Phylogeographical Analysis on Squalidus argentatus Recapitulates Historical Landscapes and Drainage Evolution on the Island of Taiwan and Mainland China


Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Genetic Diversity of Squalidus argentatus on the Island of Taiwan and Mainland China
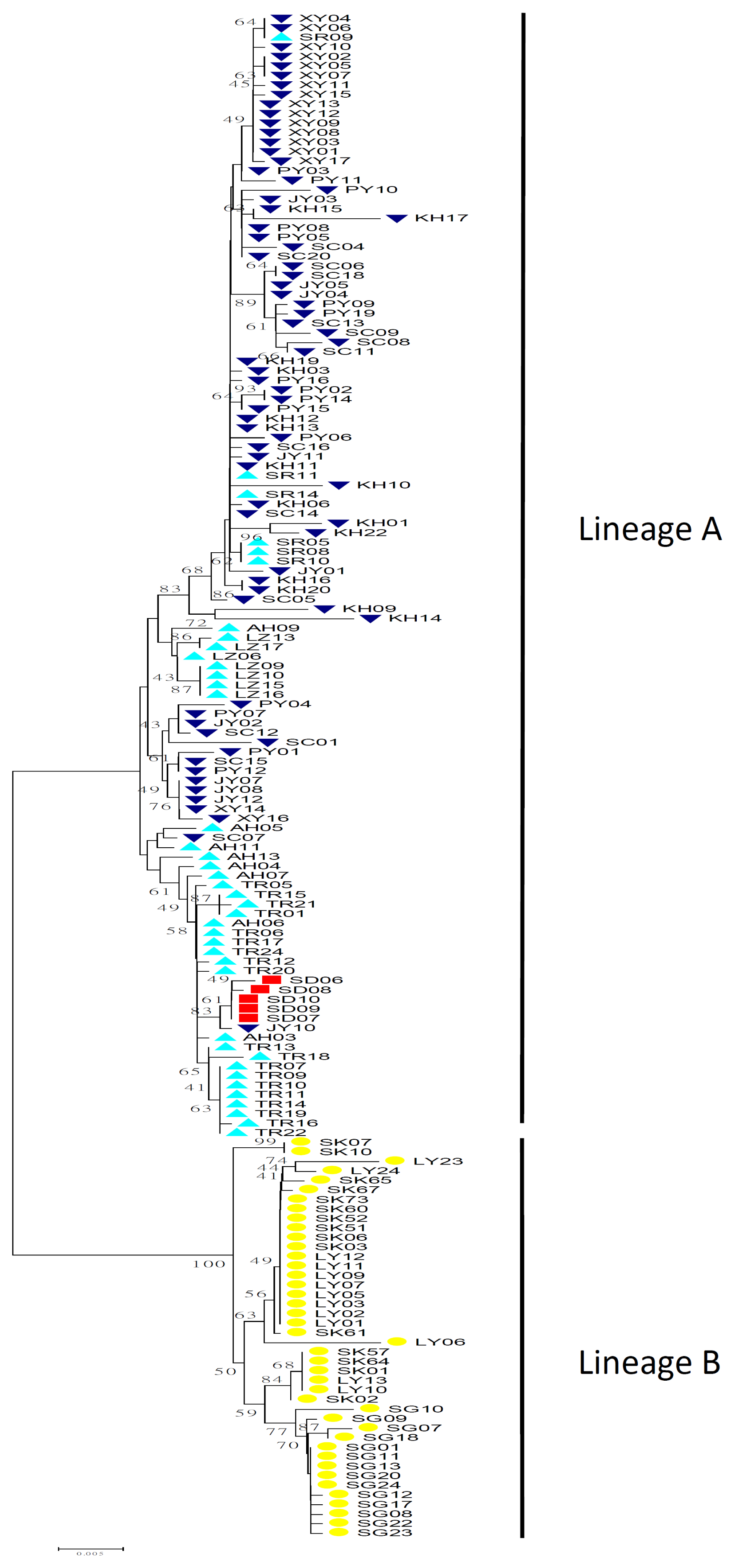
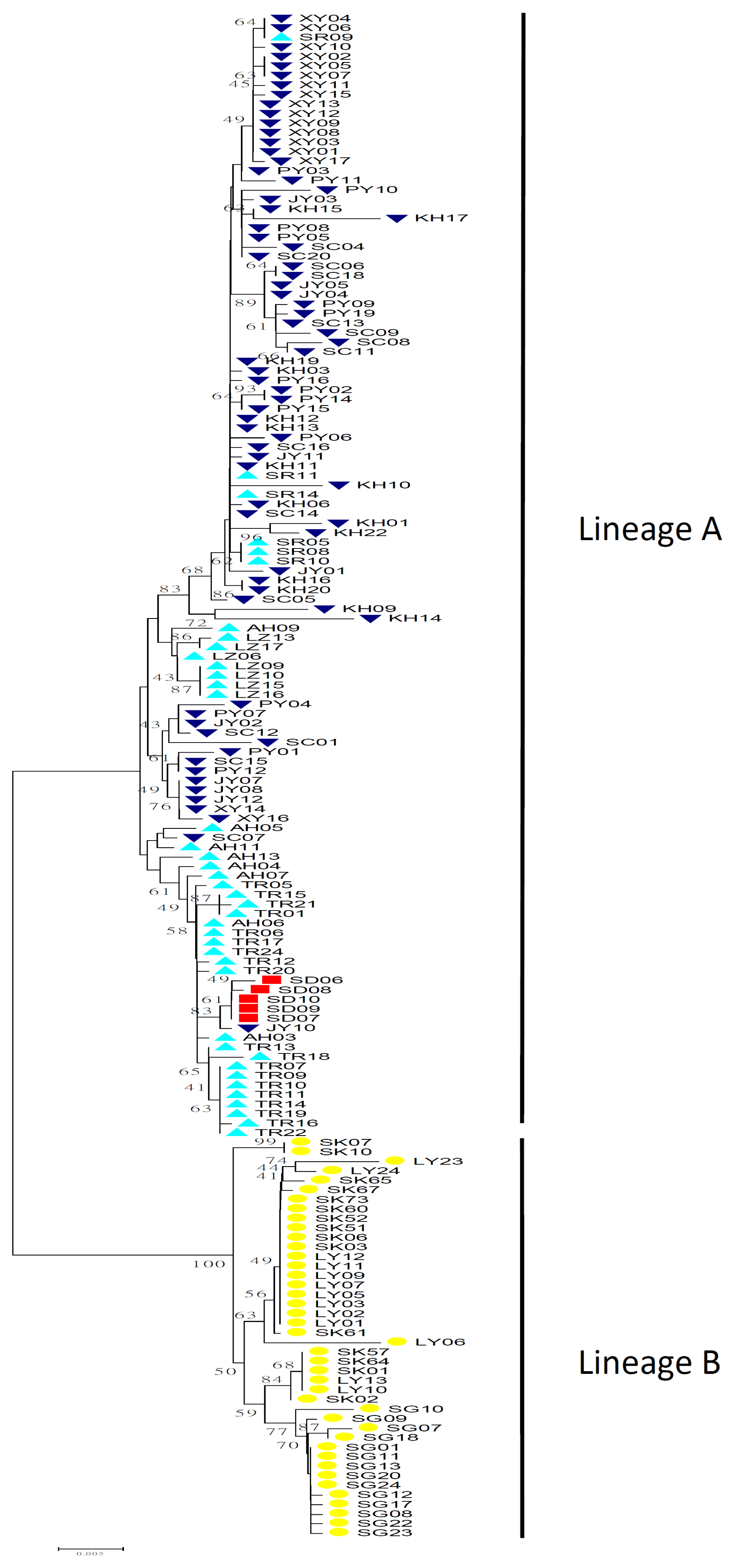
2.1.2. Phylogenetic Analysis
2.1.3. Population Genetic Analysis
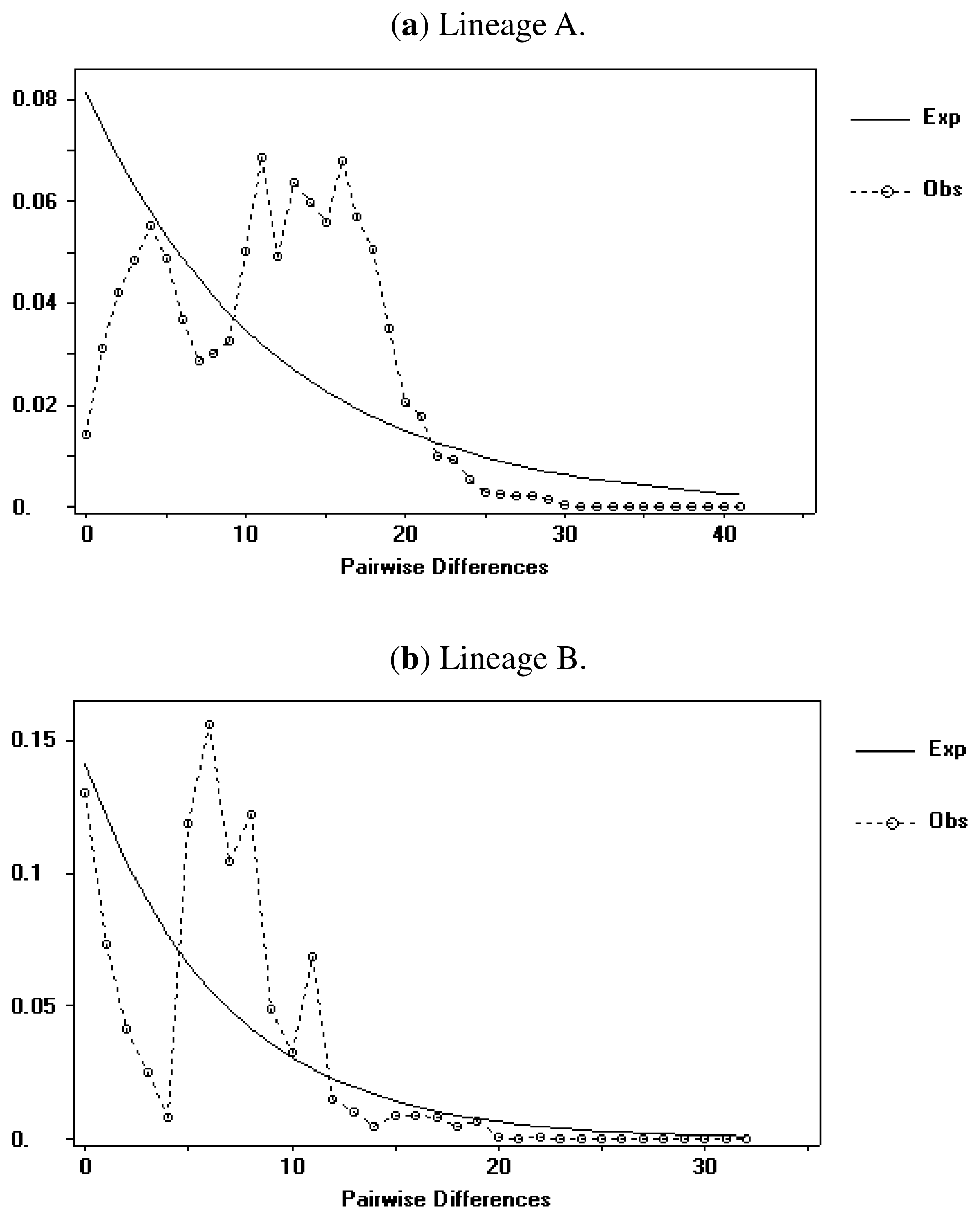
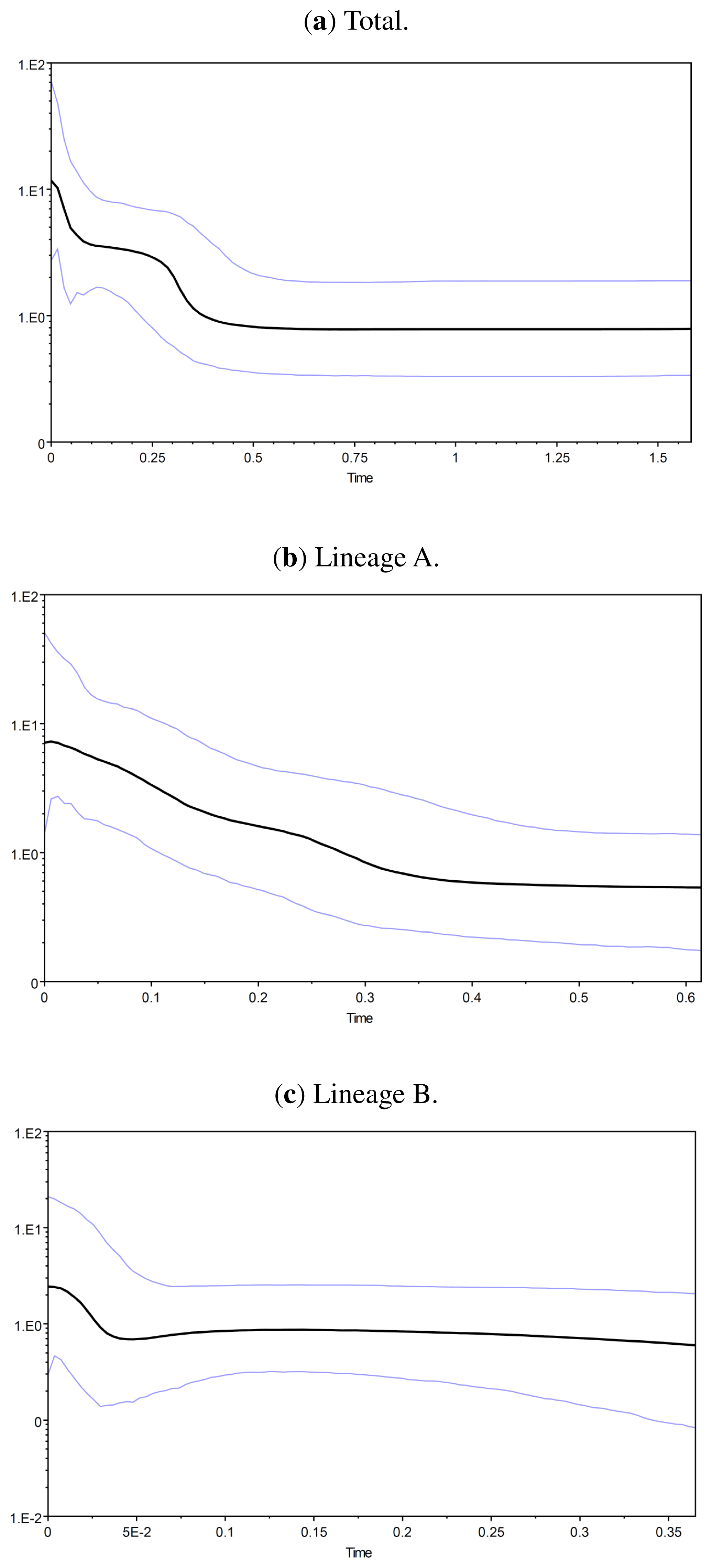
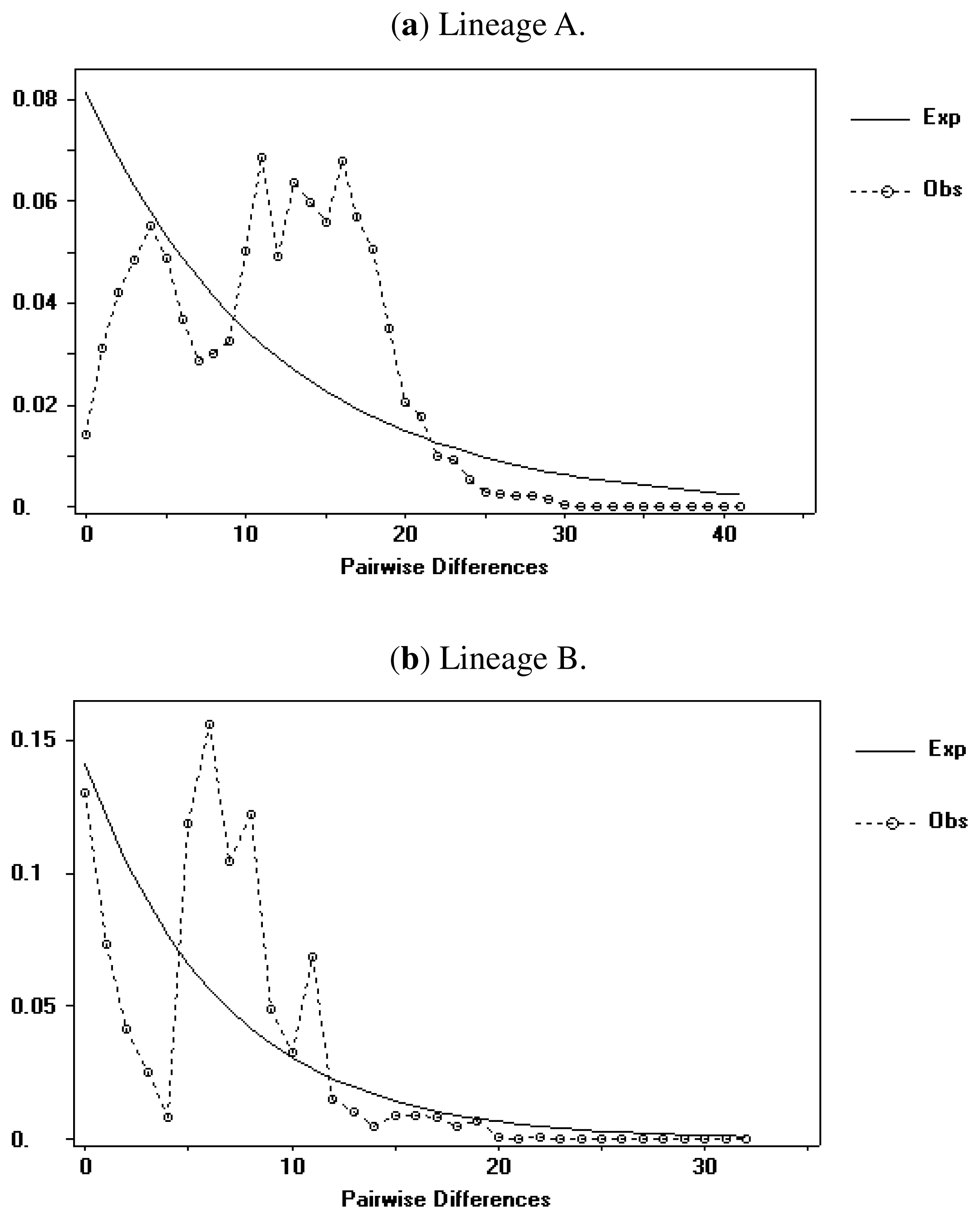
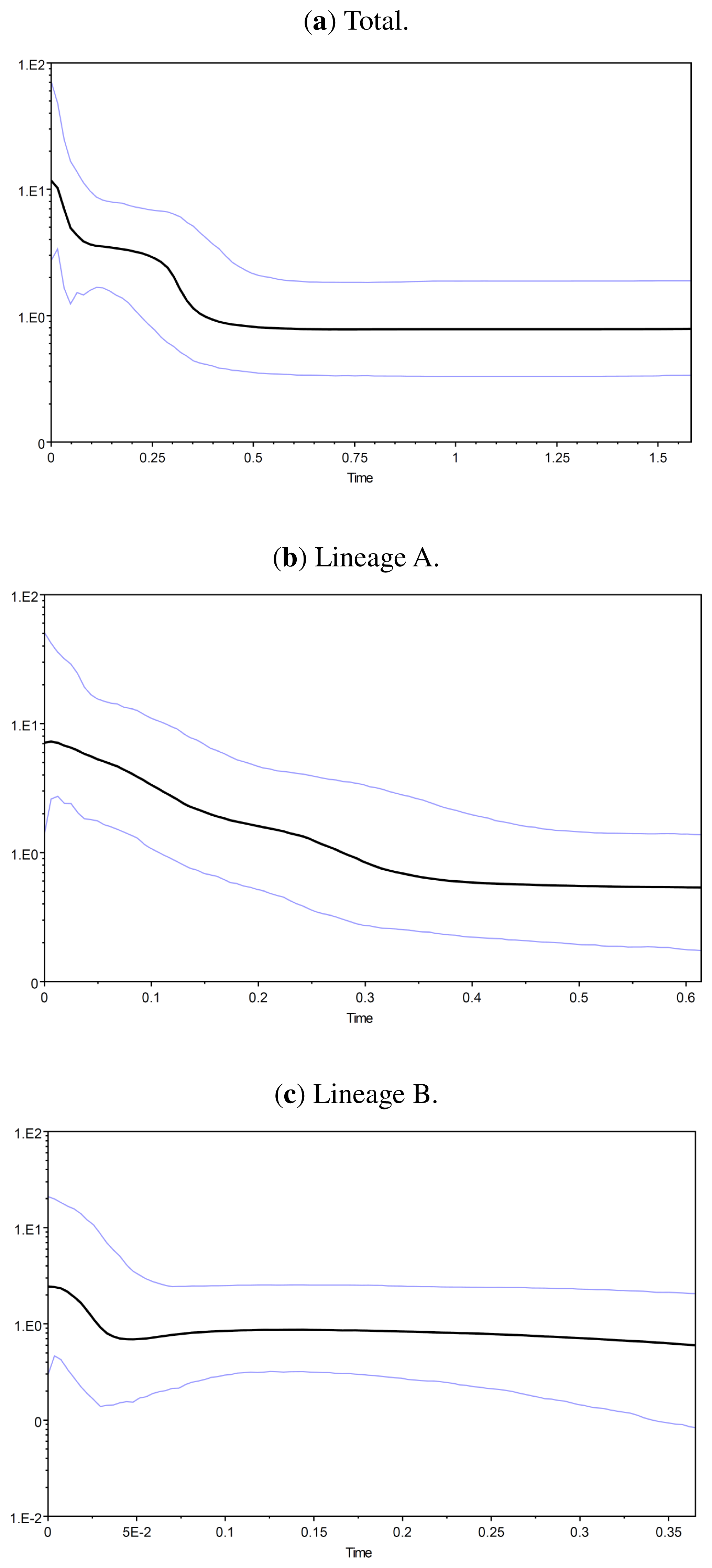
