Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review

Abstract
:1. Introduction
1.1. Prodrugs
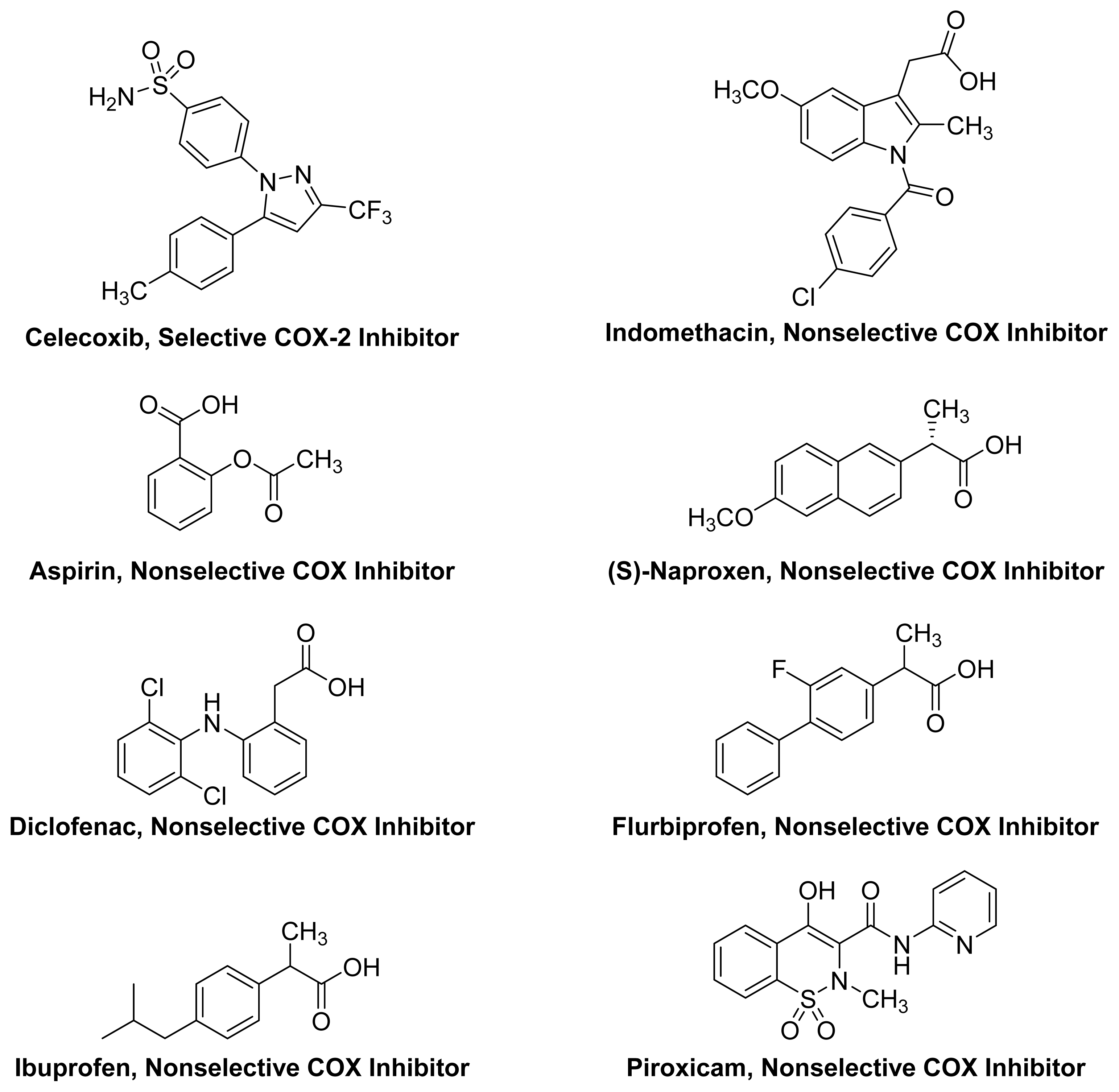
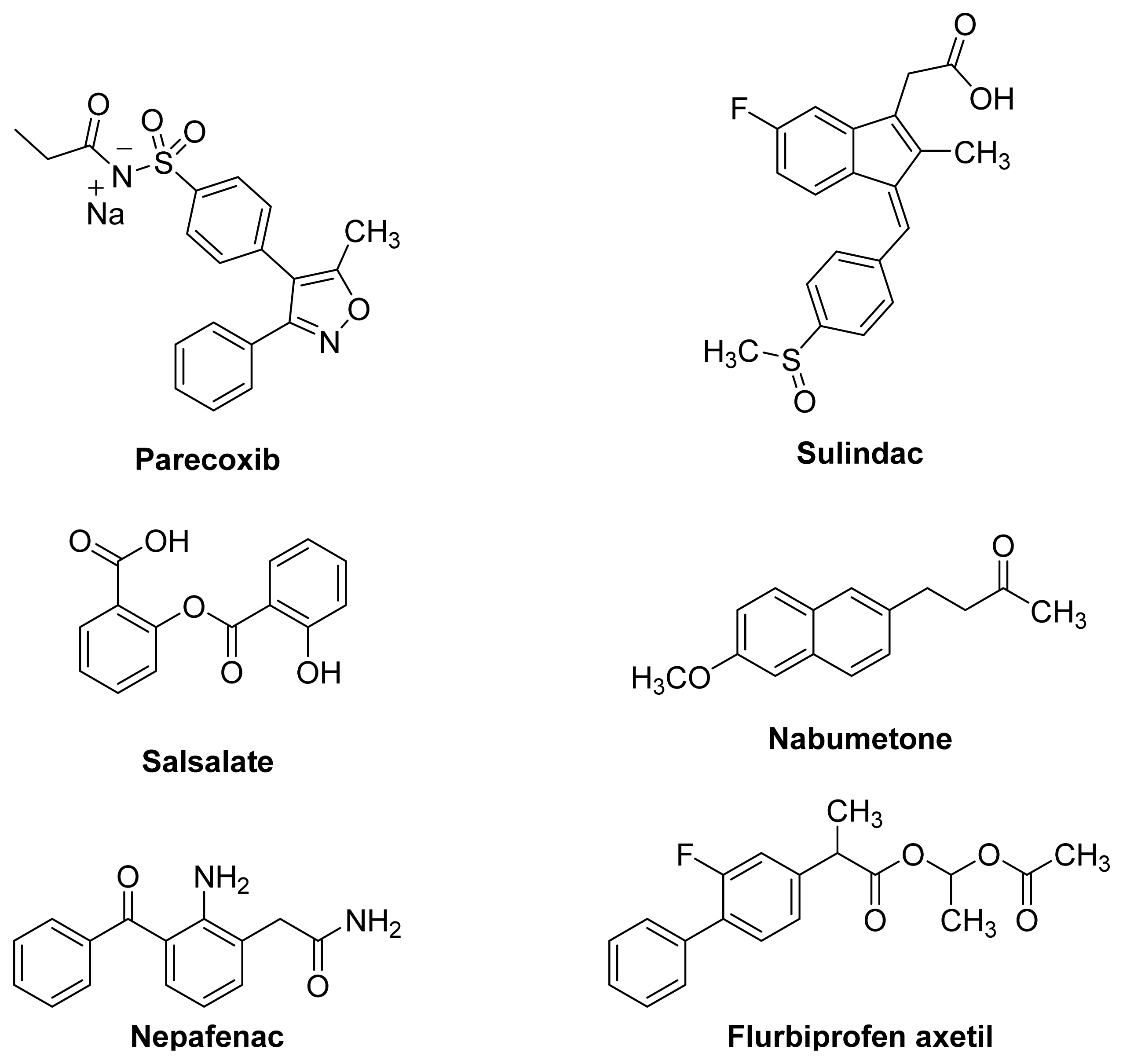
1.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
1.3. NSAID Prodrugs
2. NO-NSAIDs
2.1. Introduction
2.2. NO-NSAIDs with Intrinsic Pharmacological Activity
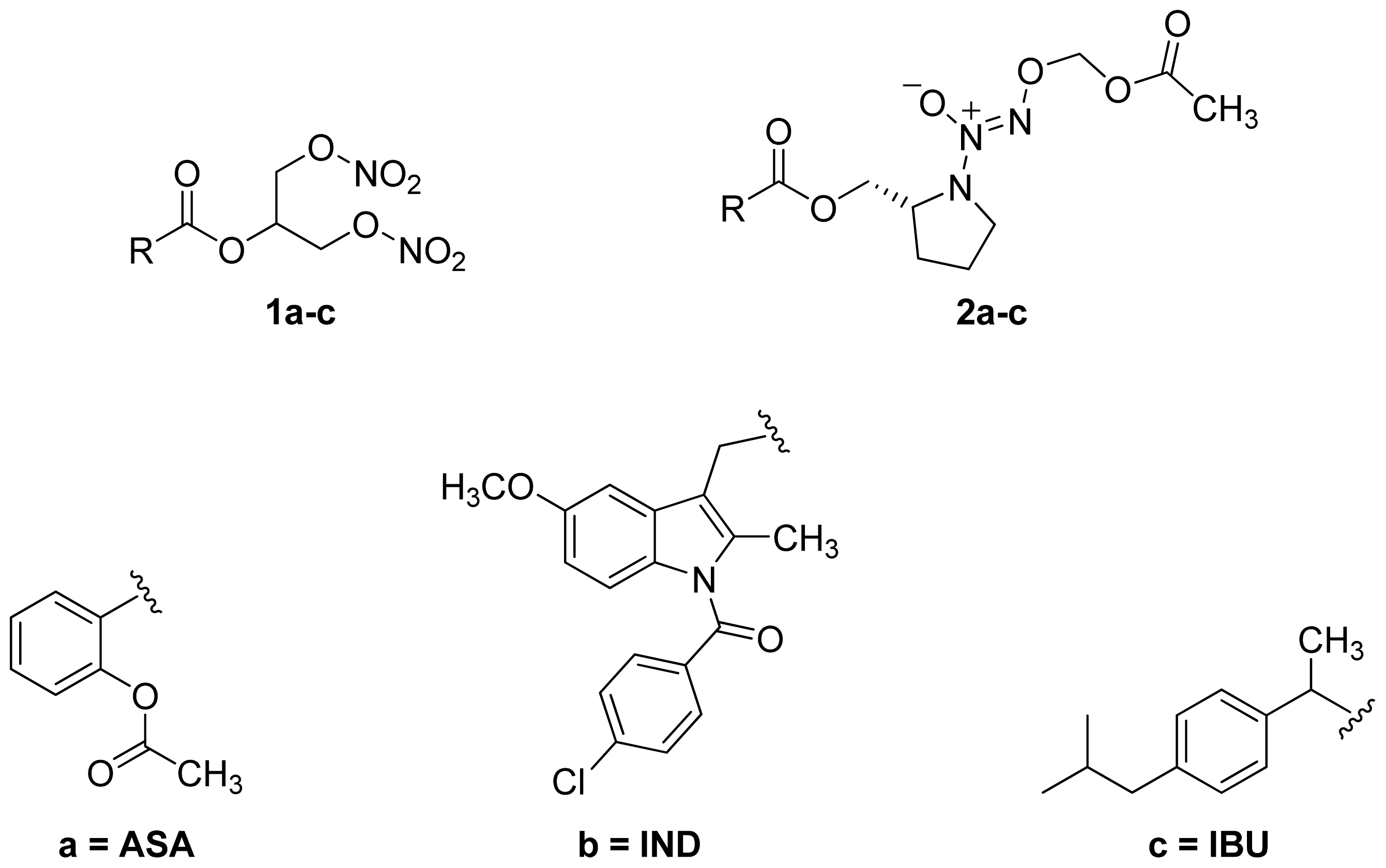
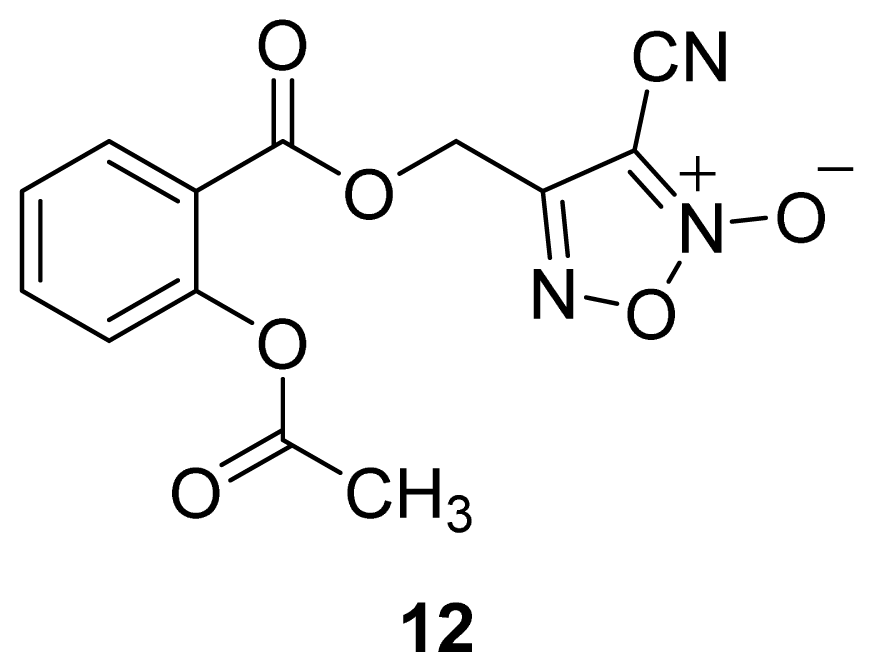
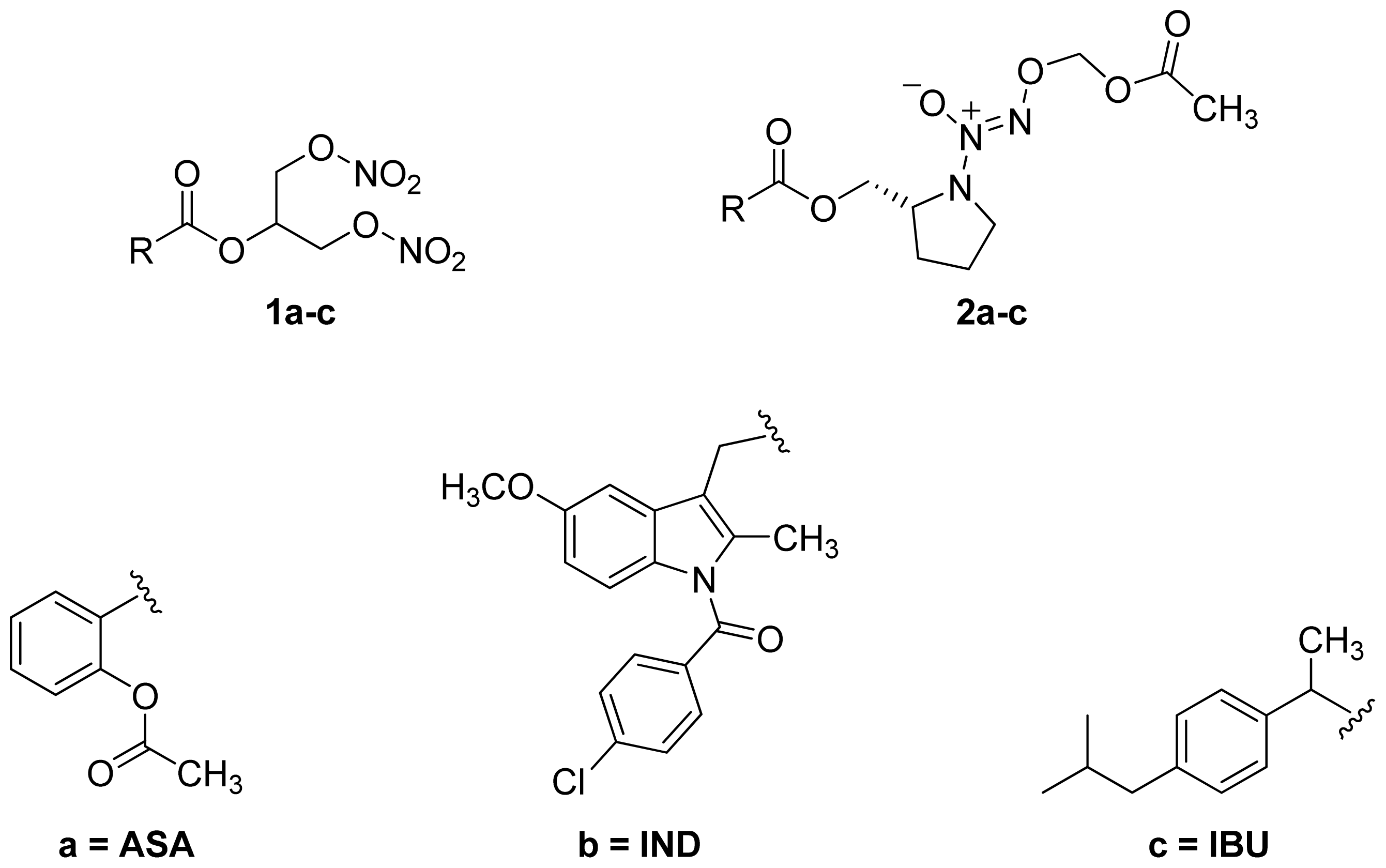
- A group of glyceryl dinitrate esters (1a–c) and NONOate-containing prodrugs (2a–c) of aspirin (a = ASA), indomethacin (b = IND) and ibuprofen (c = IBU) (Figure 7) have been synthesized and evaluated in vivo and in vitro for their anti-inflammatory activity [52]. Although the ibuprofen and indomethacin esters showed in vivo activity comparable to their parent NSAIDs, the aspirin esters showed less than half the activity of aspirin. Furthermore, none of them showed in vitro inhibitory activity against COX-1; rather, they all showed inhibitory activity against COX-2 (in vitro IC50 = 0.6–9.3 μM) that was comparable to or greater than that of their parent NSAIDs. These results pose two questions: was the in vivo biological activity of these compounds due solely to the parent NSAIDs? And, second, was the GI-sparing profile only the result of NO release?
