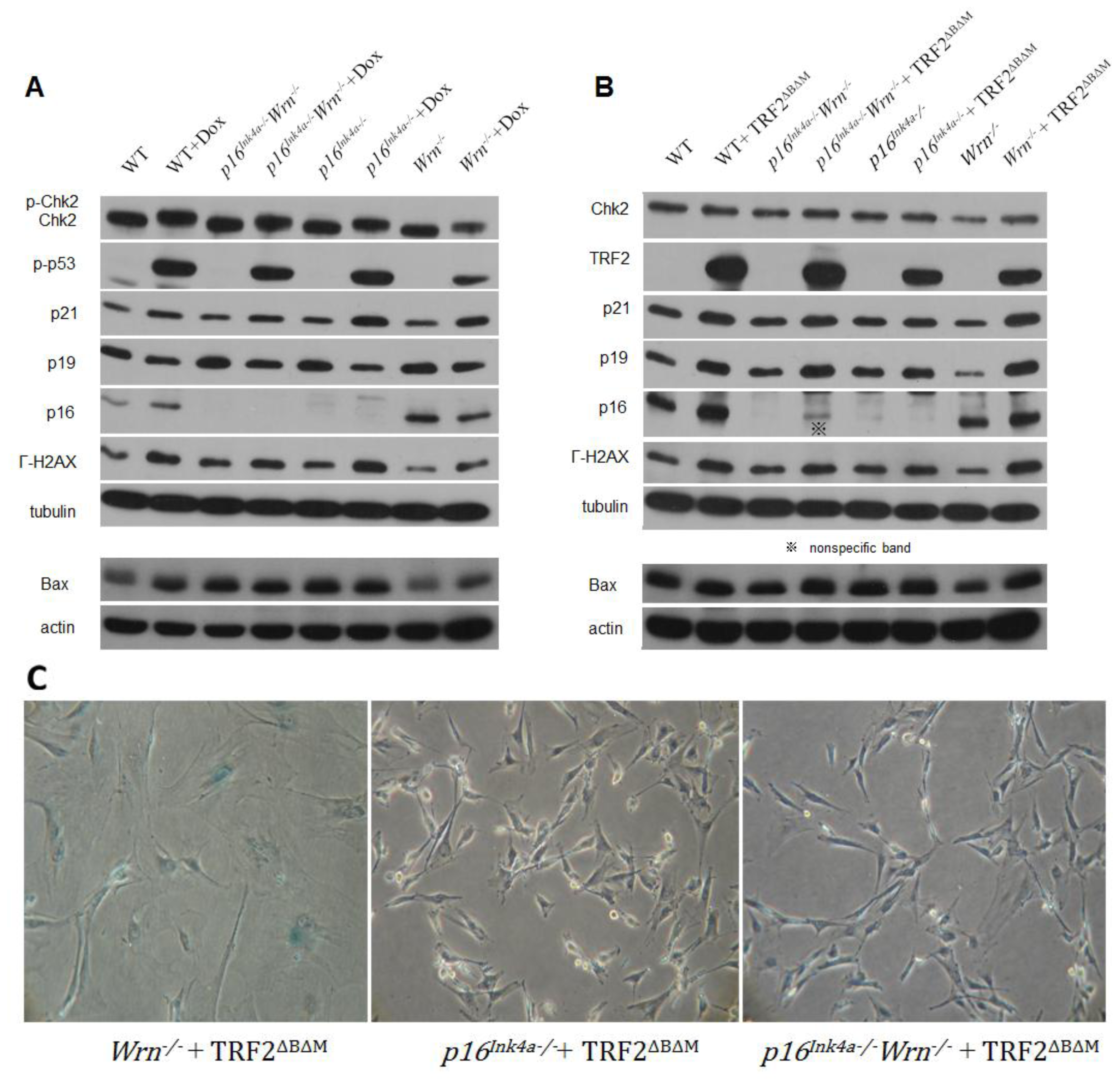
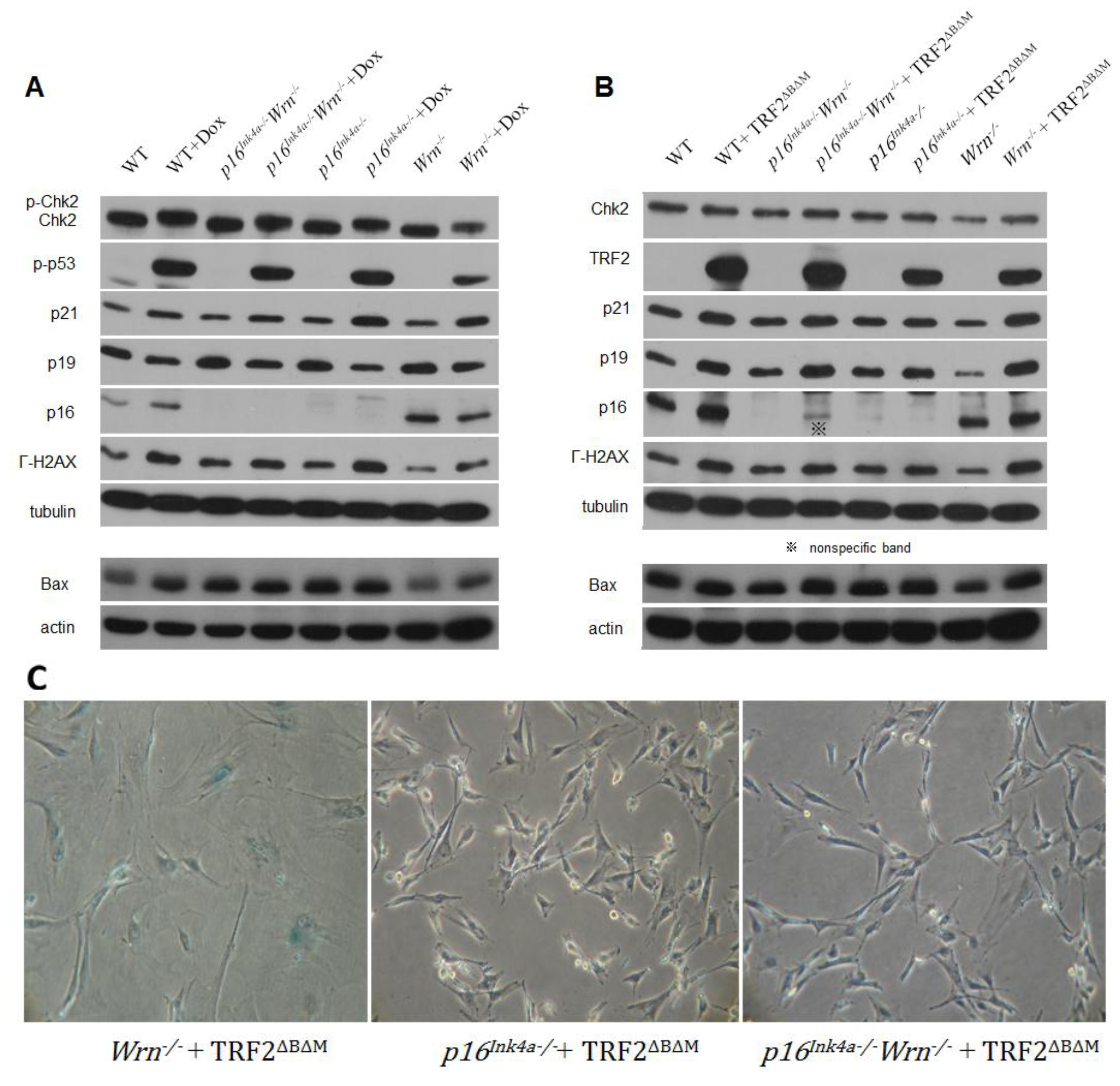
2.2. DNA Damage Response to Genotoxin or Telomere Dysfunction in Cells Lacking Wrn or p16Ink4a Function
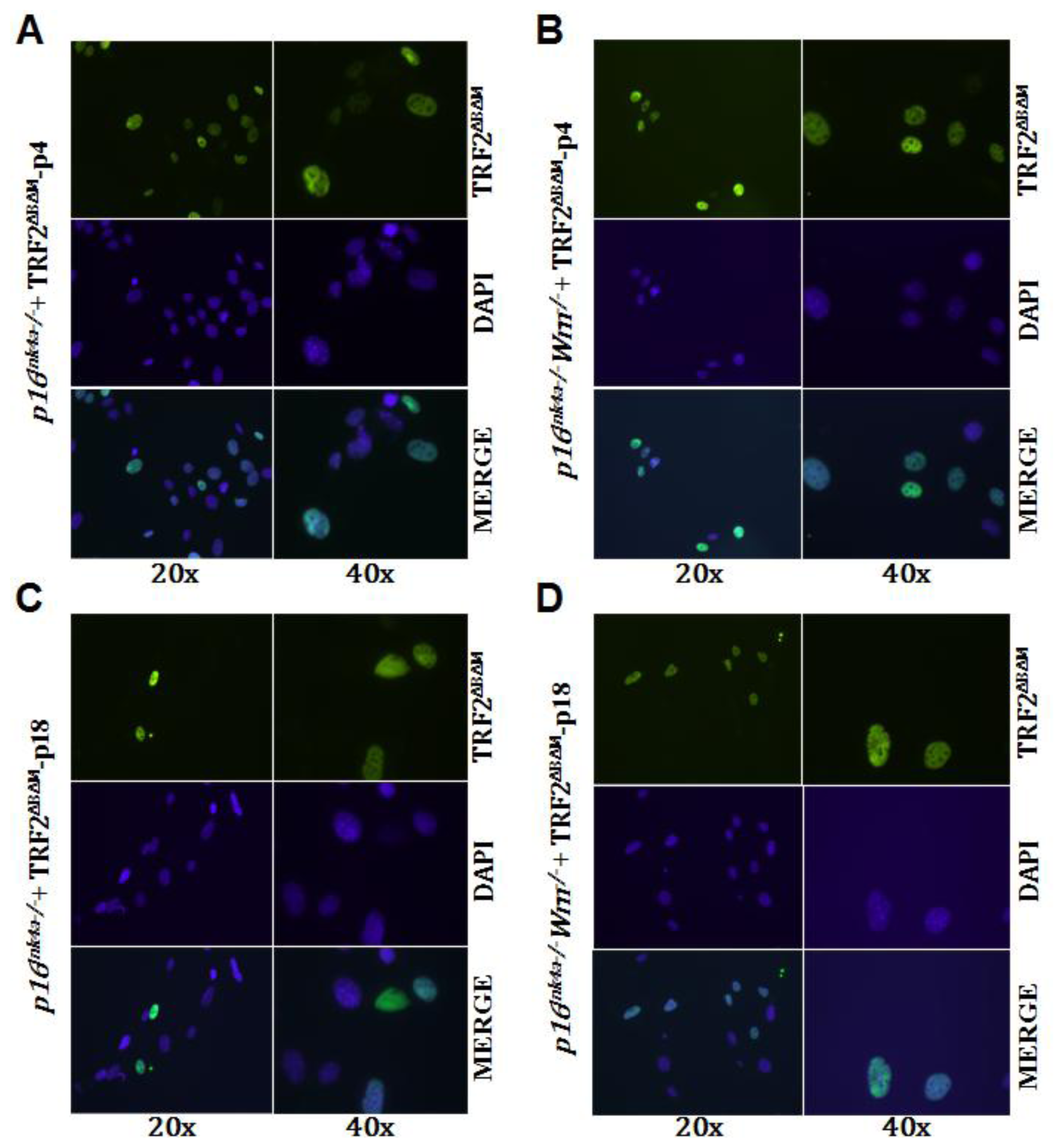
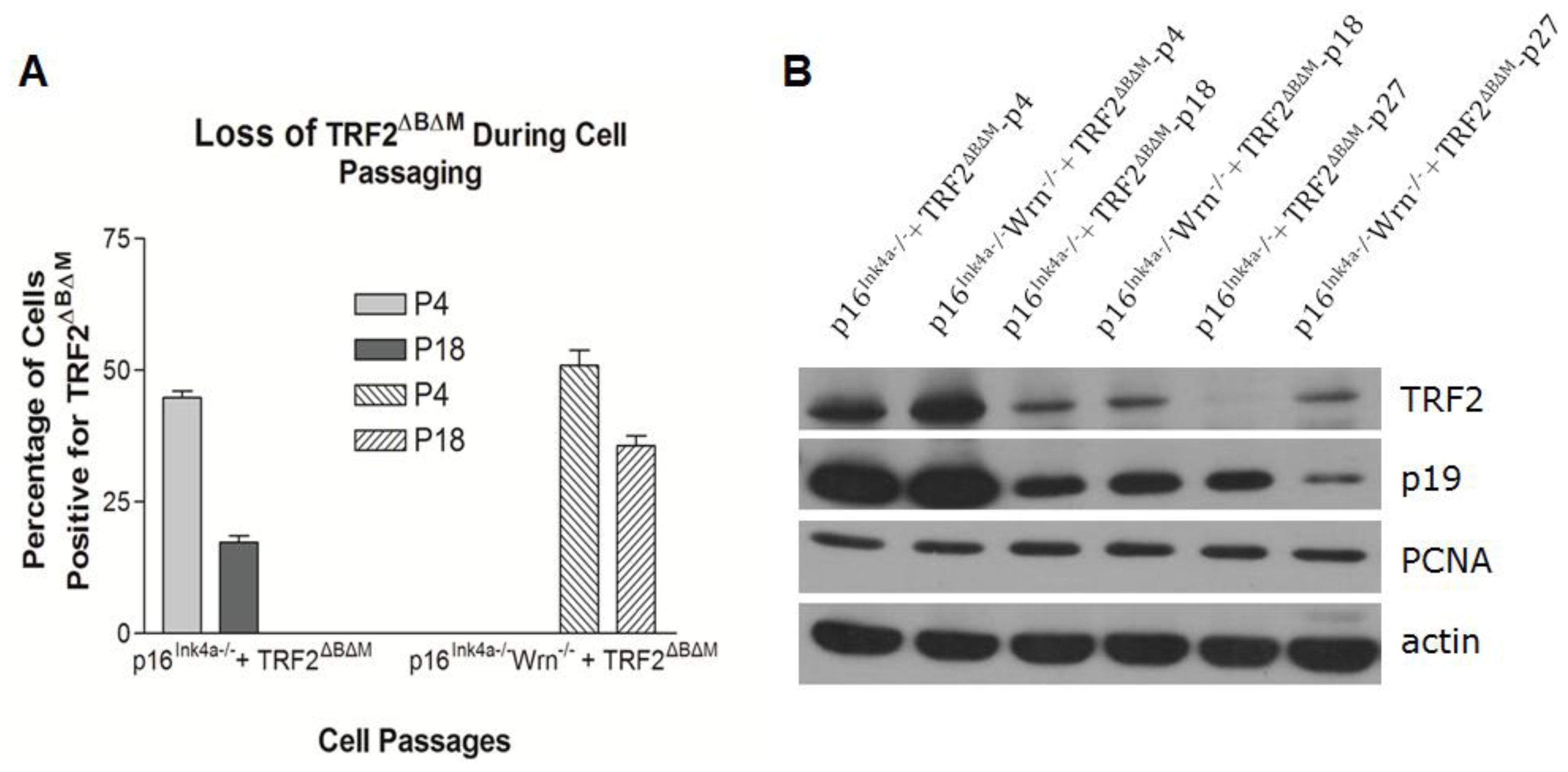
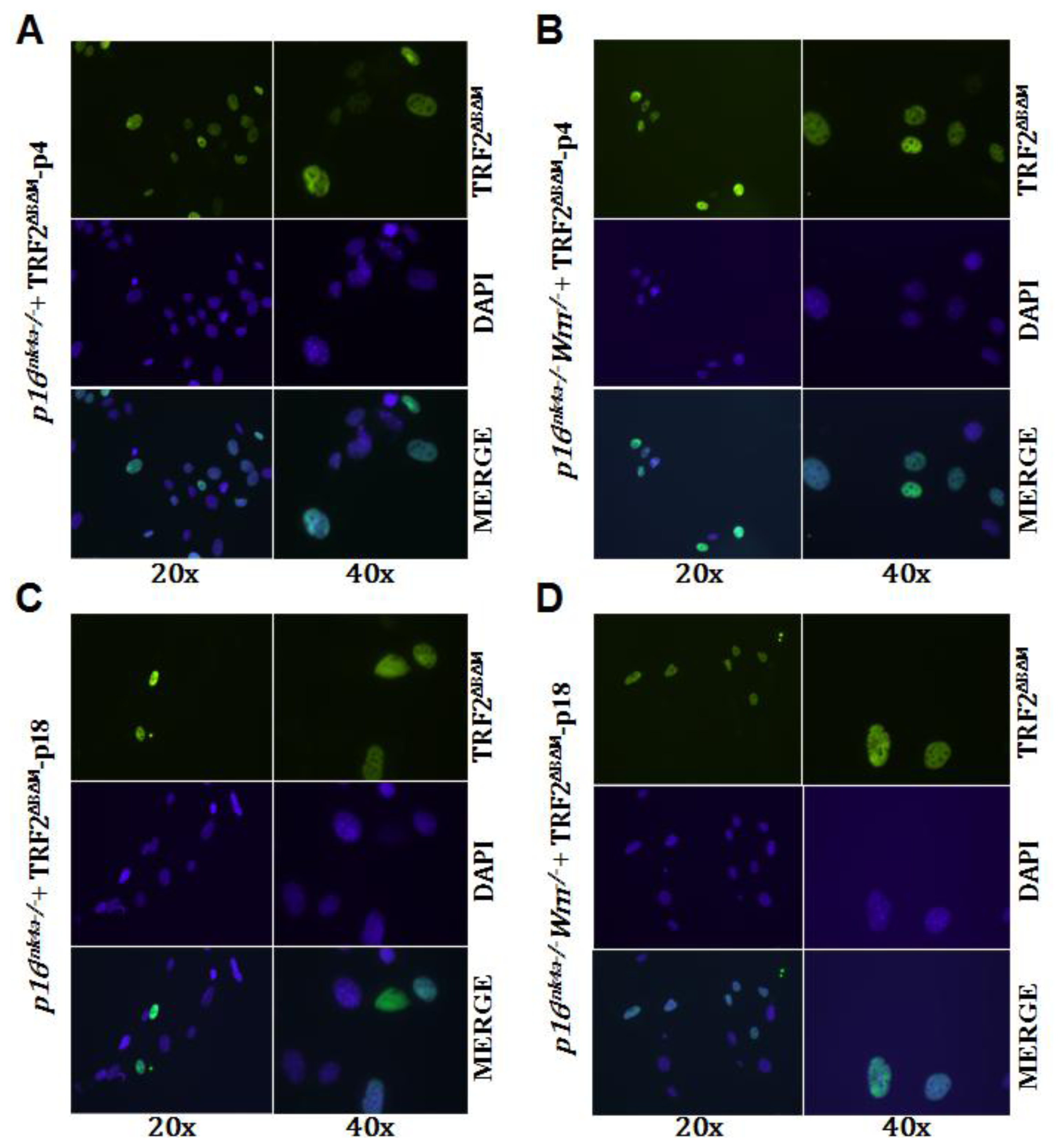
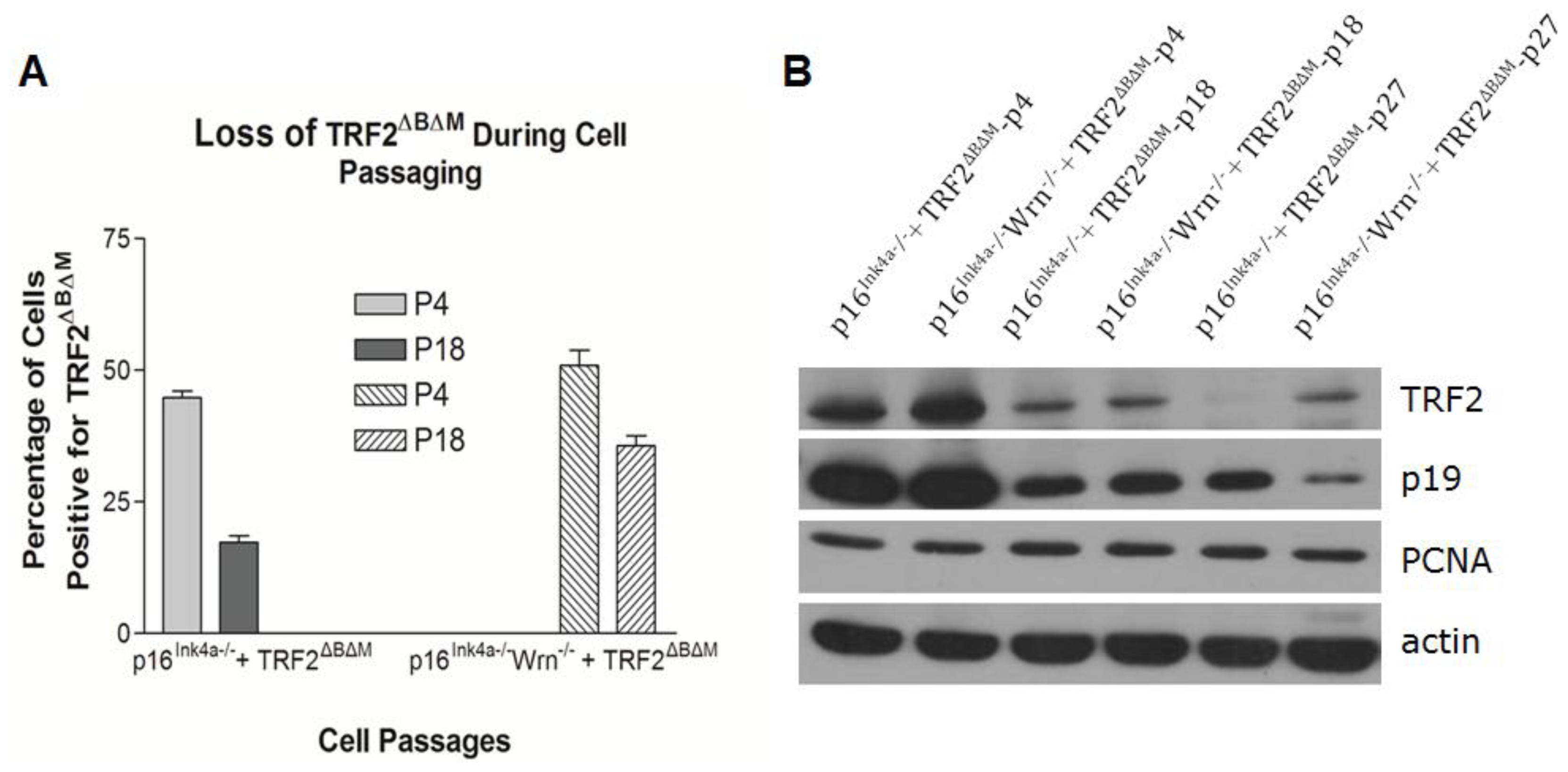
2.3. Loss of p16Ink4a Function Facilitated the Elimination of Exogenous TRF2ΔBΔM in MEFs
3. Experimental Section
3.1. Cell Lines, Constructs and Antibodies
3.2. Growth Curve Assay
3.3. Immunostaining Assay
3.4. SA-β-Gal Staining
4. Conclusions
Supplementary Information
ijms-13-05866-s001.pdfAcknowledgments
References
- Wright, W.E.; Shay, J.W. Telomere positional effects and the regulation of cellular senescence. Trends Genet 1992, 8, 193–197. [Google Scholar]