2.1.4. Demographic History
2.1.5. Molecular Dating
2.2. Discussion
2.2.1. Genetic Variation within Squalidus argentatus
2.2.2. Population Differentiation
2.2.3. Phylogeography of Squalidus argentatus
2.2.4. Demographic History
2.2.5. Implications for Conservation
3. Experimental Section
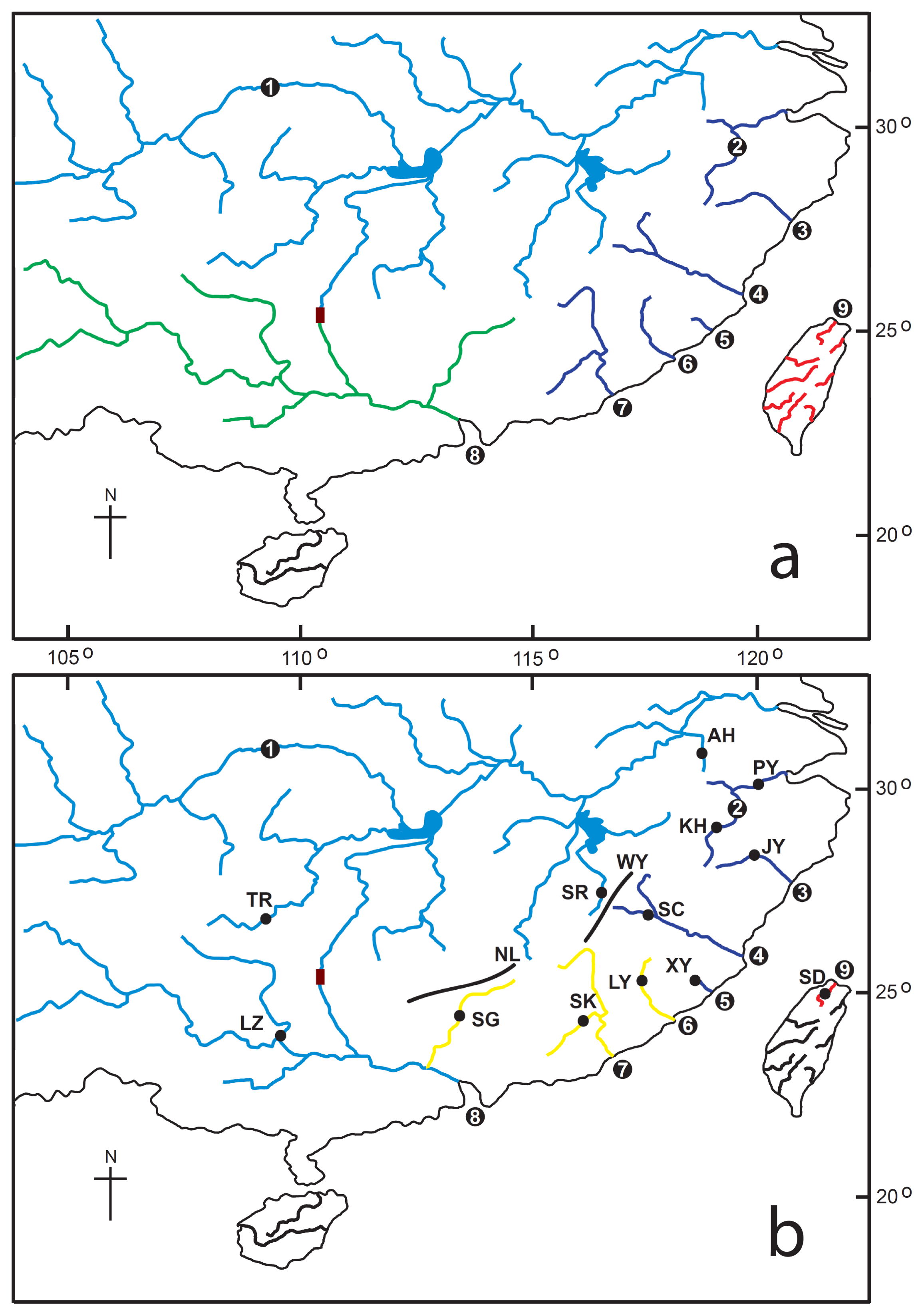
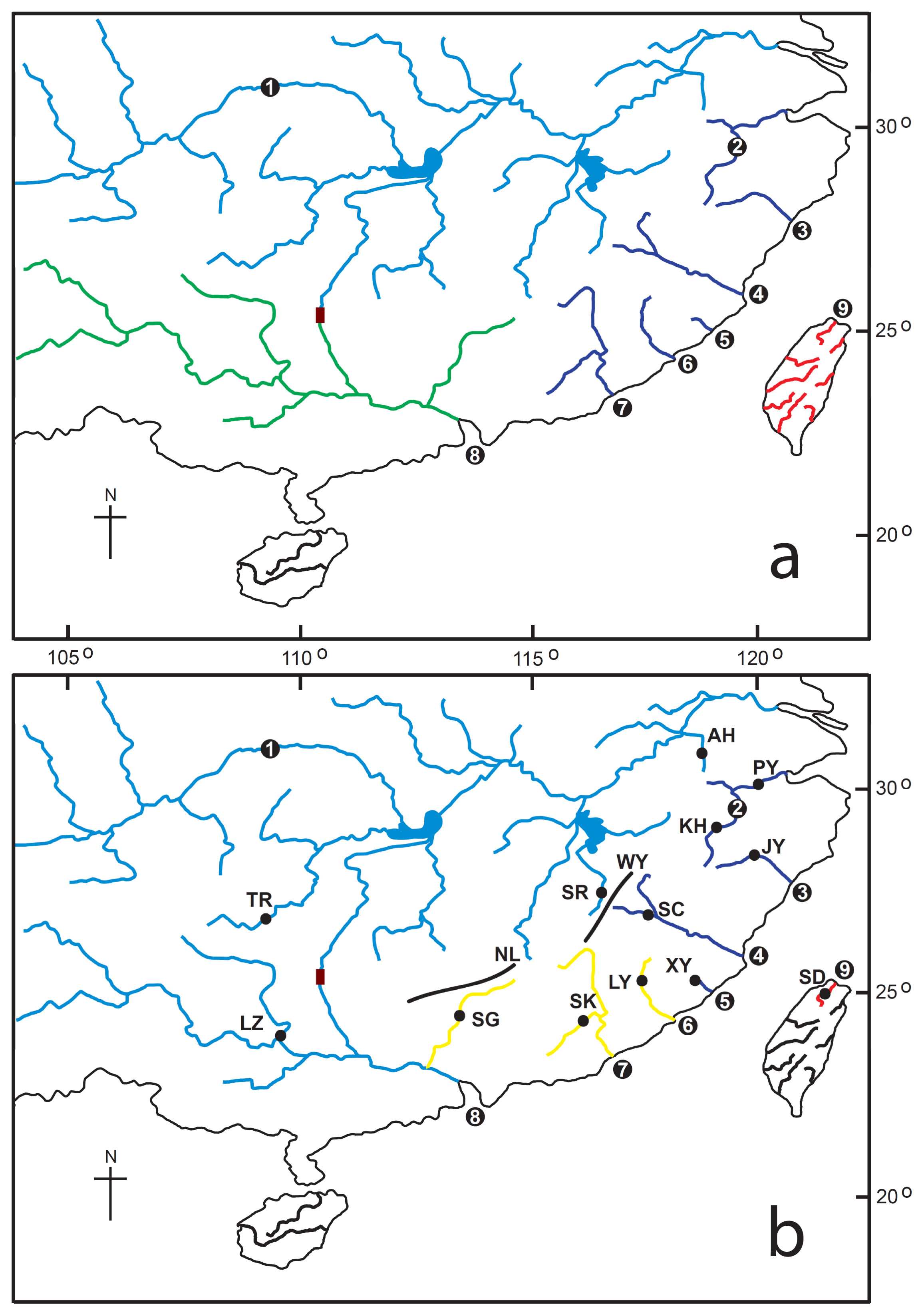
3.1. Sampling
3.2. Molecular Analyses
3.3. Data Analyses
3.3.1. Genetic Diversity, Phylogenetic and Phylogeographic Analysis
3.3.2. Historical Demography
3.3.3. Population Genetic Differentiation
3.3.4. Molecular Dating
4. Conclusions
Acknowledgments
References
- Nelson, J.S. Fishes of the World, 4th ed; John Wiley and Sons, Inc: Hoboken, NJ, USA, 2006. [Google Scholar]
- Fauna Sinica, Osteichthyes, Cypriniformes II (in Chinese); Chen, Y.Y. (Ed.) Science Press: Beijing, China, 2000.