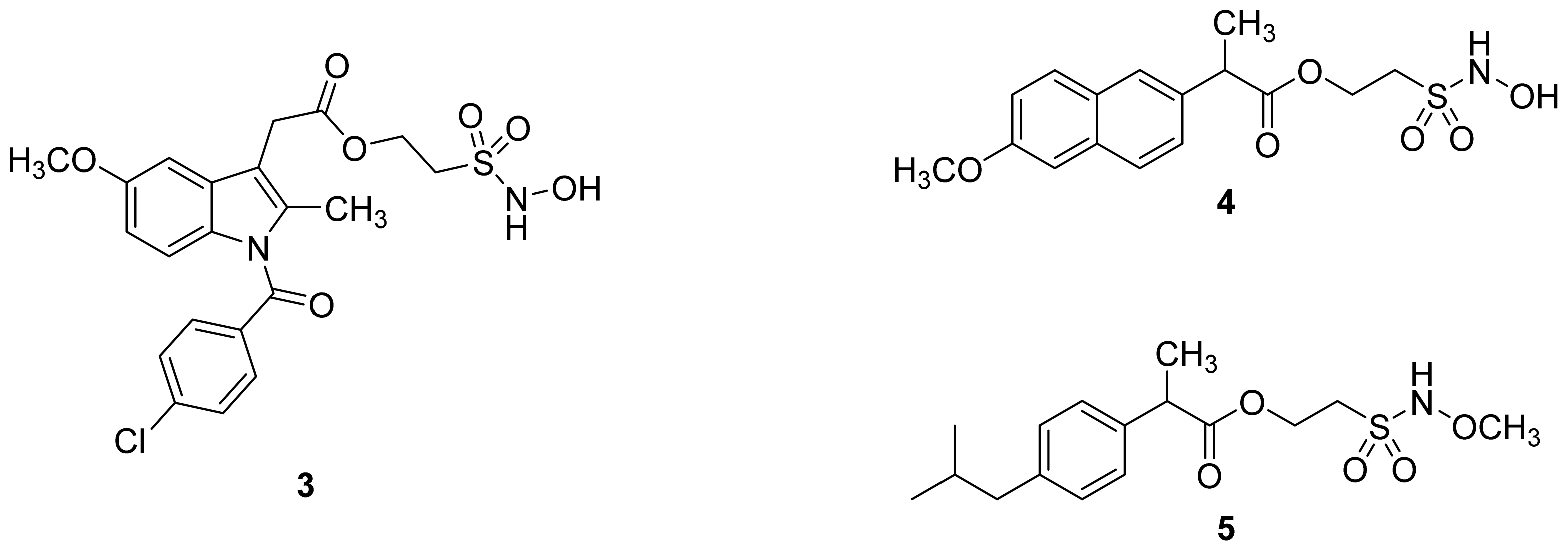
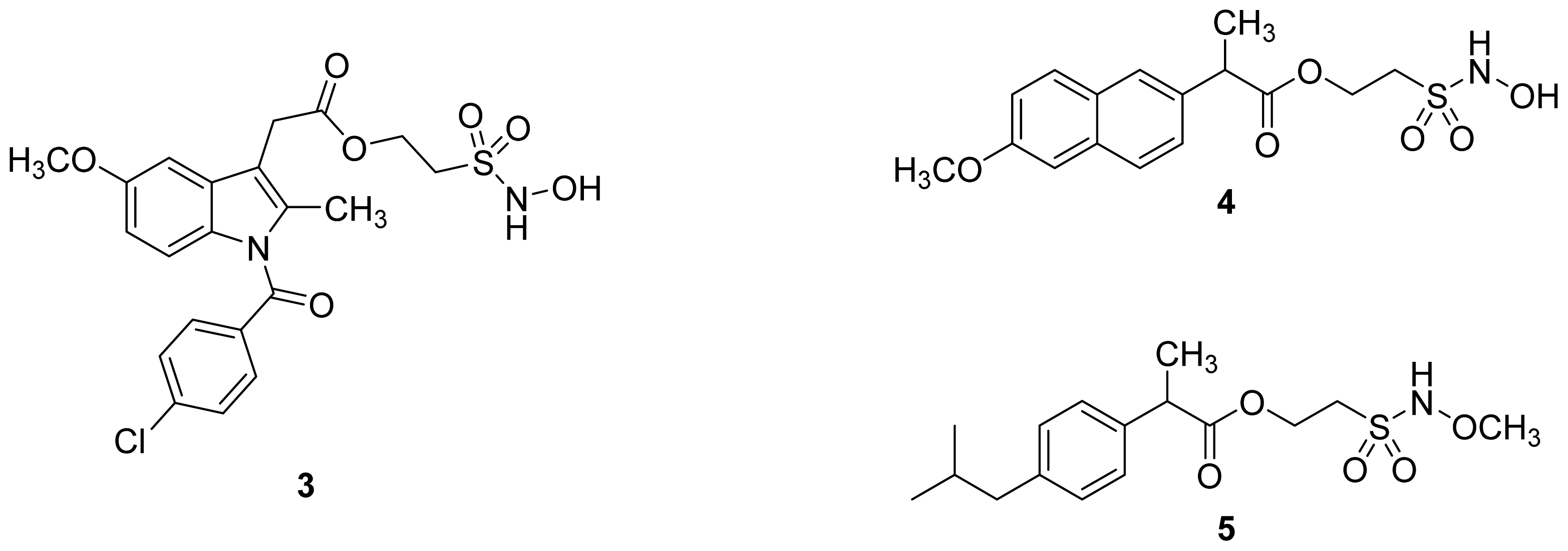
- The ethanesulfohydroxamic acid esters of indomethacin (3) and naproxen (4) were synthesized and evaluated as NO-NSAIDs [47] (Figure 8). The indomethacin ester 3 was a selective COX-2 inhibitor (IC50 = 0.42 μM) in vitro, with an in vivo ID50 of 19.1 μmol/kg compared to 11.7 μmol/kg for indomethacin. Furthermore, the non-NO-releasing hydroxamic acid ester of ibuprofen 5 showed comparable in vivo potency (78.9% inhibition of inflammation at 327 μmol/kg oral dose) to its NO-releasing hydroxamic acid (79.5% inhibition of inflammation at 327 μmol/kg oral dose). It is worth mentioning that the IC50 of 5 against COX-2 was 0.63 μM. This adds more skepticism to the accepted notion(s) that NO-releasing esters of NSAIDs are mere prodrugs and/or that NO release is essential for their GI-sparing profile. The possibility that compound 5 is a prodrug still exists, but no investigation of this possibility was performed.
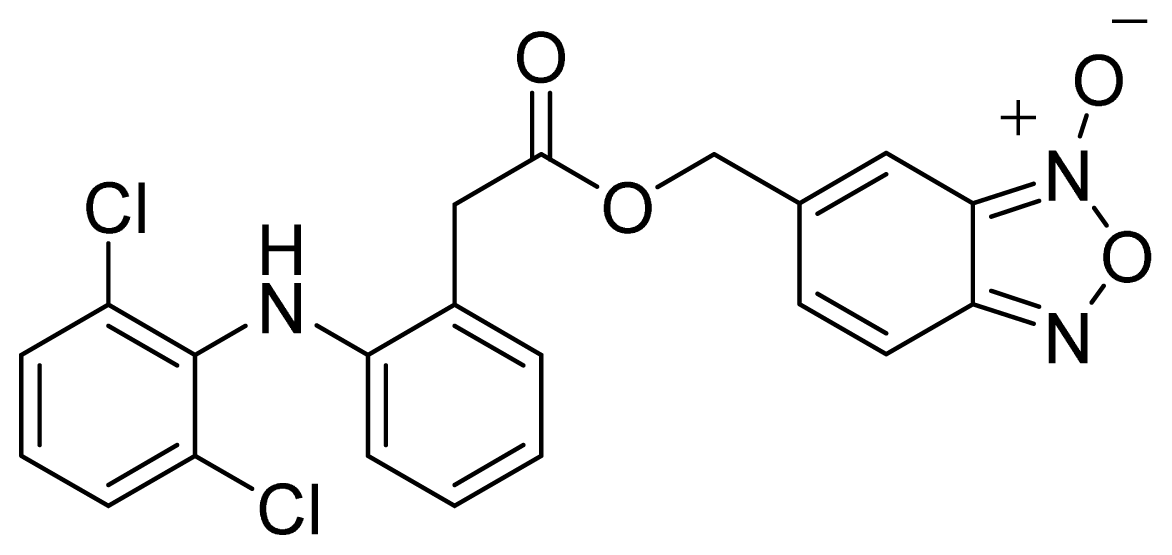
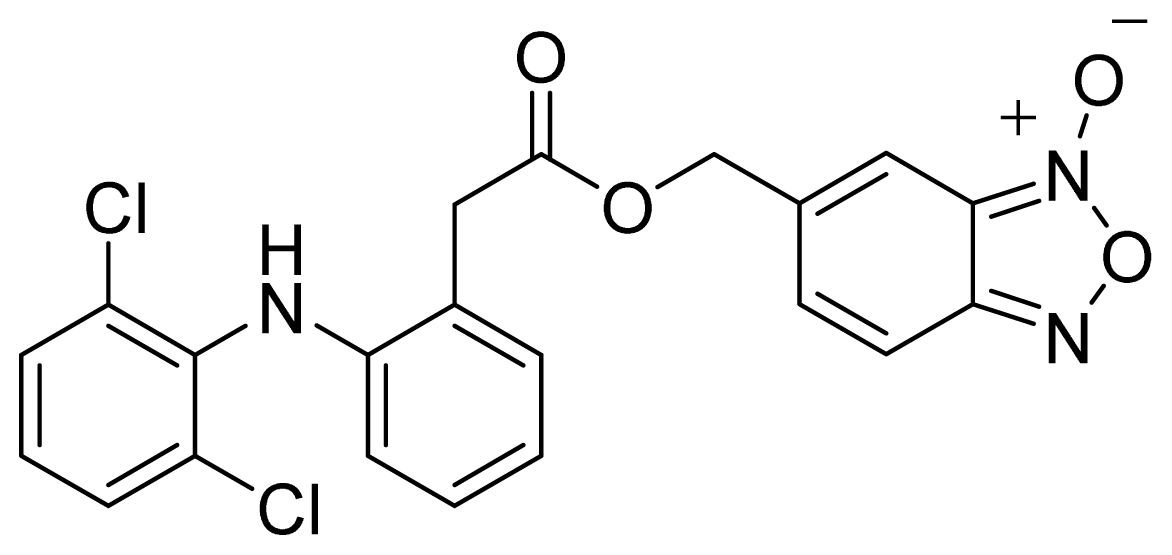
- The ester of diclofenac (6) (Figure 9) contains benzofuroxan as the NO-releasing group. Ester 6 was synthesized and evaluated in vitro and in vivo for the reduction of PGE2 and TXB2 levels in plasma. This ester was found to be more effective in inhibiting PGE2 synthesis than TXB2 synthesis in vitro. This finding suggests that this NO-diclofenac ester is more selective for COX-2 [46].
- 2-Hydroxysulfamoylbenzoic acid 7 and its ethyl benzoate ester 8 (Figure 10), which are nitric oxide-releasing analogs of aspirin, were reported by Kaur et al. (2012) [53]. Although not prodrugs, these compounds show that the simple backbone of aspirin, a nonselective COX inhibitor, can be modified to become a selective COX-2 inhibitor [53]. The pharmacological evaluation of compounds 7 and 8 revealed that hydroxamic acid 8 is as a potent and a much more selective COX-2 inhibitor than celecoxib (IC50 = 0.09 μM, with more than 1000-fold selectivity). In addition, hydroxamic acid 8 was found to be a 5-lipoxygenase (5-LOX) inhibitor (IC50 = 0.4 μM). 5-LOX is an essential enzyme in the biosynthetic pathway of leukotrienes from arachidonic acid. Leukotrienes have an important role in the inflammatory process, and hence, inhibitors of 5-LOX exert an anti-inflammatory action [54]. By contrast, hydroxamic acid 7 was 10 times less potent than both celecoxib and hydroxamic acid 8, with a COX-2 selectivity comparable to that of celecoxib. Both hydroxamic acid derivatives were effective anti-inflammatory agents in vivo, with ED50 values of 23.1 μM and 24.5 μM for compounds 7 and 8, respectively, compared to 10.8 μM for celecoxib and 128 μM for aspirin. It is worth mentioning that these compounds also exhibited a time-dependent release of nitric oxide, which leads to a GIT-safe profile.