- Wright, W.E.; Shay, J.W. Time, telomeres and tumours: Is cellular senescence more than an anticancer mechanism? Trends Cell Biol 1995, 5, 293–297. [Google Scholar]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar]
- Stewart, S.A.; Weinberg, R.A. Telomeres: Cancer to human aging. Annu. Rev. Cell Dev. Biol 2006, 22, 531–557. [Google Scholar]
- De Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Gene Dev 2005, 19, 2100–2110. [Google Scholar]
- Artandi, S.E.; DePinho, R.A. Mice without telomerase: What can they teach us about human cancer? Nat. Med 2000, 6, 852–855. [Google Scholar]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. P53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar]
- Zhang, X.F.; Tang, W.R.; Luo, Y. Aging or tumor: The crosstalk between telomerase and p53 (in Chinese). Yi Chuan 2009, 31, 451–456. [Google Scholar]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential activation of the mek-extracellular signal-regulated kinase and mkk3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell Biol 2002, 22, 3389–3403. [Google Scholar]
- Palmero, I.; McConnell, B.; Parry, D.; Brookes, S.; Hara, E.; Bates, S.; Jat, P.; Peters, G. Accumulation of p16ink4a in mouse fibroblasts as a function of replicative senescence and not of retinoblastoma gene status. Oncogene 1997, 15, 495–503. [Google Scholar]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16cdkn2 expression and its implications for cell immortalization and senescence. Mol. Cell Biol 1996, 16, 859–867. [Google Scholar]
- Jacobs, J.J.; de Lange, T. Significant role for p16ink4a in p53-independent telomere-directed senescence. Curr. Biol 2004, 14, 2302–2308. [Google Scholar]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. P53- and atm-dependent apoptosis induced by telomeres lacking trf2. Science 1999, 283, 1321–1325. [Google Scholar]