- Li, S.Z. Studies on Zoogeographical Divisions for Fresh Water Fishes of China; Science Press: Beijing, China, 1981. [Google Scholar]
- Fu, J.; Zeng, X. How many species are in the genus Batrachuperus? A phylogeographical analysis of the stream salamanders (family Hynobiidae) from southwestern China. Mol. Ecol 2008, 17, 1469–1488. [Google Scholar]
- Zhang, M.; Rao, D.; Yang, J.; Yu, G.; Wilkinson, J.A. Molecular phylogeography and population structure of a mid-elevation montane frog Leptobrachium ailaonicum in a fragmented habitat of southwest China. Mol. Phylogenet. Evol 2009, 54, 47–58. [Google Scholar]
- Chen, X.L.; Chiang, T.Y.; Lin, H.D.; Zheng, H.S.; Shao, K.T.; Zhang, Q.; Hsu, K.C. Mitochondrial DNA phylogeography of Glyptothorax fokiensis and Glyptothorax hainanensis in Asia. J. Fish Biol 2007, 70, 75–93. [Google Scholar]
- Huang, Z.H.; Liu, N.F.; Liang, W.; Zhang, Y.Y.; Liao, X.J. Phylogeography of Chinese bamboo partridge Bambusicola thoracica thoracica (Aves: Galliformes) in south China: Inference from mitochondrial DNA control-region sequences. Mol. Phylogenet. Evol 2010, 56, 273–280. [Google Scholar]
- Perdices, A.; Cunha, C.; Coelho, M.M. Phylogenetic structure of Zacco platypus (Teleostei, Cyprinidae) populations on the upper and middle Chang Jiang (=Yangtze) drainage inferred from cytochrome b sequences. Mol. Phylogenet. Evol 2004, 31, 192–203. [Google Scholar]
- Perdices, A.; Sayanda, D.; Coelho, M.M. Mitochondrial diversity of Opsariichthys bidens (Teleostei, Cyprinidae) in three Chinese drainages. Mol. Phylogenet. Evol 2005, 37, 920–927. [Google Scholar]
- Perdices, A.; Coelho, M.M. Comparative phylogeography of Zacco platypus and Opsariichthys bidens (Teleostei, Cyprinidae) in China based on cytochrome b sequences. J. Zool. Syst. Evol. Res 2006, 44, 330–338. [Google Scholar]
- Li, G.Y.; Wang, X.Z.; Zhao, Y.H.; Zhang, J.; Zhang, C.G.; He, S.P. Speciation and phylogeography of Opsariichthys bidens (Pisces: Cypriniformes: Cyprinidae) in China: Analysis of the cytochrome b gene of mtDNA from diverse populations. Zool. Stud 2009, 48, 569–583. [Google Scholar]
- Lin, C.; Lin, H.; Wang, J.; Chao, S.; Chiang, T. Phylogeography of Hemibarbus labeo (Cyprinidae) sondary contact of ancient lineages of mtDNA. Zool. Scr 2010, 39, 23–35. [Google Scholar]
- Whittaker, R.J. Island Biogeography: Ecology, Evolution, and Conservation; Oxford University Press: Oxford, UK; New York, NY, USA, 1998. [Google Scholar]
- Teng, L.S. Geotectonic evolution of late Cenozoic arc–continent collision in Taiwan. Tectonophysics 1990, 183, 57–76. [Google Scholar]
- Liu, T.K.; Chen, Y.G.; Chen, W.S.; Jiang, S.H. Rates of cooling and denudation of the early Penglai Orogeny, Taiwan, as assessed by fission-track constraints. Tectonophysics 2000, 320, 69–82. [Google Scholar]
- Oshima, M. Studies on the distribution of the fresh-water fishes of Taiwan and discuss the geographical relationship of the Taiwan island and the adjacent area (in Japanese). Zool. Mag 1923, 35, 1–49. [Google Scholar]
- Tzeng, C.S. Distribution of freshwater fishes of Taiwan. J. Taiwan Mus 1986, 39, 127–146. [Google Scholar]
- Shih, H.T.; Fang, S.H.; Ng, P.K.L. Phylogeny of the freshwater crabs genus Somanniathelphusa Bott (Decapoda: Parathelphusidae) from Taiwan and the coastal regions of China, with notes on their biogeography. Invertebr. Syst 2007, 21, 29–37. [Google Scholar]
- Hsu, K.C.; Tsai, K.C.; Shao, K.T.; Wang, J.P.; Lin, H.D. Phylogeography and population genetic structure of Sinibrama macrops based on mtDNA (in Chinese). BioFormosa 2005, 40, 58–67. [Google Scholar]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Moritz, C. Defining “Evolutionarily Significant Units” for conservation. Trends Ecol. Evol 1994, 9, 373–375. [Google Scholar]
- Zhang, D.X.; Hewitt, G.M. Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol. Evol 1996, 11, 247–251. [Google Scholar]
- Chiang, T.Y.; Lin, H.D.; Shao, K.T.; Hsu, K.C. Multiple causations shaping phylogeography of Chinese spiny loach (Cobitis sinensis) in Taiwan inferred from mitochondrial DNA variations. J. Fish Biol 2010, 76, 1173–1189. [Google Scholar]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered vs. unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar]