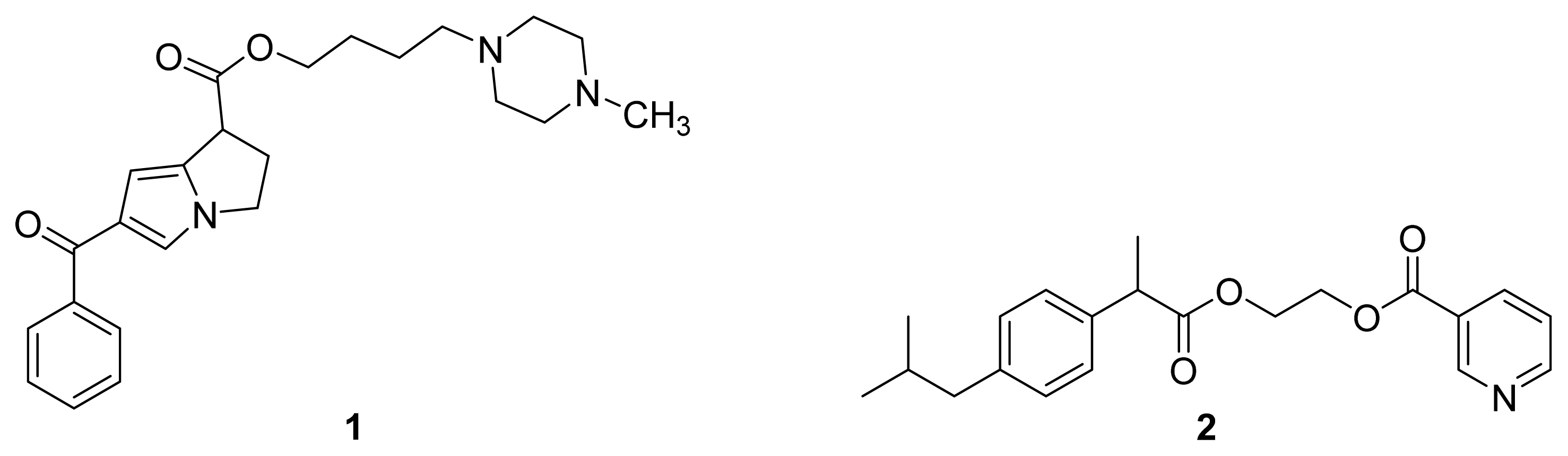
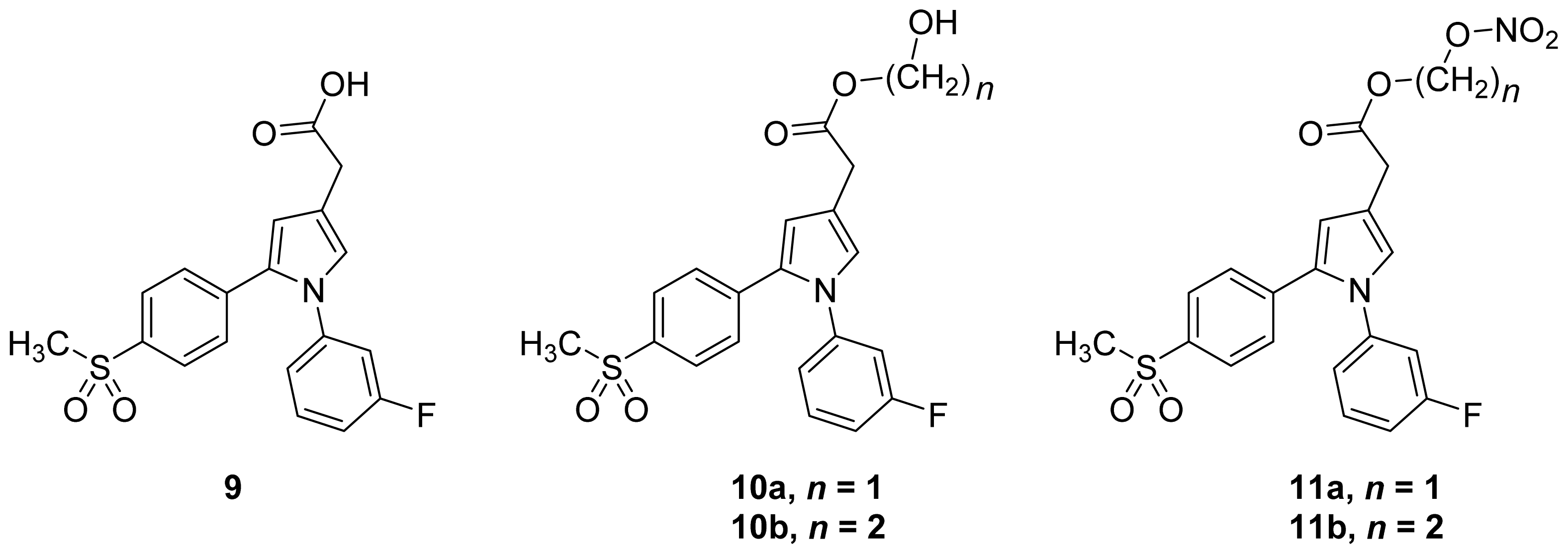
- Biava et al. (2012) reported a novel class of diarylpyrrole acetic acid derivatives, which possess the typical scaffold of classic selective COX-2 inhibitors (Figure 11). The researchers showed that 9, its hydroxyethyl and hydroxypropyl ester intermediates (10a and 10b), and their corresponding nitrate esters (11a and 11b) were all potent selective COX-2 inhibitors in vitro (IC50 = 0.019-0.083 μM) and considerably more selective than celecoxib. None of the esters (10a, 10b, 11a, and 11b) were claimed by the authors to be prodrugs of 9. Furthermore, molecular docking experiments have shown that the alcohol group in 10a and 10b and one of the oxygen atoms of the nitrates in 11a and 11b form one or more hydrogen bonds with amino acid side chains in the active site of COX-2. Furthermore, the nitrate ester 11a exhibited an in vitro vasorelaxing effect comparable to that of nitroglycerin [55]. This vasorelaxing effect can be considered the rationale behind designing NO-Coxibs.
2.3. The Role of the Linker in NO-NSAIDs
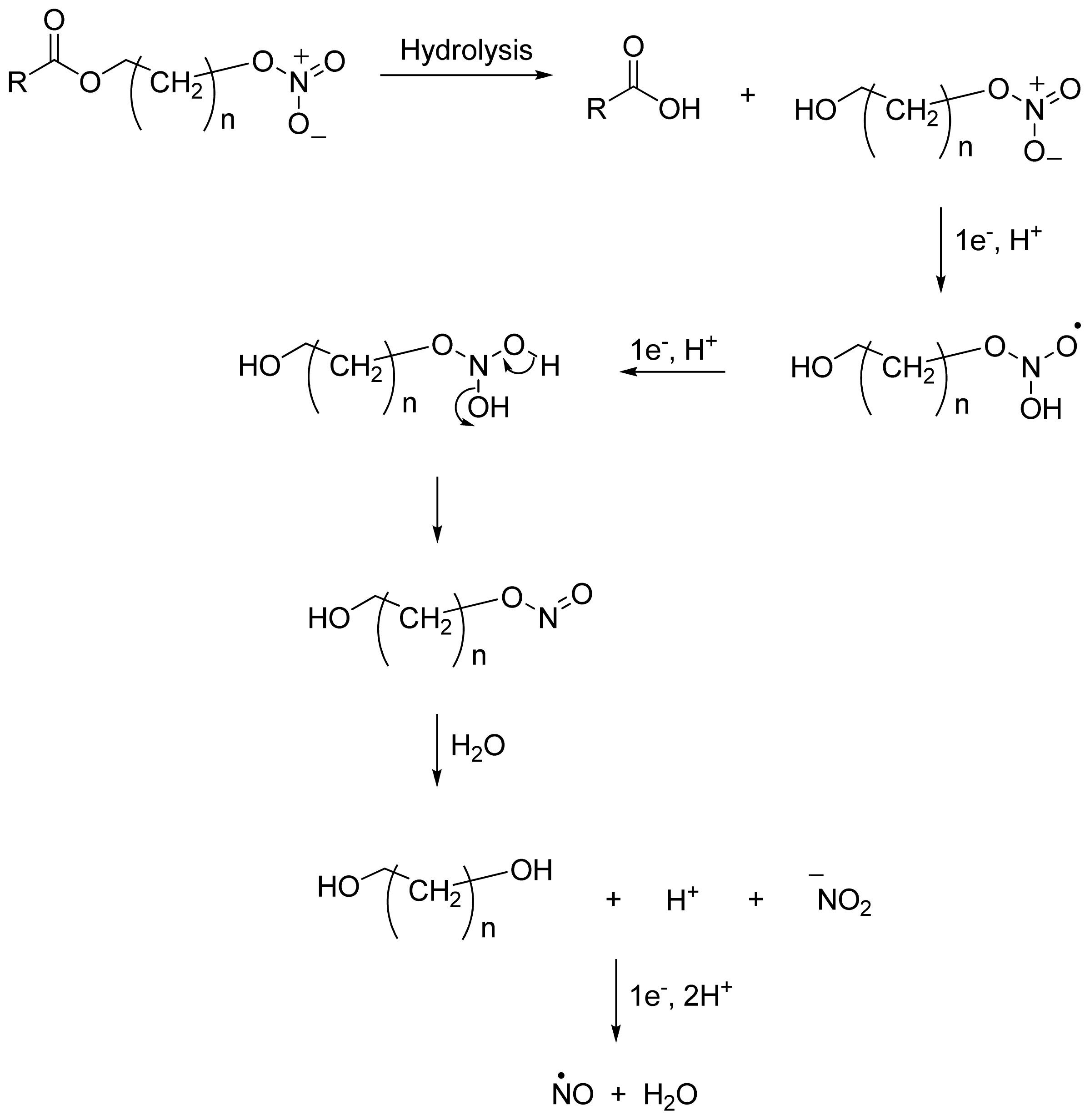
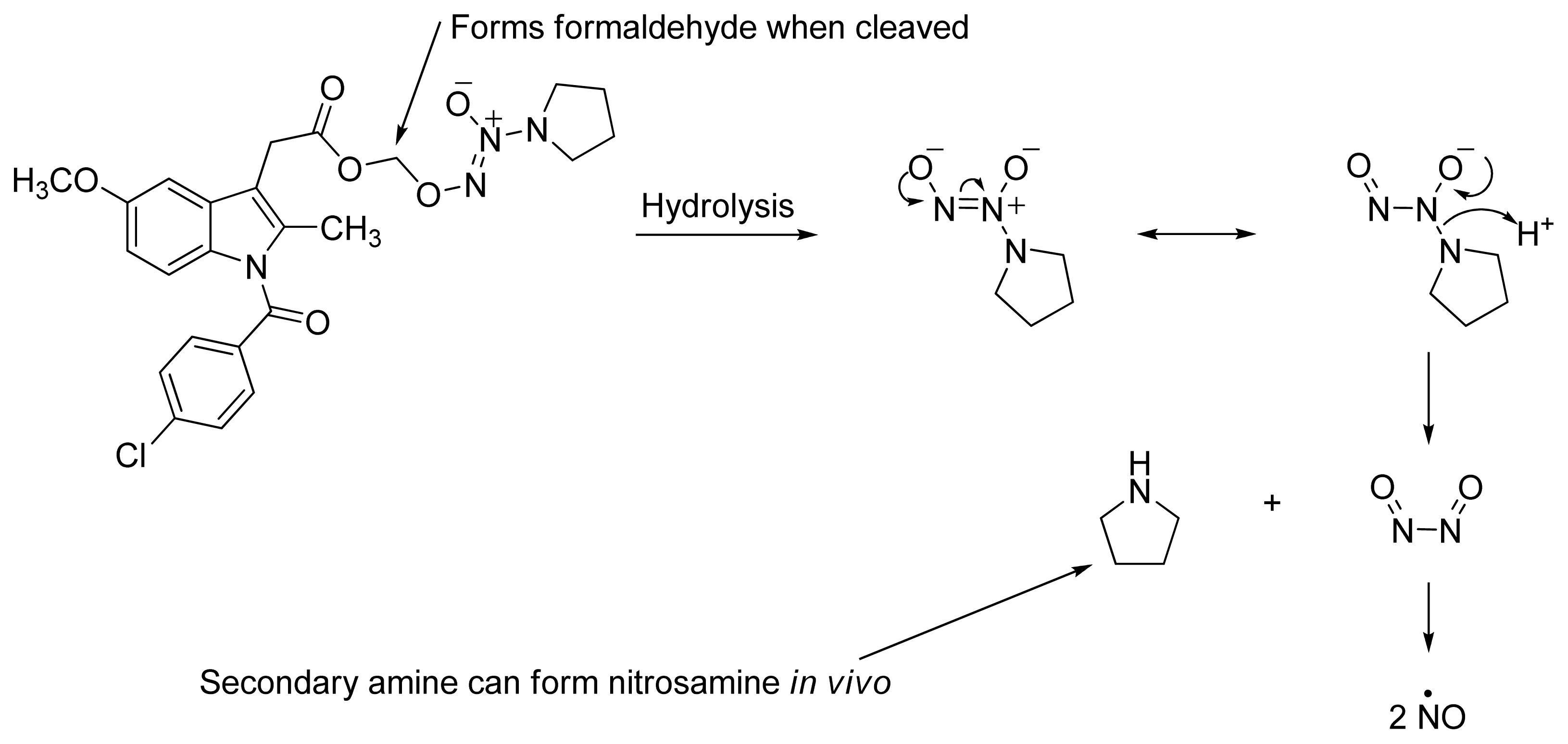
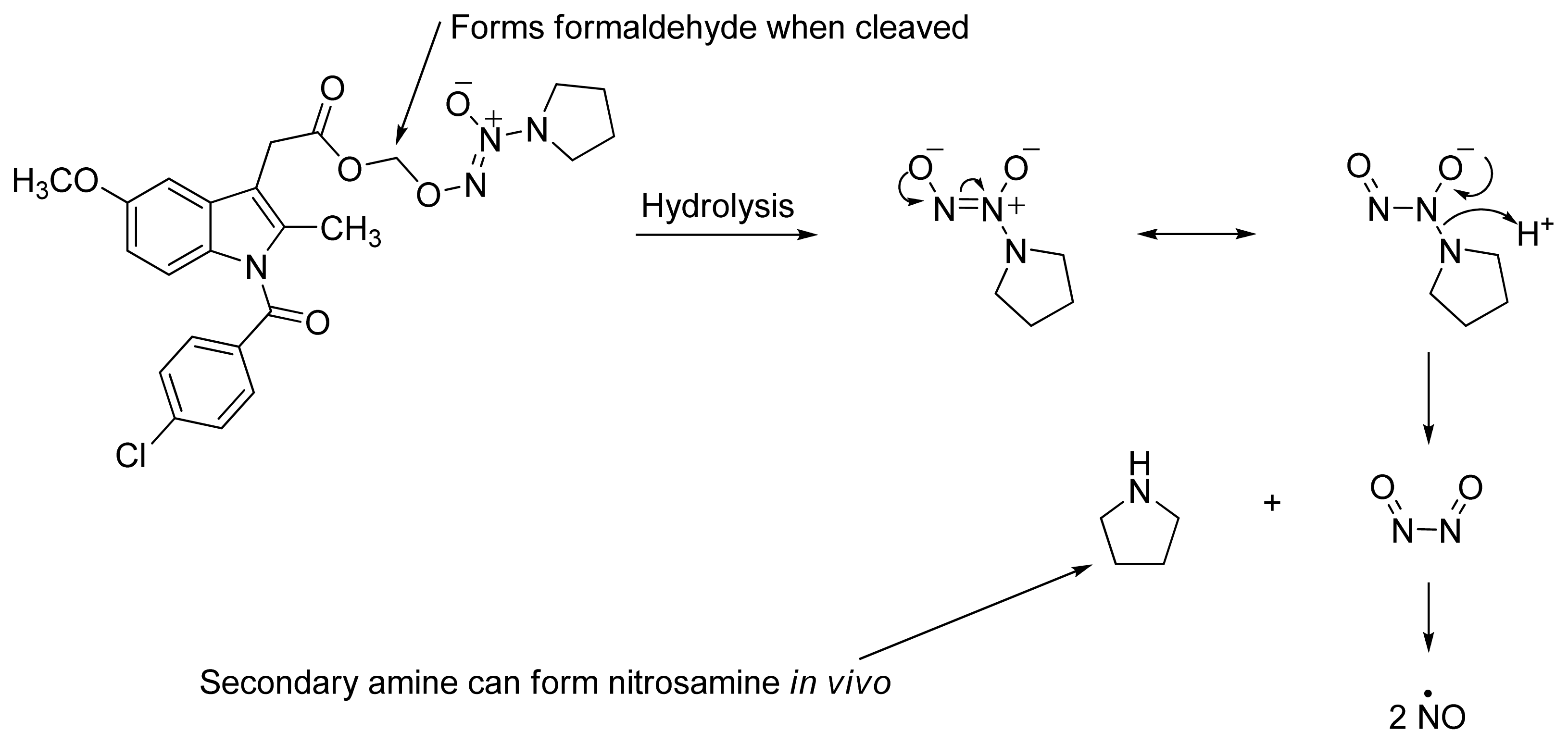
2.4. NO-NSAIDs and in Vivo Hydrolysis
3. Anticholinergic NSAIDS and AChEI-NSAIDs
3.1. Introduction
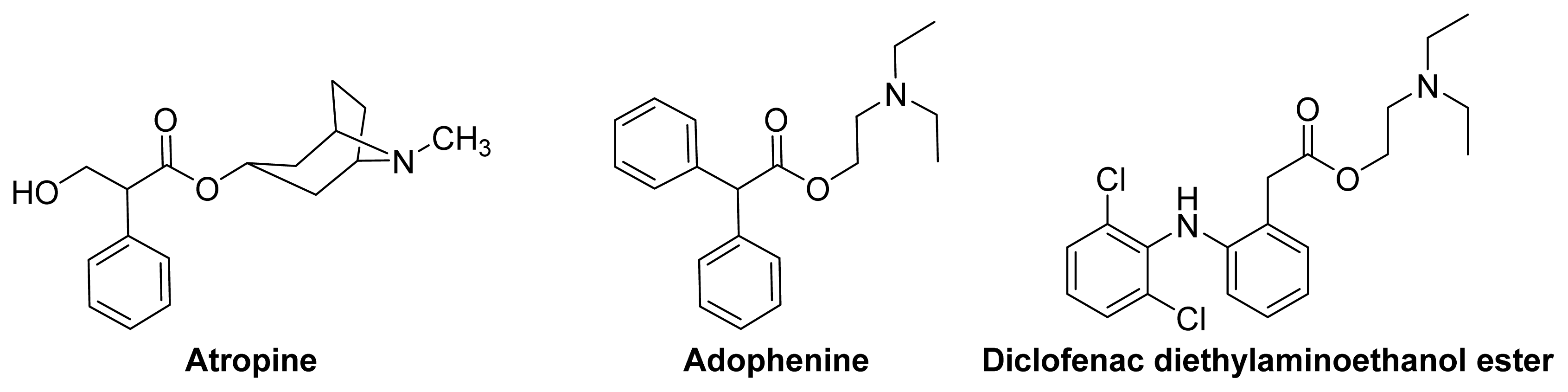
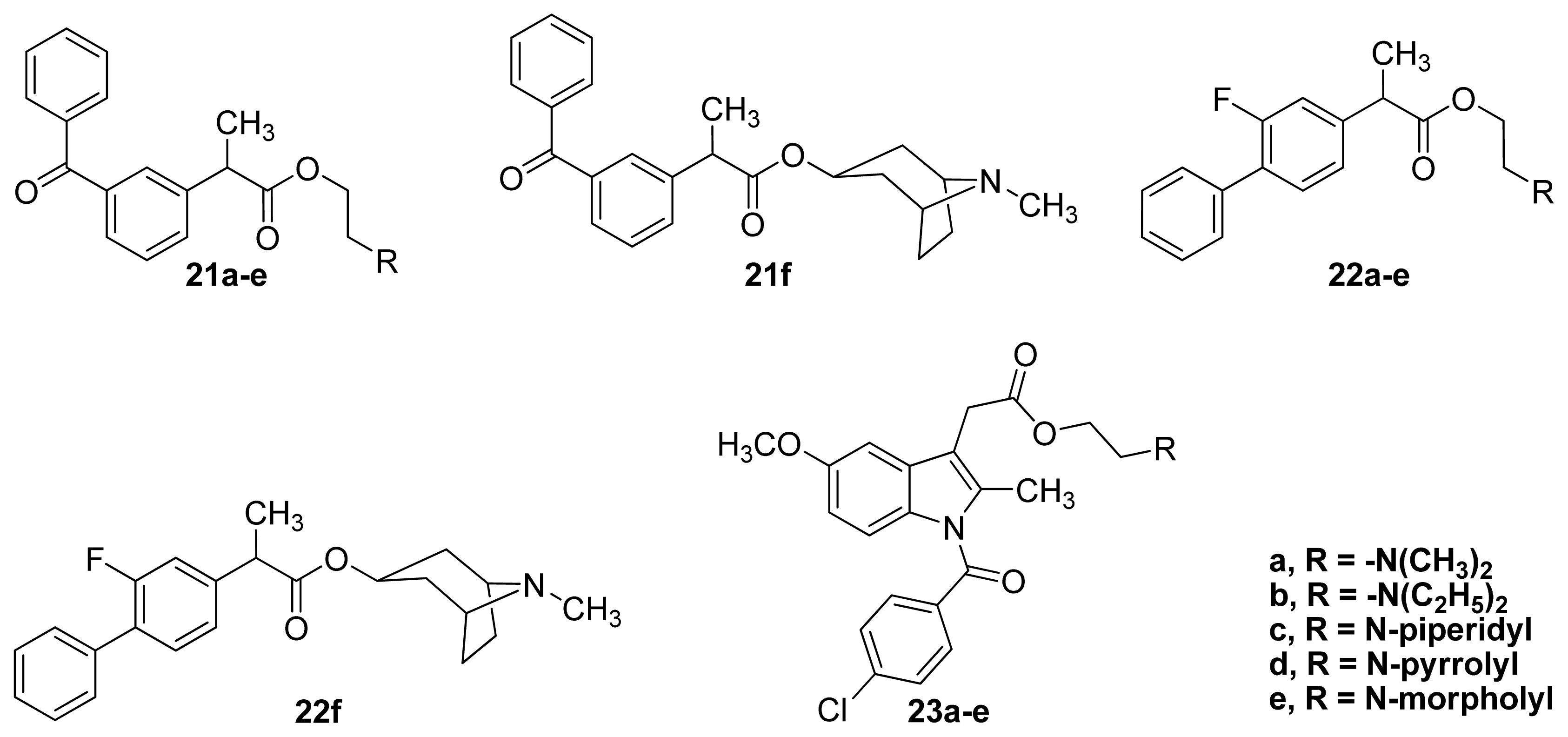
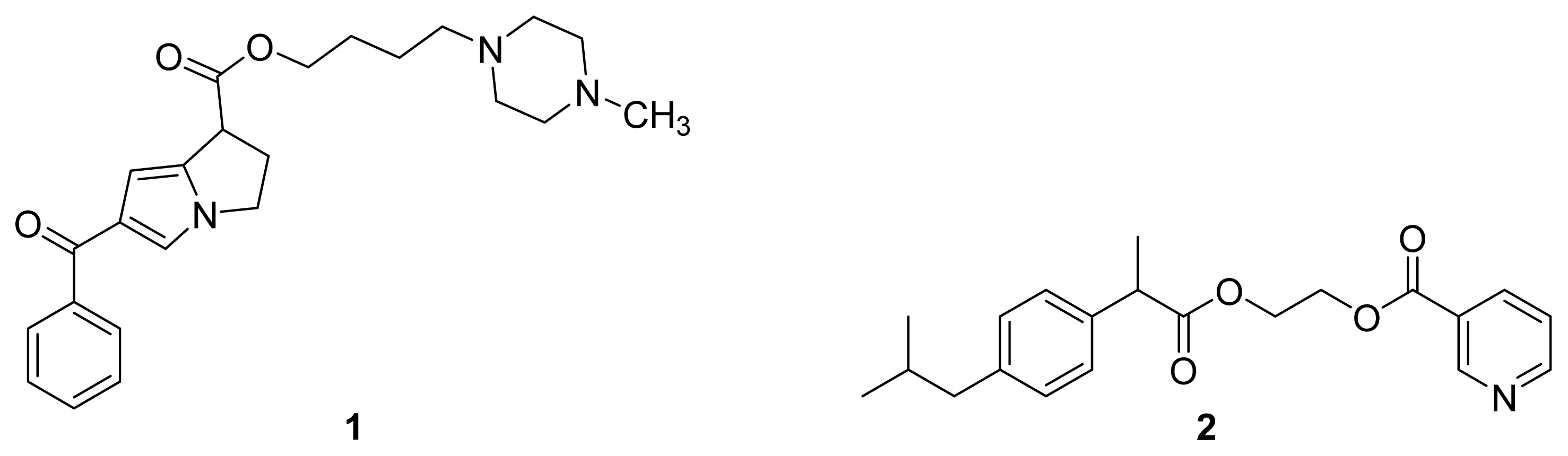
3.2. Anticholinergic NSAIDs
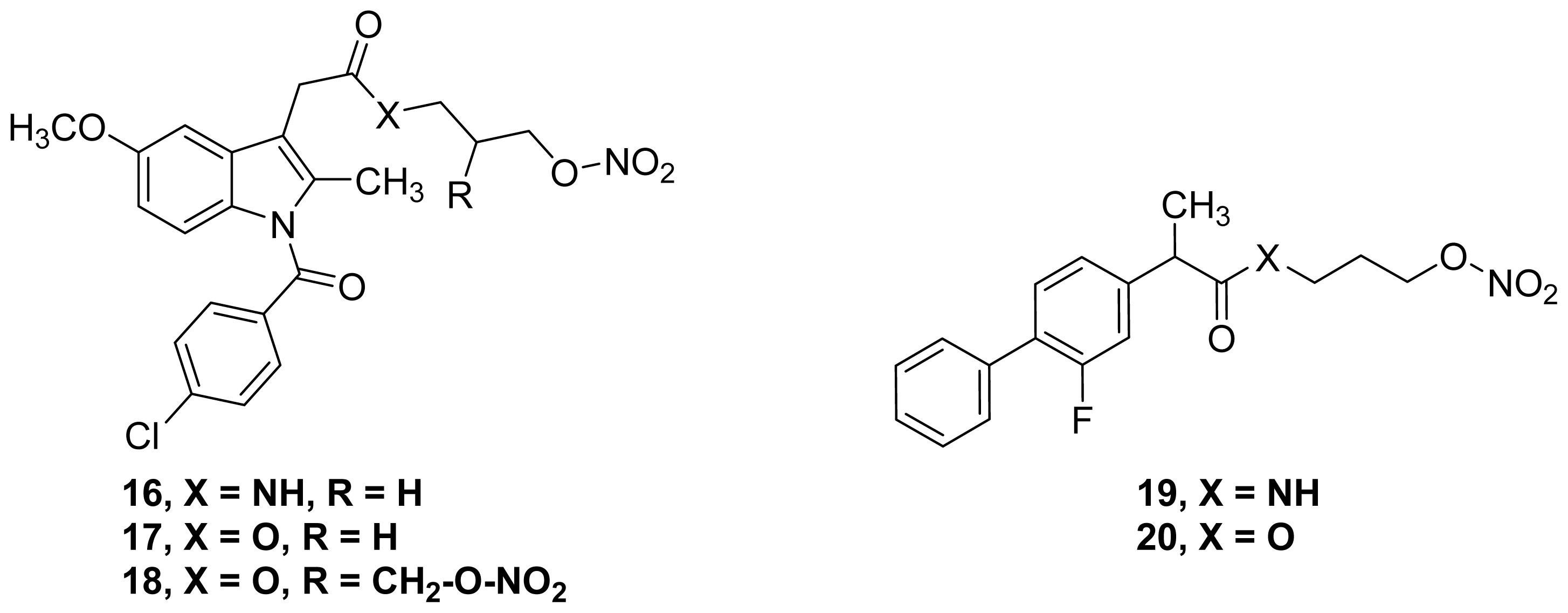
3.3. AChEI-NSAIDs
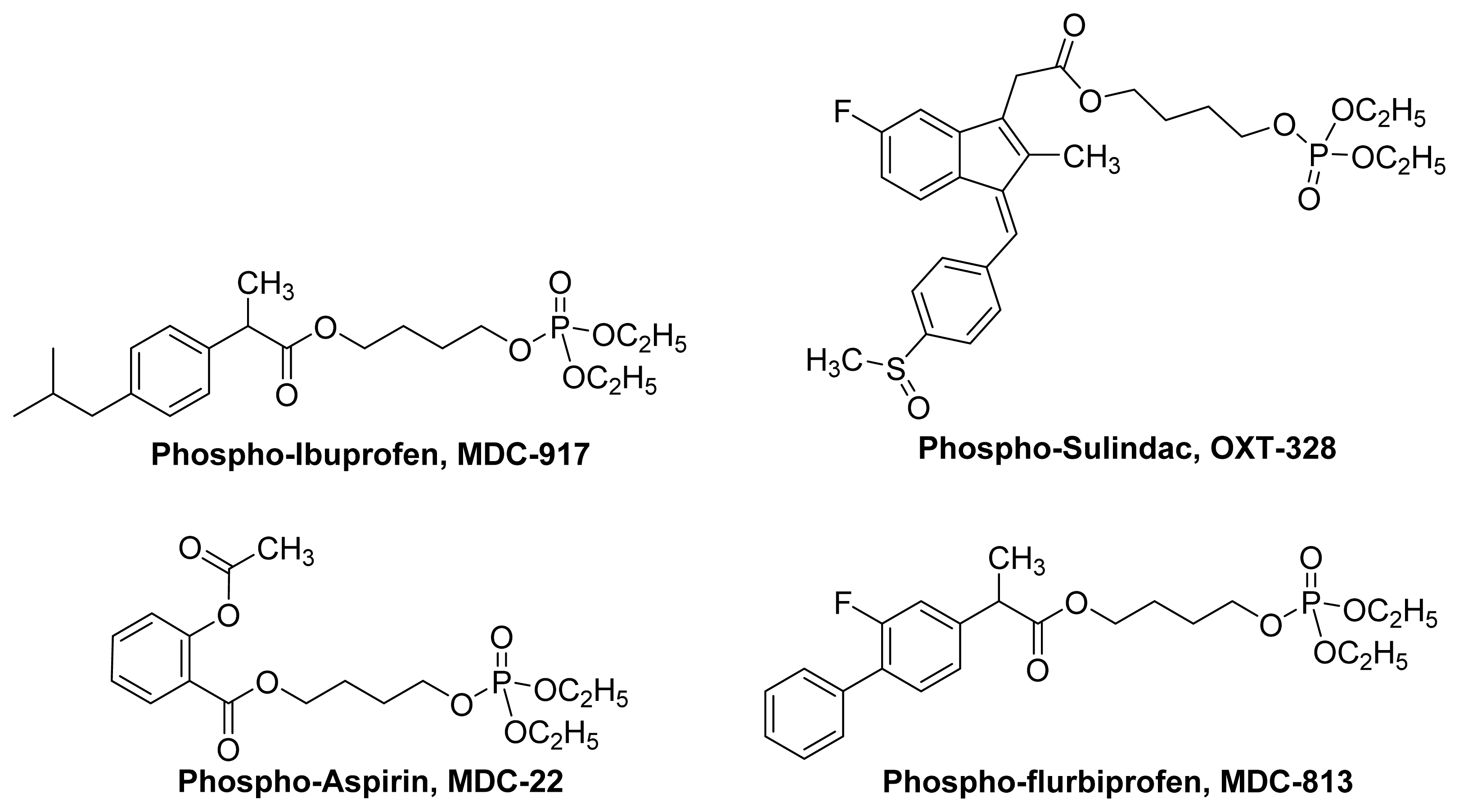
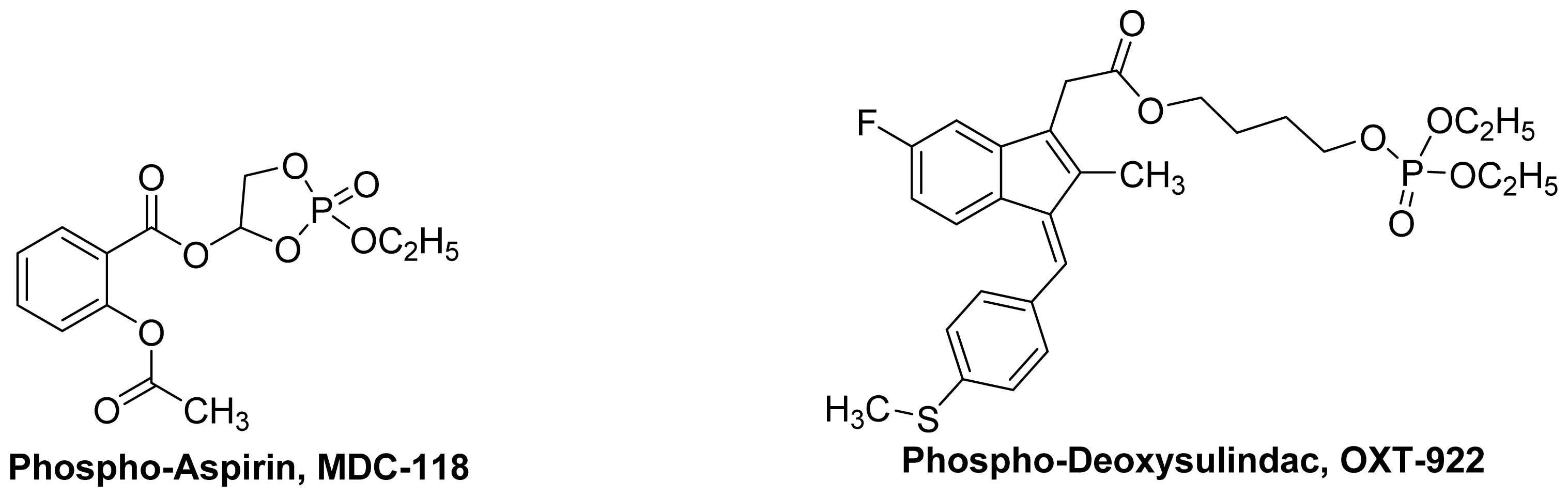
4. Phospho-NSAIDs
4.1. Introduction
4.2. Mechanism of Action Phospho-NSAIDs
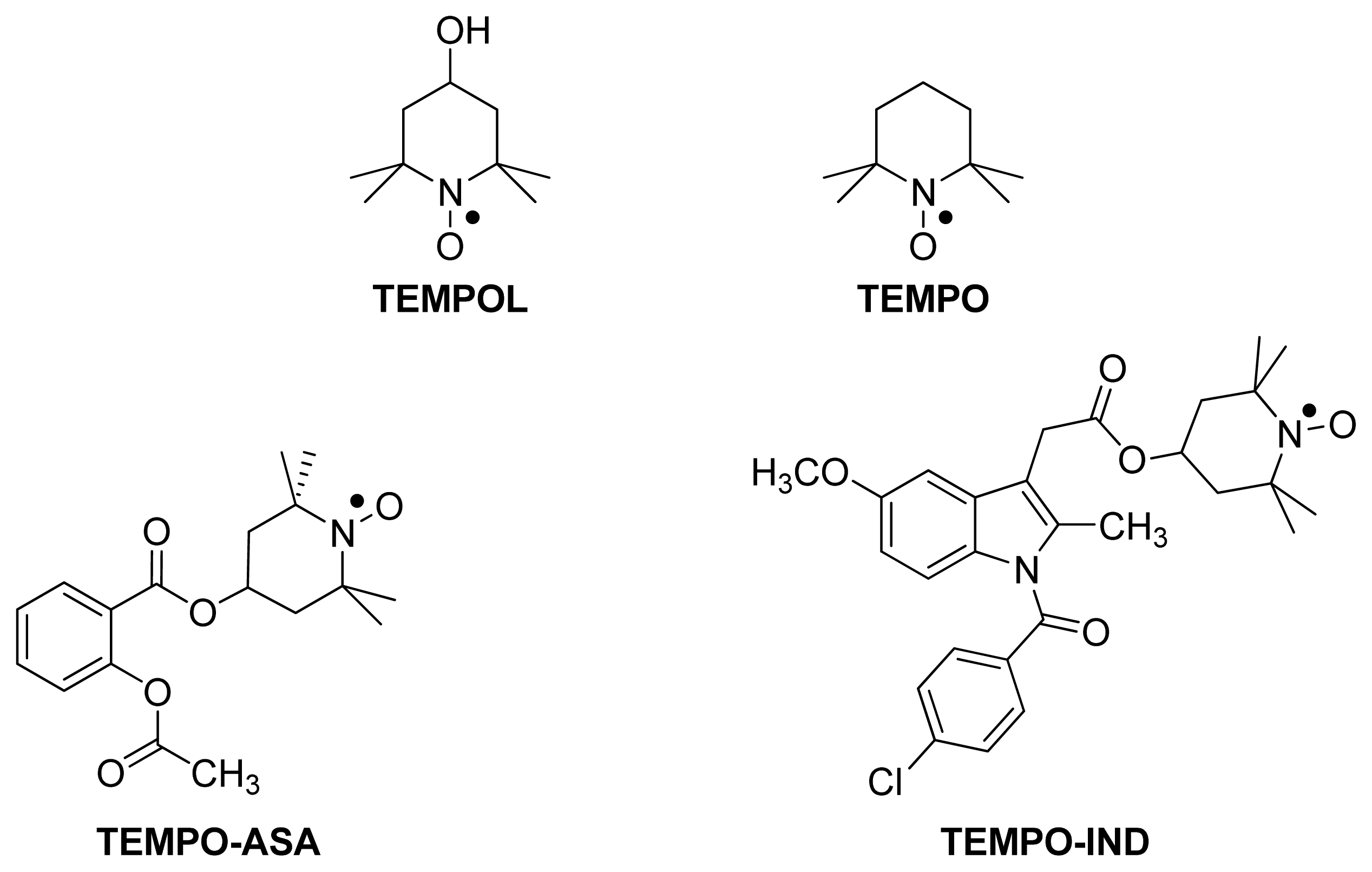
5. Miscellaneous Agents
5.1. TEMPO-NSAIDs
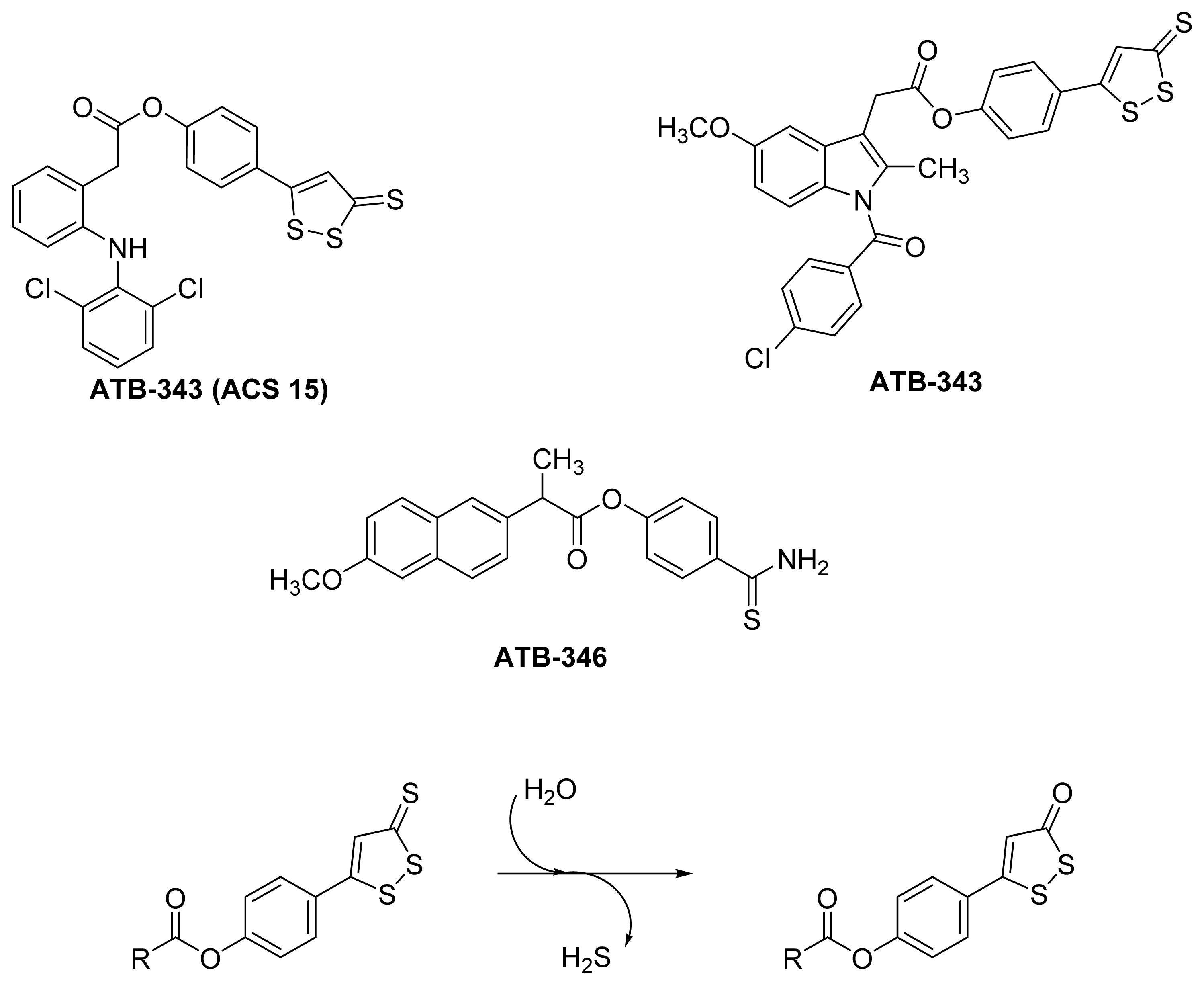
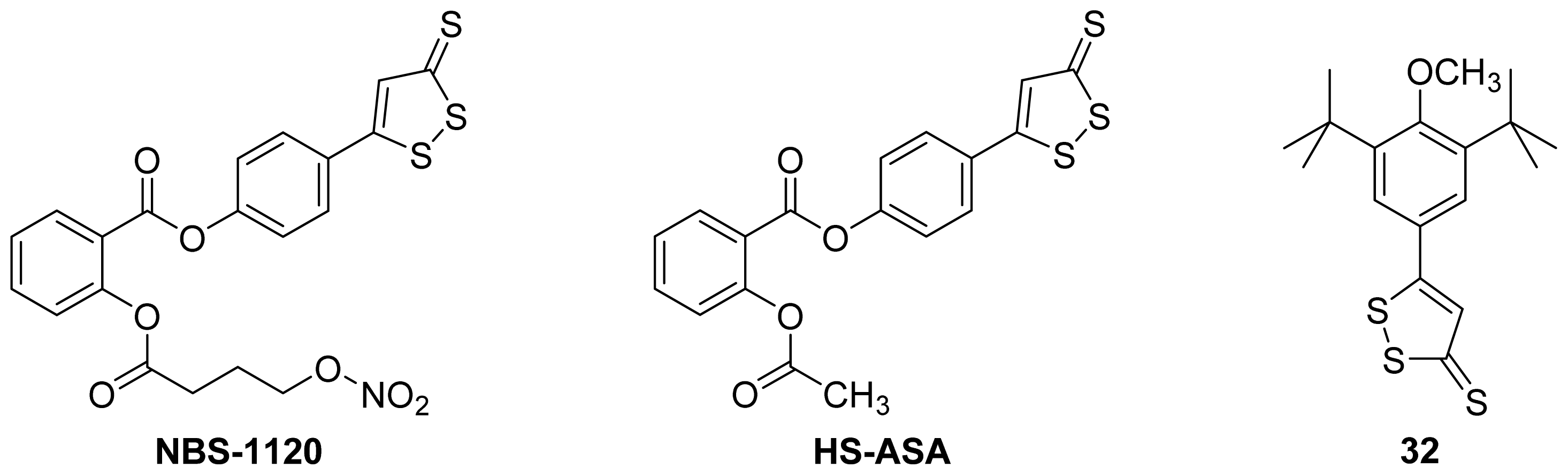
5.2. HS-NSAIDs
6. Conclusions
References
- Albert, A. Chemical Aspects of Selective Toxicity. Nature 1958, 182, 421–423. [Google Scholar]
- Li, X. Oral Bioavailability: Basic Principles, Advanced Concepts, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011; p. 448. [Google Scholar]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov 2008, 7, 255–270. [Google Scholar]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev 2011, 63, 750–771. [Google Scholar]
- Testa, B. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol 2009, 13, 338–344. [Google Scholar]
- Das, N.; Dhanawat, M.; Dash, B.; Nagarwal, R.C.; Shrivastava, S.K. Codrug: An efficient approach for drug optimization. Eur. J. Pharm. Sci 2010, 41, 571–588. [Google Scholar]
- Qandil, A.; Al-Nabulsi, S.; Al-Taani, B.; Tashtoush, B. Synthesis of piperazinylalkyl ester prodrugs of ketorolac and their in vitro evaluation for transdermal delivery. Drug Dev. Ind. Pharm 2008, 34, 1054–1063. [Google Scholar]
- Abu Zanat, F.Z.; Qandil, A.M.; Tashtoush, B.M. A promising codrug of nicotinic acid and ibuprofen for managing dyslipidemia. I: Synthesis and in vitro evaluation. Drug Dev. Ind. Pharm 2011, 37, 1090–1099. [Google Scholar]
- Chung, M.C.; Bosquesi, P.L.; dos Santos, J.L. A Prodrug approach to improve the physico-chemical properties and decrease the genotoxicity of nitro compounds. Curr. Pharm. Des 2011, 17, 3515–3526. [Google Scholar]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons Learned from Marketed and Investigational Prodrugs. J. Med. Chem 2004, 47, 2393–2404. [Google Scholar]
- Stella, V.J. Prodrugs: Some thoughts and current issues. J. Pharm. Sci 2010, 99, 4755–4765. [Google Scholar]
- Qandil, A.M.; Rezigue, M.M.; Tashtoush, B.M. Synthesis, characterization and in vitro hydrolysis of a gemfibrozil-nicotinic acid codrug for improvement of lipid profile. Eur. J. Pharm. Sci 2011, 43, 99–108. [Google Scholar]
- Dhaneshwar, S.; Vadnerkar, G. Rational Design and Development of Colon-Specific Prodrugs. Curr. Top. Med. Chem 2011, 11, 2318–2345. [Google Scholar]
- Jana, S.; Mandlekar, S.; Marathe, P. Prodrug design to improve pharmacokinetic and drug delivery properties: Challenges to the discovery scientists. Curr. Med. Chem 2010, 17, 3874–3908. [Google Scholar]