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner Syndrome protein is a DNA helicase. Nat. Genet 1997, 17, 100–103. [Google Scholar]
- Epstein, C.J.; Martin, G.M.; Schultz, A.L.; Motulsky, A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966, 45, 177–221. [Google Scholar]
- Davis, T.; Bachler, M.A.; Wyllie, F.S.; Bagley, M.C.; Kipling, D. Evaluating the role of p38 map kinase in growth of Werner Syndrome fibroblasts. Ann. N. Y. Acad. Sci 2010, 1197, 45–48. [Google Scholar]
- Wyllie, F.S.; Jones, C.J.; Skinner, J.W.; Haughton, M.F.; Wallis, C.; Wynford-Thomas, D.; Faragher, R.G.; Kipling, D. Telomerase prevents the accelerated cell ageing of Werner Syndrome fibroblasts. Nat. Genet 2000, 24, 16–17. [Google Scholar]
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential role of limiting telomeres in the pathogenesis of Werner Syndrome. Nat. Genet 2004, 36, 877–882. [Google Scholar]
- Du, X.; Shen, J.; Kugan, N.; Furth, E.E.; Lombard, D.B.; Cheung, C.; Pak, S.; Luo, G.; Pignolo, R.J.; DePinho, R.A.; et al. Telomere shortening exposes functions for the mouse werner and bloom syndrome genes. Mol. Cell Biol 2004, 24, 8437–8446. [Google Scholar]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective telomere lagging strand synthesis in cells lacking wrn helicase activity. Science 2004, 306, 1951–1953. [Google Scholar]
- Opresko, P.L.; Otterlei, M.; Graakjaer, J.; Bruheim, P.; Dawut, L.; Kolvraa, S.; May, A.; Seidman, M.M.; Bohr, V.A. The Werner Syndrome helicase and exonuclease cooperate to resolve telomeric d-loops in a manner regulated by trf1 and trf2. Mol. Cell 2004, 14, 763–774. [Google Scholar]
- Lebel, M.; Leder, P. A deletion within the murine Werner Syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc. Natl. Acad. Sci. USA 1998, 95, 13097–13102. [Google Scholar]
- Jacobs, J.J.; de Lange, T. P16ink4a as a second effector of the telomere damage pathway. Cell Cycle 2005, 4, 1364–1368. [Google Scholar]