- Dowling, T.E.; Tibbets, C.A.; Minckley, W.L.; Smith, G.R. Evolutionary relationships of the plagopterins (Teleostei: Cyprinidae) from cytochrome b sequences. Copeia 2002, 2002, 665–678. [Google Scholar]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J. Hered 1998, 89, 415–426. [Google Scholar]
- Nunes, F.; Norris, R.D.; Knowlton, N. Implications of isolation and low genetic diversity in peripheral population so fanamphi-Atlanticcoral. Mol. Ecol 2009, 18, 4283–4297. [Google Scholar]
- Frankham, R. Relationship of genetic variation to population size in wildlife. Conserv. Biol 1996, 10, 1500–1508. [Google Scholar]
- Tew, K.S.; Han, C.C.; Chou, W.R.; Fang, L.S. Habitat and fish fauna structure in a subtropical mountain stream in Taiwan before and after a catastrophic typhoon. Environ. Biol. Fish 2002, 65, 457–462. [Google Scholar]
- Berrebi, P.; Xavier, R.; Fang, F.; Zhang, C.G. Population structure and systematics of Opsariichthys bidens (Osteichthyes: Cyprinidae) in south-east China using a new nuclear marker: the introns (EPIC-PCR). Biol. J. Linn. Soc 2006, 87, 155–166. [Google Scholar]
- Zhong, F. Preliminary study of geological geomorphological conditions of Ling Canal. Carsologica Sin 1990, 9, 88–93. [Google Scholar]
- Tchang, H.C.; Chen, T.C.; Hung, K.C. The New Geological Structures in Coastal Line of South China; Earthquake Press: Beijing, China, 1990. [Google Scholar]
- Zheng, H.S. Freshwater Fish Fauna and Biogeography of Eight Rivers in East Guangdong, China. M.Sc. Thesis, Institute of Zoology, South China Normal University, Guangzhou, China, 2004. [Google Scholar]
- Wang, J.P.; Lin, H.D.; Huang, S.; Pan, C.H.; Chen, X.L.; Chiang, T.Y. Phylogeography of Varicorhinus barbatulus (Cyprinidae) in Taiwan based on nucleotide variation of mtDNA and allozymes. Mol. Phylogenet. Evol 2004, 31, 1143–1156. [Google Scholar]
- Wang, T.Y.; Tzeng, C.S.; Teng, H.Y.; Chang, T. Phylogeography and identification of a 187-bp-long duplication within the mitochondrial control region of Formosania lacustre (Teleostei: Balitoridae). Zool. Stud 2007, 46, 569–582. [Google Scholar]
- Okazaki, T.; Jeon, S.R.; Watanabe, M.; Kitagawa, T. Genetic relationships of Japanese and Korean bagrid catfishes inferred from mitochondrial DNA analysis. Zool. Sci 1999, 16, 363–373. [Google Scholar]
- Blin, N.; Stafford, D.W. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res 1976, 3, 2303–2308. [Google Scholar]
- Xiao, W.; Zhang, Y.; Liu, H. Molecular systematics of Xenocyprinae (Teleostei: Cyprinidae): taxonomy, biogeography and coevolution of a special group restricted in East Asia. Mol. Phylogenet. Evol. 2001, 18, 163–173. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997, 25, 4876–4882. [Google Scholar]
- Nei, M.; Tajima, F. Maximum likelihood estimation of the number of nucleotide substitutions from restriction sites data. Genetics 1983, 105, 207–217. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. In Mammalian Protein Metabolism; Munroled, H.N., Ed.; Academic Press: New York, NY, USA, 1969; pp. 31–132. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol 2007, 24, 1596–1599. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar]
- Nylander, J.A.A. MrModeltest v2; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol 2002, 19, 2092–2100. [Google Scholar]
- Fu, Y. Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Kuhner, M.K. LAMARC 2.0: Maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 2006, 22, 768–770. [Google Scholar]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol 2005, 22, 1185–1192. [Google Scholar]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol 1992, 9, 552–569. [Google Scholar]
- Harpending, H. Signature of ancient population growth in a low resolution mitochondrial DNA mismatch distribution. Hum. Biol 1994, 66, 591–600. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 2007, 7, 214. [Google Scholar]
- Rambaut, A.; Drummond, A.J. Tracer version 1.5. 2007. Available online: http://beast.bio.ed.ac.uk/Tracer accessed on 9 December 2011.