- Teagarden, D.L.; Nema, S. Case Study: Parecoxib: A Prodrug of Valdecoxib Prodrugs; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 1335–1346. [Google Scholar]
- Melnikova, I. Pain market. Nat. Rev. Drug Discov 2010, 9, 589–590. [Google Scholar]
- Laine, L. The gastrointestinal effects of nonselective NSAIDs and COX-2—selective inhibitors. Semin. Arthritis Rheumatism 2002, 32, 25–32. [Google Scholar]
- Gwaltney-Brant, S.M. Nonsteroidal Anti-Inflammatory Drug-Induced Toxicity. In Comprehensive Toxicology, 2nd ed; Charlene, A.M., Ed.; Elsevier: Oxford, UK, 2010; pp. 159–161. [Google Scholar]
- Lanas, A.; Garcia-Tell, G.; Armada, B.; Oteo-Alvaro, A. Prescription patterns and appropriateness of NSAID therapy according to gastrointestinal risk and cardiovascular history in patients with diagnoses of osteoarthritis. BMC Med 2011, 9, 38. [Google Scholar]
- Marnett, L.J. Mechanisms of cyclooxygenase-2 inhibition and cardiovascular side effects—The plot thickens. Cancer Prevention Res 2009, 2, 288–290. [Google Scholar]
- Bäck, M.; Yin, L.; Ingelsson, E. Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur. Heart J 2011, 33, 1928–1933. [Google Scholar]
- Duggan, D.E. Sulindac: Therapeutic Implications of the Prodrug/Pharmacophore Equilibrium. Drug Metab. Rev 1981, 12, 325–337. [Google Scholar]
- Sloan, K.B.; Wasdo, S. Designing for topical delivery: Prodrugs can make the difference. Med. Res. Rev 2003, 23, 763–793. [Google Scholar]
- Maag, H. Prodrugs of Carboxylic Acids. In Prodrugs; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 703–729. [Google Scholar]
- Pavan, B.; Dalpiaz, A.; Ciliberti, N.; Biondi, C.; Manfredini, S.; Vertuani, S. Progress in drug delivery to the central nervous system by the prodrug approach. Molecules 2008, 13, 1035–1065. [Google Scholar]
- Halen, P.K.K.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Prodrug designing of NSAIDs. Mini Rev. Med. Chem 2009, 9, 124–139. [Google Scholar]
- Müller, C.E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodiversity 2009, 6, 2071–2083. [Google Scholar]
- Koc, E.; Kucukguzel, S.G. Medicinal chemistry and anti-inflammatory activity of nitric oxide-releasing NSAI drugs. Mini Rev. Med. Chem 2009, 9, 611–619. [Google Scholar]
- Karim, A.; Laurent, A.; Slater, M.; Kuss, M.; Qian, J.; Crosby-Sessoms, S.; Hubbard, R. A Pharmacokinetic study of intramuscular (i.m) Parecoxib Sodium in normal subjects. Clin. Pharmacol 2001, 41, 1111–1119. [Google Scholar]
- Mamidi, R.; Mullangi, R.; Kota, J.; Bhamidipati, R.; Khan, A.; Katneni, K.; Datla, S.; Singh, S.; Rao, K.; Rao, C.; et al. Pharmacological and pharmacokinetic evaluation of celecoxib prodrugs in rats. Biopharm. Drug Dispos 2002, 23, 273–282. [Google Scholar]
- Qandil, A.M.; El Mohtadi, F.H.; Tashtoush, B.M. Chemical and in vitro enzymatic stability of newly synthesized celecoxib lipophilic and hydrophilic amides. Int. J. Pharm 2011, 416, 85–96. [Google Scholar]
- Lanas, A. Role of nitric oxide in the gastrointestinal tract. Arthritis Res. Ther 2008, 10, 1–6. [Google Scholar]
- Daff, S. NO synthase: Structures and mechanisms. Nitric Oxide 2010, 23, 1–11. [Google Scholar]
- Ruan, C.-H.; So, S.-P.; Ruan, K.-H. Inducible COX-2 dominates over COX-1 in prostacyclin biosynthesis: Mechanisms of COX-2 inhibitor risk to heart disease. Life Sci 2011, 88, 24–30. [Google Scholar]
- Sharma, S.K.; Al-Hourani, B.J.; Wuest, M.; Mane, J.Y.; Tuszynski, J.; Baracos, V.; Suresh, M.; Wuest, F. Synthesis and evaluation of fluorobenzoylated di- and tripeptides as inhibitors of cyclooxygenase-2 (COX-2). Bioorg. Med. Chem 2012, 20, 2221–2226. [Google Scholar]
- Ongini, E.; Bolla, M. Nitric-oxide based nonsteroidal anti-inflammatory agents. Drug Discovery Today 2006, 3, 395–400. [Google Scholar]
- Wong, P.S.-Y.; Fukuto, J.M. Reaction of organic nitrate esters ands-nitrosothiols with reduced flavins: A possible mechanism of bioactivation. Drug Metab. Dispos 1999, 27, 502–509. [Google Scholar]