- Minty, F.; Thurlow, J.K.; Harrison, P.R.; Parkinson, E.K. Telomere dysfunction in human keratinocytes elicits senescence and a novel transcription profile. Exp. Cell Res 2008, 314, 2434–2447. [Google Scholar]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving atm, p53, and p21(cip1), but not p16(ink4a). Mol. Cell 2004, 14, 501–513. [Google Scholar]
- Smogorzewska, A.; de Lange, T. Different telomere damage signaling pathways in human and mouse cells. EMBO J 2002, 21, 4338–4348. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MEFs’ Genotype | metaphases counted | breakage | fusion | Diploid 2N = 40 | Aneuploid 2N > 40 | Aneuploid 2N < 40 |
|---|---|---|---|---|---|---|
| p16Ink4a−/− + TRF2ΔBΔM-p4 | 30 | 1 | 15 | 14 | ||
| p16Ink4a−/−Wrn−/− + TRF2ΔBΔM-p4 | 30 | 14 | 14 | 2 | ||
| p16Ink4a−/− + TRF2ΔBΔM-p18 | 30 | 1 | 7 | 20 | 2 | |
| p16Ink4a−/−Wrn−/− + TRF2ΔBΔM-p18 | 30 | 15 | 13 | 2 | ||
| p16Ink4a−/− + TRF2ΔBΔM-p27 | 30 | 2 | 1 | 27 | ||
| p16Ink4a−/−Wrn−/− + TRF2ΔBΔM-p27 | 30 | 12 | 1 | 16 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.; Wu, X.; Tang, W.; Luo, Y. Loss of p16Ink4a Function Rescues Cellular Senescence Induced by Telomere Dysfunction. Int. J. Mol. Sci. 2012, 13, 5866-5877. https://doi.org/10.3390/ijms13055866
Zhang X, Wu X, Tang W, Luo Y. Loss of p16Ink4a Function Rescues Cellular Senescence Induced by Telomere Dysfunction. International Journal of Molecular Sciences. 2012; 13(5):5866-5877. https://doi.org/10.3390/ijms13055866
Chicago/Turabian StyleZhang, Xiufeng, Xiaoming Wu, Wenru Tang, and Ying Luo. 2012. "Loss of p16Ink4a Function Rescues Cellular Senescence Induced by Telomere Dysfunction" International Journal of Molecular Sciences 13, no. 5: 5866-5877. https://doi.org/10.3390/ijms13055866