- Excoffier, L.; Lischer, H.E.L. Arlequin suite version 3.5: A new series of programs toperform population genetics analyses under Linux and Windows. Mol. Ecol. Res 2010, 10, 564–567. [Google Scholar]
- Dupanloup, I.; Schneidera, S.; Excoffier, N.L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol 2002, 11, 2571–2581. [Google Scholar]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res 1967, 27, 209–220. [Google Scholar]
- Miller, M.P. Alleles in space: Computer software for the joint analysis of inter-individual spatial and genetic information. J. Hered 2005, 96, 722–724. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region subregion | Populations (Abbreviation) | Sample size | Haplotype numbers | Haplotype Diversity (h) | Nucleotide diversity (π) | Nucleotide diversity (θ) | Tajima’s D * | Fu’s FS | R2 | g |
|---|---|---|---|---|---|---|---|---|---|---|
| Mainland China | 155 | 92 | 0.983 | 0.02073 | 0.03295 | −1.19838 | −33.072*** | 0.0584 | ||
| Yangtze River | 33 | 20 | 0.939 | 0.00659 | 0.00908 | −0.99662 | −5.021** | 0.0584 | ||
| AnHua (AH) I | 8 | 8 | 1.000 | 0.00583 | 0.00677 | −0.72263 | −3.129* | 0.1136 | 1259.96 | |
| Tongren (TR) I | 19 | 10 | 0.854 | 0.00214 | 0.00301 | −1.04808 | −3.859* | 0.0913 | 2180.86 | |
| ShangRao (SR) I | 6 | 3 | 0.733 | 0.00140 | 0.00154 | −0.46983 | 0.615 | 0.2894 | 569.91 | |
| Qiantang River | 31 | 25 | 0.981 | 0.00834 | 0.01998 | −2.20960** | −10.765*** | 0.0584 | ||
| Kaihua (KH) II | 15 | 11 | 0.933 | 0.00836 | 0.01565 | −2.01412* | −1.083 | 0.0766 | 273.56 | |
| PingYao (PY) II | 16 | 14 | 0.983 | 0.00787 | 0.01005 | −0.90881 | −4.471* | 0.0998 | 666.16 | |
| Oujiang River | ||||||||||
| Jinyun (JY) II | 10 | 7 | 0.911 | 0.00823 | 0.00806 | 0.09713 | 0.724 | 0.1648 | 217.45 | |
| Minjiang River | ||||||||||
| Shunchang (SC) II | 15 | 14 | 0.990 | 0.00855 | 0.01052 | −0.79692 | −5.112** | 0.1015 | 580.04 | |
| Mulan River | ||||||||||
| Xianyou (XY) II | 17 | 9 | 0.860 | 0.00337 | 0.00493 | −1.24407 | −1.450 | 0.0959 | 31.26 | |
| Jiulong River | ||||||||||
| Longyan (LY) III | 13 | 5 | 0.628 | 0.00490 | 0.00820 | −1.75948 | 2.843 | 0.1268 | 90.72 | |
| Hanjiang River | ||||||||||
| Jiaoling (SK) III | 15 | 7 | 0.819 | 0.00459 | 0.00459 | 0.00760 | 0.878 | 0.1424 | 98.89 | |
| Pearl River | ||||||||||
| Xijiang River | Shaoguan (SG) III | 14 | 10 | 0.890 | 0.00222 | 0.00441 | −2.04167* | −5.409** | 0.0959 | 7760.08 |
| Beijiang River | Liuzhou (LZ) I | 7 | 4 | 0.714 | 0.00209 | 0.00179 | 0.82563 | 0.205 | 0.2209 | 504.63 |
| Taiwan | ||||||||||
| Tamsui River | Tamsui (SD) IV | 5 | 3 | 0.700 | 0.00105 | 0.00126 | −1.04849 | −0.186 | 0.2667 | 9936.337 |
| Total | 160 | 95 | 0.984 | 0.02063 | 0.03323 | −1.22243 | −36.677*** | 0.0584 | 193.36 |
| AH | TR | SR | KH | PY | JY | SC | XY | LY | SK | SG | LZ | SD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AH | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| TR | 0.300 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| SR | 0.671 | 0.858 | 0.459 | 0.126 | 0.009 | 0.036 | 0.036 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| KH | 0.486 | 0.693 | −0.007 | 0.027 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| PY | 0.427 | 0.660 | 0.053 | 0.052 | 0.162 | 0.333 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| JY | 0.279 | 0.600 | 0.236 | 0.150 | 0.028 | 0.162 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| SC | 0.410 | 0.648 | 0.098 | 0.085 | −0.001 | 0.022 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| XY | 0.661 | 0.811 | 0.251 | 0.213 | 0.194 | 0.319 | 0.246 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| LY | 0.845 | 0.904 | 0.904 | 0.839 | 0.835 | 0.833 | 0.829 | 0.897 | 0.243 | 0.000 | 0.000 | 0.000 | |
| SK | 0.848 | 0.903 | 0.905 | 0.841 | 0.837 | 0.836 | 0.831 | 0.897 | 0.008 | 0.000 | 0.000 | 0.000 | |
| SG | 0.901 | 0.939 | 0.953 | 0.877 | 0.873 | 0.880 | 0.866 | 0.931 | 0.490 | 0.463 | 0.000 | 0.000 | |
| LZ | 0.364 | 0.757 | 0.814 | 0.500 | 0.457 | 0.399 | 0.442 | 0.717 | 0.882 | 0.884 | 0.939 | 0.000 | |
| SD | 0.441 | 0.603 | 0.919 | 0.651 | 0.629 | 0.558 | 0.611 | 0.826 | 0.893 | 0.893 | 0.949 | 0.841 |
| Source of variation | Variance components | Percentage of variation |
|---|---|---|
| Among geographic district | 1.03198 | 7.96 |
| Among populations within geographic district | 9.06970 *** | 69.97 |
| Within populations | 2.86151 *** | 22.07 |
| Source of variation | Variance components | Percentage of variation |
|---|---|---|
| Among groups | 9.83421 * | 77.40 |
| Among populations within zoogeographic zone | 0.01060 *** | 0.08 |
| Within populations | 2.86151 *** | 22.52 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, J.-Q.; Tang, W.-Q.; Liao, T.-Y.; Sun, Y.; Zhou, Z.-C.; Han, C.-C.; Liu, D.; Lin, H.-D. Phylogeographical Analysis on Squalidus argentatus Recapitulates Historical Landscapes and Drainage Evolution on the Island of Taiwan and Mainland China. Int. J. Mol. Sci. 2012, 13, 1405-1425. https://doi.org/10.3390/ijms13021405
Yang J-Q, Tang W-Q, Liao T-Y, Sun Y, Zhou Z-C, Han C-C, Liu D, Lin H-D. Phylogeographical Analysis on Squalidus argentatus Recapitulates Historical Landscapes and Drainage Evolution on the Island of Taiwan and Mainland China. International Journal of Molecular Sciences. 2012; 13(2):1405-1425. https://doi.org/10.3390/ijms13021405
Chicago/Turabian StyleYang, Jin-Quan, Wen-Qiao Tang, Te-Yu Liao, Yang Sun, Zhuo-Cheng Zhou, Chiao-Chuan Han, Dong Liu, and Hung-Du Lin. 2012. "Phylogeographical Analysis on Squalidus argentatus Recapitulates Historical Landscapes and Drainage Evolution on the Island of Taiwan and Mainland China" International Journal of Molecular Sciences 13, no. 2: 1405-1425. https://doi.org/10.3390/ijms13021405