- Minamiyama, Y.; Takemura, S.; Akiyama, T.; Imaoka, S.; Inoue, M.; Funae, Y.; Okada, S. Isoforms of cytochrome P450 on organic nitrate-derived nitric oxide release in human heart vessels. FEBS Lett 1999, 452, 165–169. [Google Scholar]
- Wallace, J.L.; Reuter, B.; Cicala, C.; McKnight, W.; Grisham, M.B.; Cirino, G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology 1994, 107, 173–179. [Google Scholar]
- Bandarage, U.K.; Chen, L.; Fang, X.; Garvey, D.S.; Glavin, A.; Janero, D.R.; Letts, L.G.; Mercer, G.J.; Saha, J.K.; Schroeder, J.D.; et al. Nitrosothiol esters of diclofenac: Synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs. J. Med. Chem 2000, 43, 4005–4016. [Google Scholar]
- Gilmer, J.F.; Moriarty, L.M.; McCafferty, D.F.; Clancy, J.M. Synthesis, hydrolysis kinetics and anti-platelet effects of isosorbide mononitrate derivatives of aspirin. Eur. J. Pharm. Sci 2001, 14, 221–227. [Google Scholar]
- Cena, C.; Lolli, M.L.; Lazzarato, L.; Guaita, E.; Morini, G.; Coruzzi, G.; McElroy, S.P.; Megson, I.L.; Fruttero, R.; Gasco, A. Antiinflammatory, gastrosparing, and antiplatelet properties of new no-donor esters of aspirin. J. Med. Chem 2003, 46, 747–754. [Google Scholar]
- Velázquez, C.; Rao, P.N.P.; Knaus, E.E. Novel nonsteroidal antiinflammatory drugs possessing a nitric oxide donor diazen-1-ium-1,2-diolate moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem 2005, 48, 4061–4067. [Google Scholar]
- Ranatunge, R.R.; Augustyniak, M.E.; Dhawan, V.; Ellis, J.L.; Garvey, D.S.; Janero, D.R.; Letts, L.G.; Richardson, S.K.; Shumway, M.J.; Trocha, A.M.; et al. Synthesis and anti-inflammatory activity of a series of N-substituted naproxen glycolamides: Nitric oxide-donor naproxen prodrugs. Bioorg. Med. Chem 2006, 14, 2589–2599. [Google Scholar]
- Velázquez, C.; Chen, Q.-H.; Citro, M.L.; Keefer, L.K.; Knaus, E.E. Second-Generation aspirin and indomethacin prodrugs possessing an O2-(acetoxymethyl)-1-(2-carboxypyrrolidin-1-yl)diazenium-1,2-diolate nitric oxide donor moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem 2008, 51, 1954–1961. [Google Scholar]
- De Carvalho, P.S.; Maróstica, M.; Gambero, A.; Pedrazzoli, J., Jr. Synthesis and pharmacological characterization of a novel nitric oxide-releasing diclofenac derivative containing a benzofuroxan moiety. Eur. J. Med. Chem. 2010, 45, 2489–2493. [Google Scholar]
- Huang, Z.; Velázquez, C.; Abdellatif, K.; Chowdhury, M.A.; Reisz, J.; DuMond, J.; King, S.B.; Knaus, E.E. Ethanesulfohydroxamic acid ester prodrugs of nonsteroidal anti-inflammatory drugs (nsaids): Synthesis, nitric oxide and nitroxyl release, cyclooxygenase inhibition, anti-inflammatory, and ulcerogenicity index studies. J. Med. Chem 2011, 54, 1356–1364. [Google Scholar]
- Chattopadhyay, M.; Velazquez, C.A.; Pruski, A.; Nia, K.V.; Abdellatif, K.R.; Keefer, L.K.; Kashfi, K. Comparison between 3-nitrooxyphenyl acetylsalicylate (NO-ASA) and O2-(acetylsalicyloxymethyl)-1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate (NONO-ASA) as safe anti-Inflammatory, analgesic, antipyretic, antioxidant prodrugs. J. Pharmacol. Exp. Ther 2010, 335, 443–450. [Google Scholar]
- Jain, S.; Tran, S.; El Gendy, M.A.M.; Kashfi, K.; Jurasz, P.; Velázquez-Martínez, C.A. Nitric oxide release is not required to decrease the ulcerogenic profile of nonsteroidal anti-inflammatory drugs. J. Med. Chem 2011, 55, 688–696. [Google Scholar]
- Maragos, C.M.; Morley, D.; Wink, D.A.; Dunams, T.M.; Saavedra, J.E.; Hoffman, A.; Bove, A.A.; Isaac, L.; Hrabie, J.A.; Keefer, L.K. Complexes of .NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem 1991, 34, 3242–3247. [Google Scholar]
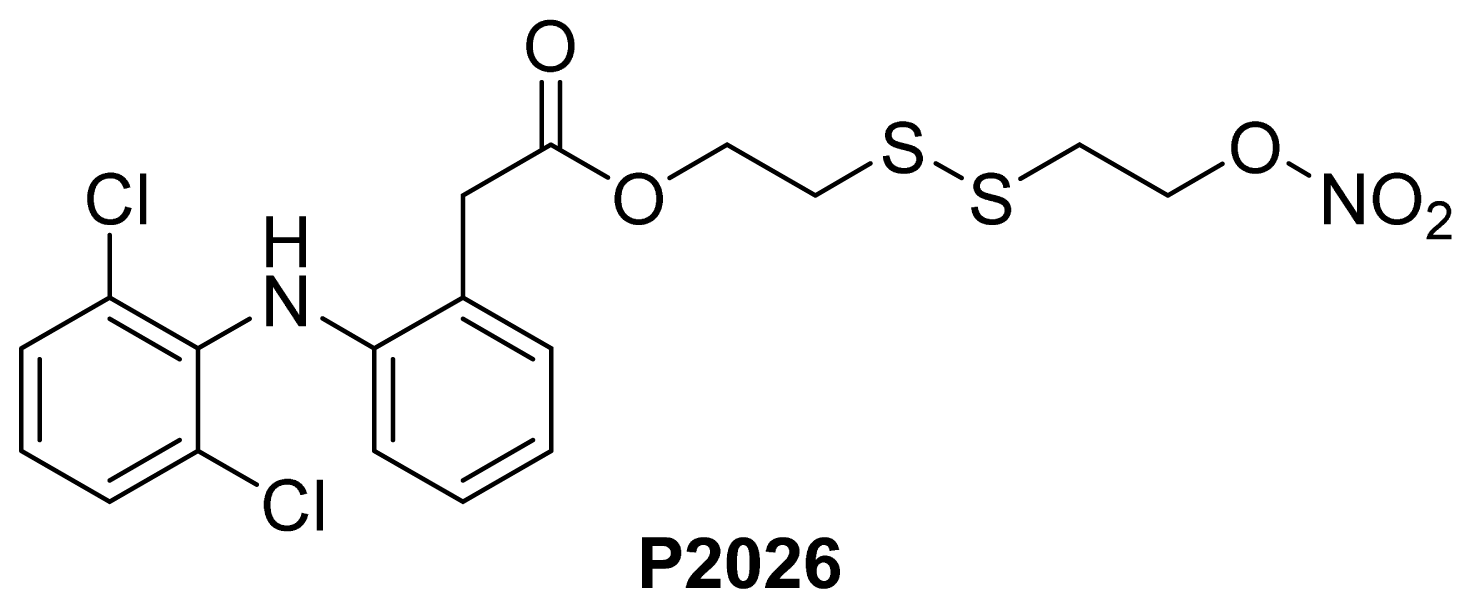
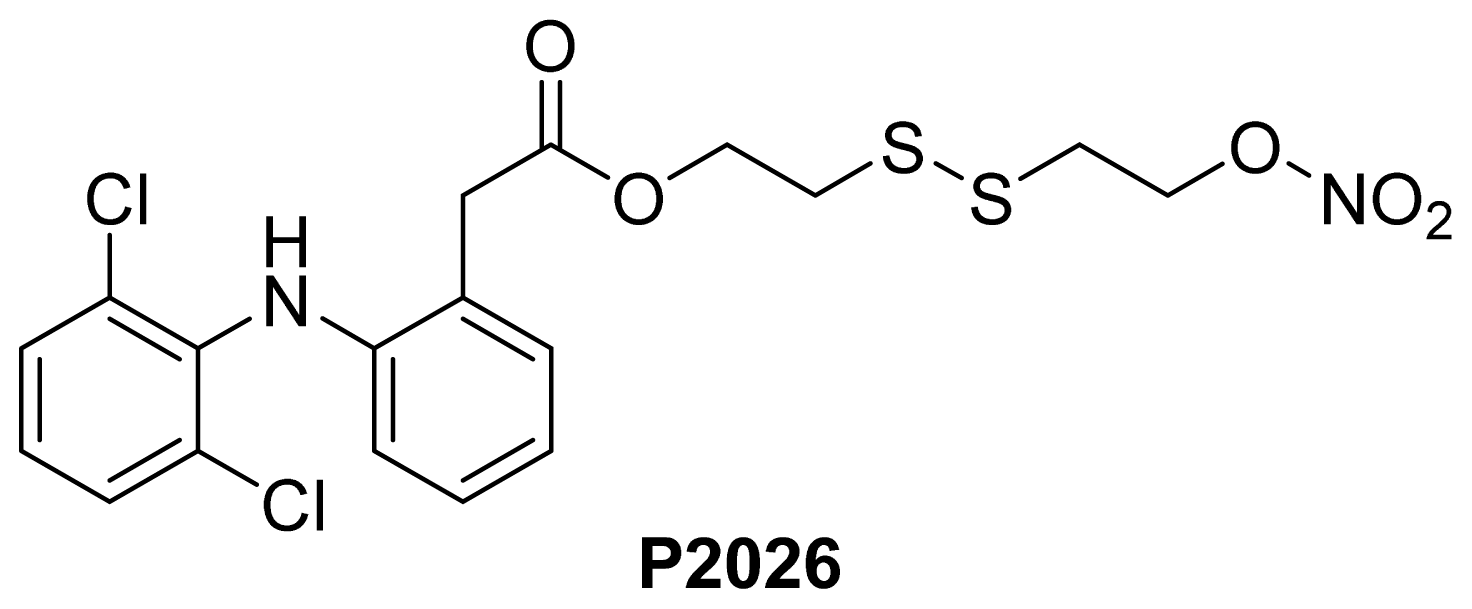
- Pathan, A.; Karwa, M.; Pamidiboina, V.; Deshattiwar, J.; Deshmukh, N.; Gaikwad, P.; Mali, S.; Desai, D.; Dhiman, M.; Mariappan, T.T.; et al. Oral bioavailability, efficacy and gastric tolerability of P2026, a novel nitric oxide-releasing diclofenac in rat. Inflammopharmacology 2010, 18, 157–168. [Google Scholar]
- Abdellatif, K.R.; Chowdhury, M.A.; Dong, Y.; Das, D.; Yu, G.; Velazquez, C.A.; Suresh, M.R.; Knaus, E.E. Dinitroglyceryl and diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of aspirin, indomethacin and ibuprofen: synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett 2009, 19, 3014–3018. [Google Scholar]
- Kaur, J.; Bhardwaj, A.; Huang, Z.; Knaus, E.E. Aspirin analogues as dual cyclooxygenase-2/5- lipoxygenase inhibitors: Synthesis, nitric oxide release, molecular modeling, and biological evaluation as anti-inflammatory agents. ChemMedChem 2012, 7, 144–150. [Google Scholar]
- Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.-P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheumatic Dis 2003, 62, 501–509. [Google Scholar]
- Biava, M.; Porretta, G.C.; Poce, G.; Battilocchio, C.; Alfonso, S.; Rovini, M.; Valenti, S.; Giorgi, G.; Calderone, V.; Martelli, A.; et al. Novel analgesic/anti-inflammatory agents: Diarylpyrrole acetic esters endowed with nitric oxide releasing properties. J. Med. Chem 2011, 54, 7759–7771. [Google Scholar]
- Fabbri, F.; Brigliadori, G.; Ulivi, P.; Tesei, A.; Vannini, I.; Rosetti, M.; Bravaccini, S.; Amadori, D.; Bolla, M.; Zoli, W. Pro-apoptotic effect of a nitric oxide-donating NSAID, NCX 4040, on bladder carcinoma cells. Apoptosis 2005, 10, 1095–1103. [Google Scholar]
- Tesei, A.; Rosetti, M.; Ulivi, P.; Fabbri, F.; Medri, L.; Vannini, I.; Bolla, M.; Amadori, D.; Zoli, W. Study of molecular mechanisms of pro-apoptotic activity of NCX 4040, a novel nitric oxide-releasing aspirin, in colon cancer cell lines. J. Transl. Med 2007, 5, 52. [Google Scholar]
- Tesei, A.; Ulivi, P.; Fabbri, F.; Rosetti, M.; Leonetti, C.; Scarsella, M.; Zupi, G.; Amadori, D.; Bolla, M.; Zoli, W. In vitro and in vivo evaluation of NCX 4040 cytotoxic activity in human colon cancer cell lines. J. Transl. Med 2005, 3, 7. [Google Scholar]
- Tesei, A.; Zoli, W.; Fabbri, F.; Leonetti, C.; Rosetti, M.; Bolla, M.; Amadori, D.; Silvestrini, R. NCX 4040, an NO-donating acetylsalicylic acid derivative: Efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008, 19, 225–236. [Google Scholar]
- Rosetti, M.; Tesei, A.; Ulivi, P.; Fabbri, F.; Vannini, I.; Brigliadori, G.; Amadori, D.; Bolla, M.; Zoli, W. Molecular characterization of cytotoxic and resistance mechanisms induced by NCX 4040, a novel NO-NSAID, in pancreatic cancer cell lines. Apoptosis 2006, 11, 1321–1330. [Google Scholar]
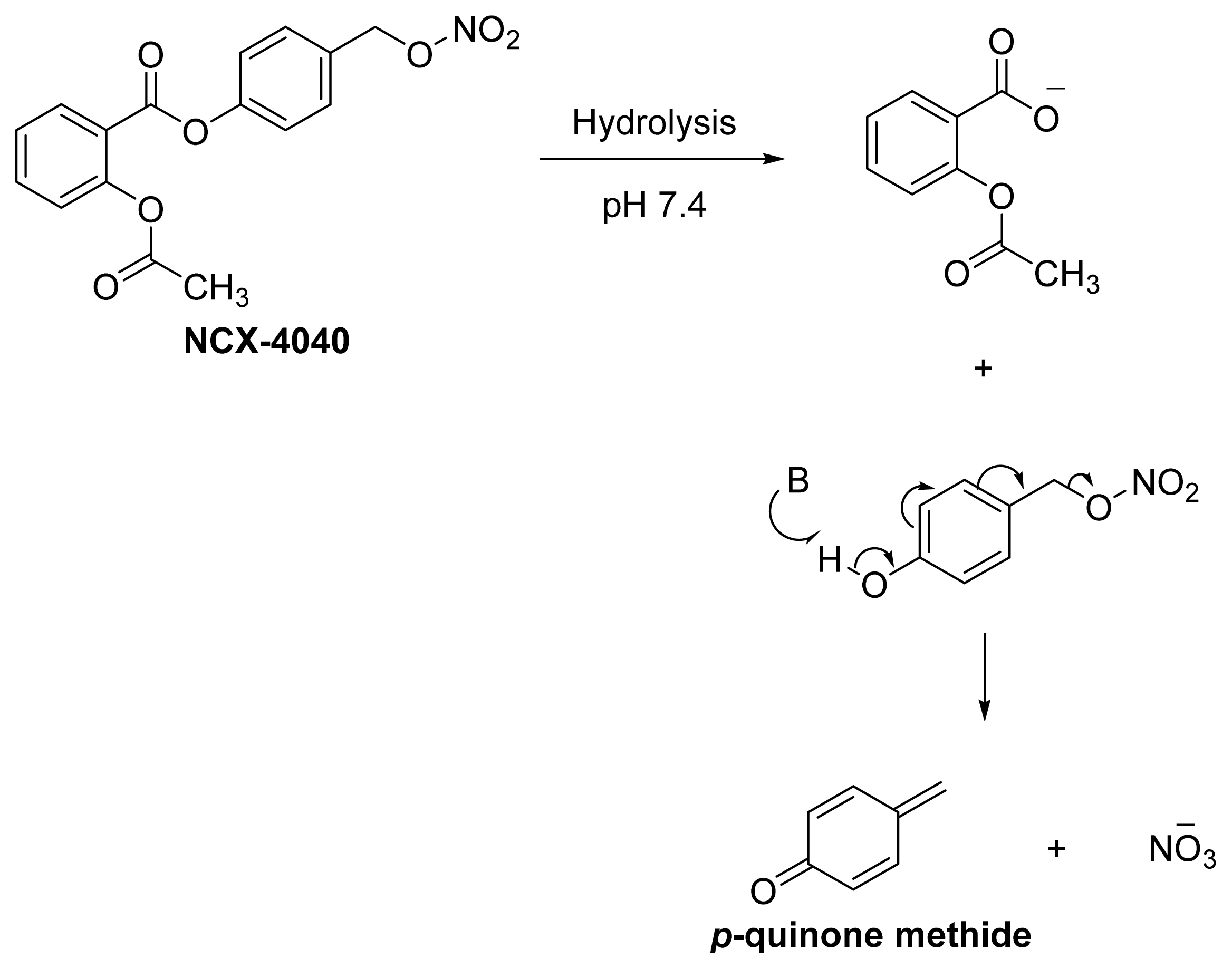
- Hulsman, N.; Medema, J.P.; Bos, C.; Jongejan, A.; Leurs, R.; Smit, M.J.; de Esch, I.J.P.; Richel, D.; Wijtmans, M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem 2007, 50, 2424–2431. [Google Scholar]
- Kashfi, K.; Rigas, B. The mechanism of action of nitric oxide-donating aspirin. Biochem. Biophys. Res. Commun 2007, 358, 1096–1101. [Google Scholar]
- Nemmani, K.V.S.; Mali, S.V.; Borhade, N.; Pathan, A.R.; Karwa, M.; Pamidiboina, V.; Senthilkumar, S.P.; Gund, M.; Jain, A.K.; Mangu, N.K.; et al. NO-NSAIDs: Gastric-sparing nitric oxide-releasable prodrugs of non-steroidal anti-inflammatory drugs. Bioorg. Med. Chem. Lett 2009, 19, 5297–5301. [Google Scholar]
- Lazzarato, L.; Chegaev, K.; Marini, E.; Rolando, B.; Borretto, E.; Guglielmo, S.; Joseph, S.; di Stilo, A.; Fruttero, R.; Gasco, A. New nitric oxide or hydrogen sulfide releasing aspirins. J. Med. Chem 2011, 54, 5478–5484. [Google Scholar]
- Wey, S.-J.; Augustyniak, M.E.; Cochran, E.D.; Ellis, J.L.; Fang, X.; Garvey, D.S.; Janero, D.R.; Letts, L.G.; Martino, A.M.; Melim, T.L.; et al. Structure-Based design, synthesis, and biological evaluation of indomethacin derivatives as cyclooxygenase-2 inhibiting nitric oxide donors. J. Med. Chem 2007, 50, 6367–6382. [Google Scholar]
- Downing, J.E.G.; Madden, J.C.; Ingram, M.J.; Rostron, C. Gastric and thymic assay of acute oral treatment of rats with nitric oxide esters of ibuprofen or indomethacin. Biochem. Biophys. Res. Commun 2005, 334, 646–653. [Google Scholar]
- Yang, C.-F.; Zhang, Y.-Y.; Yang, B.; Li, P.-F.; Zhuang, D.-Y. Synthesis of indomethacin derivatives and their anti-inflammatory activities. Chin. New Drugs J 2004, 9, 818–2106. [Google Scholar]
- Schiefer, I.T.; Abdul-Hay, S.; Wang, H.; Vanni, M.; Qin, Z.; Thatcher, G.R.J. Inhibition of amyloidogenesis by nonsteroidal anti-inflammatory drugs and their hybrid nitrates. J. Med. Chem 2011, 54, 2293–2306. [Google Scholar]
- Schubert, M.L.; Peura, D.A. Control of gastric acid secretion in health and disease. Gastroenterology 2008, 134, 1842–1860. [Google Scholar]
- Ehlert, F.J.; Pak, K.J.; Griffin, M.T. Muscarinic Agonists and Antagonists: Effects on Gastrointestinal Function Muscarinic Receptors; Fryer, A.D., Christopoulos, A., Nathanson, N.M., Eds.; Springer: Berlin, Germany, 2012; Volume 208, pp. 343–374. [Google Scholar]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Synthesis and pharmacological evaluation of some dual-acting amino-alcohol ester derivatives of flurbiprofen and 2-[1,1′-biphenyl-4-yl]acetic acid: A potential approach to reduce local gastrointestinal toxicity. Chem. Biodiversity 2006, 3, 1238–1248. [Google Scholar]
- Sato, N.; Kawano, S.; Tsuji, S.; Ogihara, T.; Yamada, S. Gastric blood flow in ulcer diseases. Scand. J. Gastroenterol 1995, 30, 14–20. [Google Scholar]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Substituted aminoalcohol ester analogs of indomethacin with reduced toxic effects. Med. Chem. Res 2007, 16, 101–111. [Google Scholar]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Combining anticholinergic and anti-inflammatory activities into a single moiety: A novel approach to reduce gastrointestinal toxicity of ibuprofen and ketoprofen. Chem. Biol. Drug Des 2007, 70, 450–455. [Google Scholar]
- Cowan, F.M.; Broomfield, C.A.; Lenz, D.E.; Smith, W.J. Putative role of proteolysis and inflammatory response in the toxicity of nerve and blister chemical warfare agents: Implications for multi-threat medical countermeasures. J. Appl. Toxicol 2003, 23, 177–186. [Google Scholar]
- Pita, R.; Vidal-Asensi, S. Cutaneous and systemic toxicology of vesicant (blister) warfare agents. Actas Dermo-Sifiliográficas (English Edition) 2010, 101, 7–18. [Google Scholar]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar]
- Green, R.D.; Fleming, W.W. Agonist-Antagonist interactions in the normal and supersensitive nictitating membrane of the spinal cat. J. Pharmacol. Exp. Ther 1967, 156, 207–214. [Google Scholar]
- Kalgutkar, A.S.; Marnett, A.B.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J. Med. Chem 2000, 43, 2860–2870. [Google Scholar]
- Rautio, J.; Nevalainen, T.; Taipale, H.; Vepsäläinen, J.; Gynther, J.; Laine, K.; Järvinen, T. Piperazinylalkyl prodrugs of naproxen improve in vitro skin permeation. Eur. J. Pharm. Sci 2000, 11, 157–163. [Google Scholar]
- Young, S.; Fabio, K.; Guillon, C.; Mohanta, P.; Halton, T.A.; Heck, D.E.; Flowers, R.A., II; Laskin, J.D.; Heindel, N.D. Peripheral site acetylcholinesterase inhibitors targeting both inflammation and cholinergic dysfunction. Bioorg. Med. Chem. Lett 2010, 20, 2987–2990. [Google Scholar]
- Young, S.; Fabio, K.; Huang, M.-T.; Saxena, J.; Harman, M.; Guillon, C.; Vetrano, A.; Heck, D.; Flowers, R.; Heindel, N.; et al. Investigation of anticholinergic and non-steroidal anti-inflammatory prodrugs which reduce chemically induced skin inflammation. J. Appl. Toxicol 2012, 32, 135–141. [Google Scholar]
- Huang, L.; Mackenzie, G.; Sun, Y.; Ouyang, N.; Xie, G.; Vrankova, K.; Komninou, D.; Rigas, B. Chemotherapeutic properties of phospho-nonsteroidal anti-inflammatory drugs, a new class of anticancer compounds. Cancer Res 2011, 71, 7617–7627. [Google Scholar]
- Huang, L.; Mackenzie, G.; Ouyang, N.; Sun, Y.; Xie, G.; Johnson, F.; Komninou, D.; Rigas, B. The novel phospho-non-steroidal anti-inflammatory drugs, OXT-328, MDC-22 and MDC-917, inhibit adjuvant-induced arthritis in rats. Br. J. Pharmacol 2011, 162, 1521–1533. [Google Scholar]
- Sun, Y.; Huang, L.; Mackenzie, G.G.; Rigas, B. Oxidative stress mediates through apoptosis the anticancer effect of phospho-nonsteroidal anti-inflammatory drugs: Implications for the role of oxidative stress in the action of anticancer agents. J. Pharmacol. Exp.Ther 2011, 338, 775–783. [Google Scholar]
- Huang, L.; Zhu, C.; Sun, Y.; Xie, G.; Mackenzie, G.; Qiao, G.; Komninou, D.; Rigas, B. Phospho-sulindac (OXT-922) inhibits the growth of human colon cancer cell lines: A redox/polyamine dependent effect. Carcinogenesis 2010, 31, 1982–1990. [Google Scholar]
- Xie, G.; Sun, Y.; Nie, T.; Mackenzie, G.G.; Huang, L.; Kopelovich, L.; Komninou, D.; Rigas, B. Phospho-Ibuprofen (MDC-917) is a novel agent against colon cancer: efficacy, metabolism, and pharmacokinetics in mouse models. J. Pharmacol. Exp. Ther 2011, 337, 876–886. [Google Scholar]
- Wong, C.C.; Cheng, K.-W.; Xie, G.; Zhou, D.; Zhu, C.-H.; Constantinides, P.P.; Rigas, B. Carboxylesterases 1 and 2 hydrolyze phospho-nonsteroidal anti-inflammatory drugs: Relevance to their pharmacological activity. J. Pharmacol. Exp. Ther 2012, 340, 422–432. [Google Scholar]
- Zhao, W.; Mackenzie, G.G.; Murray, O.T.; Zhang, Z.; Rigas, B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: A redox-dependent effect. Carcinogenesis 2009, 30, 512–519. [Google Scholar]
- Dufrasne, F.; Gelbcke, M.; Neve, J.; Kiss, R.; Kraus, J.-L. Quinone methides and their prodrugs: A subtle equilibrium between cancer promotion, prevention, and cure. Curr. Med. Chem 2011, 18, 3995–4011. [Google Scholar]
- Deguchi, H.; Yasukawa, K.; Yamasaki, T.; Mito, F.; Kinoshita, Y.; Naganuma, T.; Sato, S.; Yamato, M.; Ichikawa, K.; Sakai, K.; et al. Nitroxides prevent exacerbation of indomethacin-induced gastric damage in adjuvant arthritis rats. Free Radical Biol. Med 2011, 51, 1799–1805. [Google Scholar]
- Flores-Santana, W.; Moody, T.; Chen, W.; Gorczynski, M.J.; Shoman, M.E.; Velázquez, C.; Thetford, A.; Mitchell, J.B.; Cherukuri, M.K.; King, S.B.; et al. Nitroxide derivatives of non-steroidal anti-inflammatory drugs exert anti-inflammatory and superoxide dismutase scavenging properties in A459 cells. Br. J. Pharmacol 2012, 165, 1058–1067. [Google Scholar]
- Yokomizo, T.; Izumi, T.; Shimizu, T. Co-expression of two LTB4 receptors in human mononuclear cells. Life Sci 2001, 68, 2207–2212. [Google Scholar]
- Caliendo, G.; Cirino, G.; Santagada, V.; Wallace, J.L. Synthesis and biological effects of hydrogen sulfide (h2s): Development of h2s-releasing drugs as pharmaceuticals. J. Med. Chem 2010, 53, 6275–6286. [Google Scholar]
- Fiorucci, S.; Distrutti, E.; Cirino, G.; Wallace, J.L. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 2006, 131, 259–271. [Google Scholar]
- Wallace, J.L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci 2007, 28, 501–505. [Google Scholar]
- Zanatta, S.D.; Jarrott, B.; Williams, S.J. Synthesis and preliminary pharmacological evaluation of aryl dithiolethiones with cyclooxygenase-2-selective inhibitory activity and hydrogen sulfide-releasing properties. Aust. J. Chem 2010, 63, 946–957. [Google Scholar]
- Kodela, R.; Chattopadhyay, M.; Kashfi, K. NOSH-Aspirin: A novel nitric oxide–hydrogen sulfide-releasing hybrid: A new class of anti-inflammatory pharmaceuticals. ACS Med. Chem. Lett 2012, 3, 257–262. [Google Scholar]
- Chattopadhyay, M.; Kodela, R.; Nath, N.; Dastagirzada, Y.M.; Velázquez-Martínez, C.A.; Boring, D.; Kashfi, K. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: A general property and evidence of a tissue type-independent effect. Biochem. Pharmacol 2012, 83, 715–722. [Google Scholar]
- Chattopadhyay, M.; Kodela, R.; Olson, K.R.; Kashfi, K. NOSH–aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem. Biophy. Res. Commun 2012, 419, 523–528. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | NO donor moiety | Equivalents of NO released | Examples |
|---|---|---|---|
| Nitrates | –ONO2 | 1 | 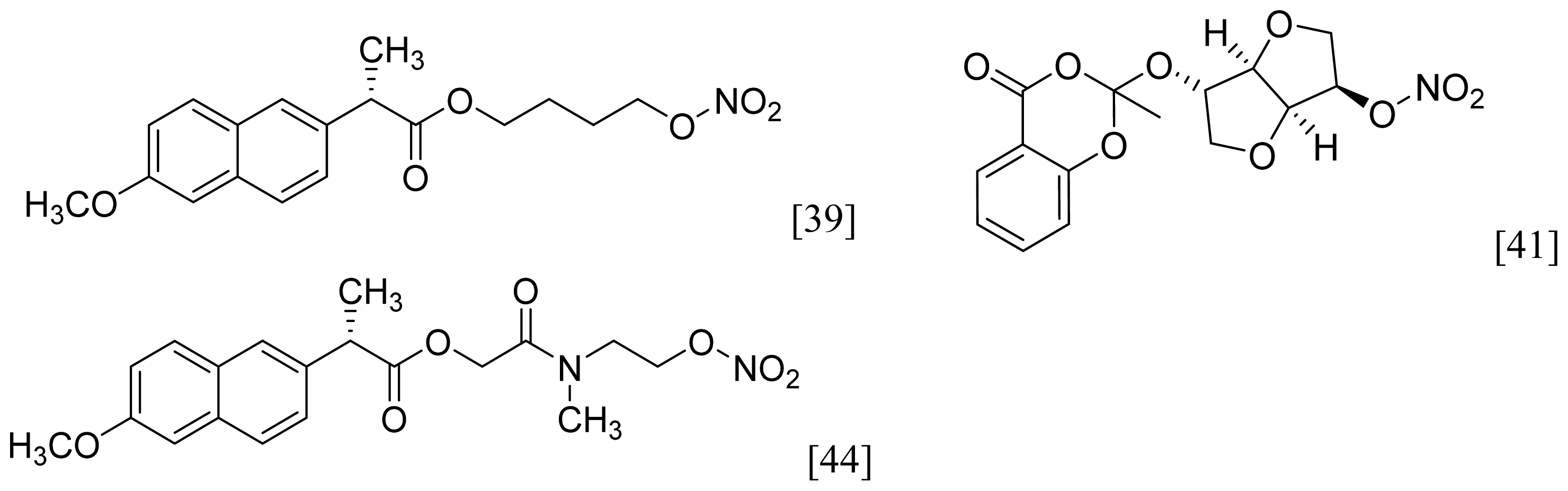 |
| NONOate | 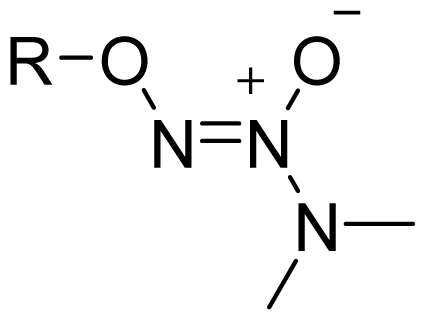 | 2 | 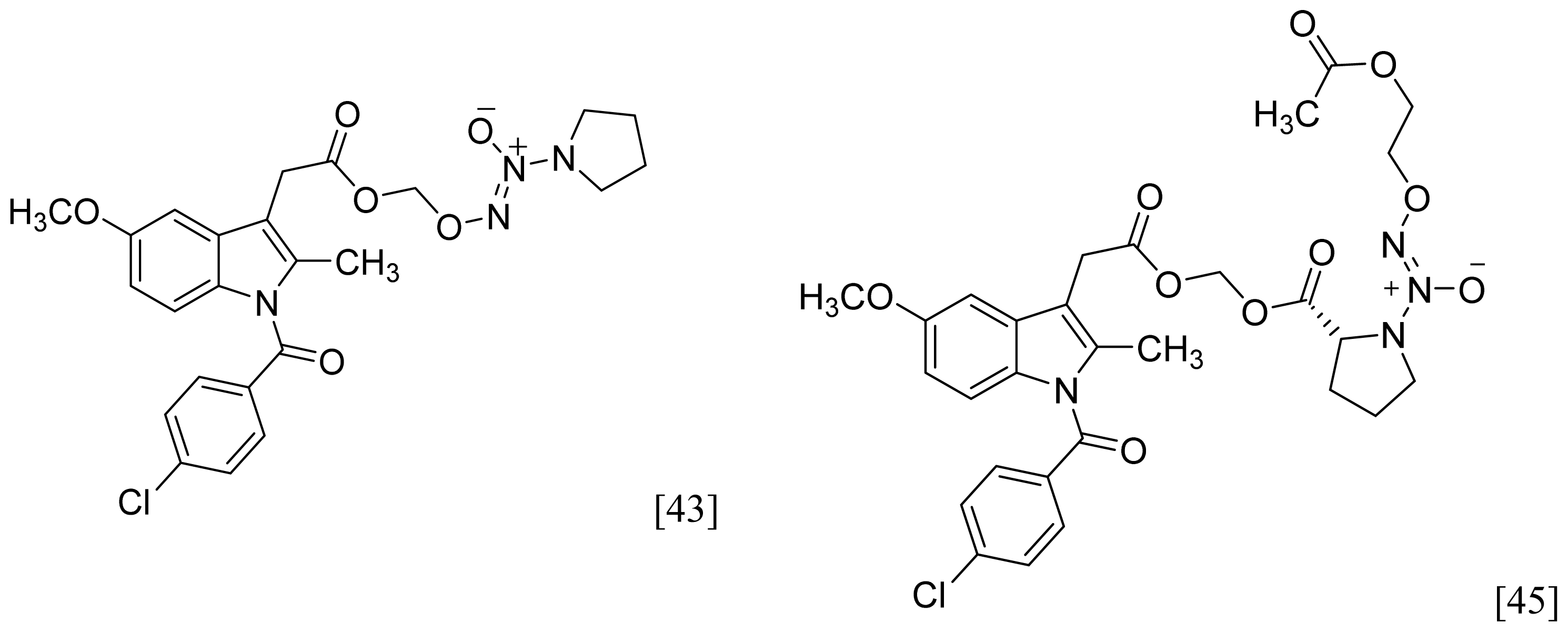 |
| Furoxan and Benzofuroxan | 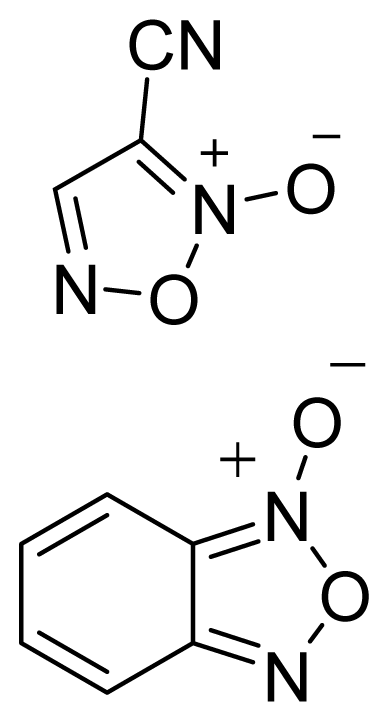 | 1 | 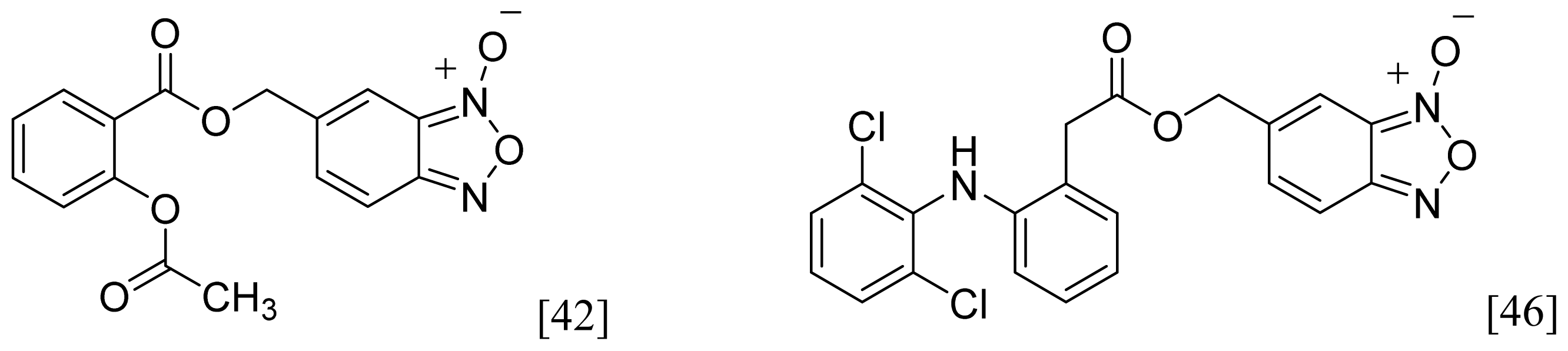 |
| Sulfohydroxamic Acid | 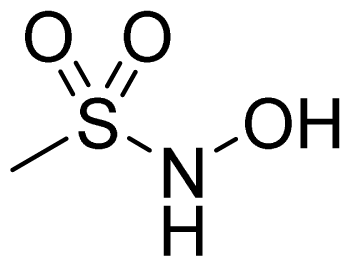 | 1 | 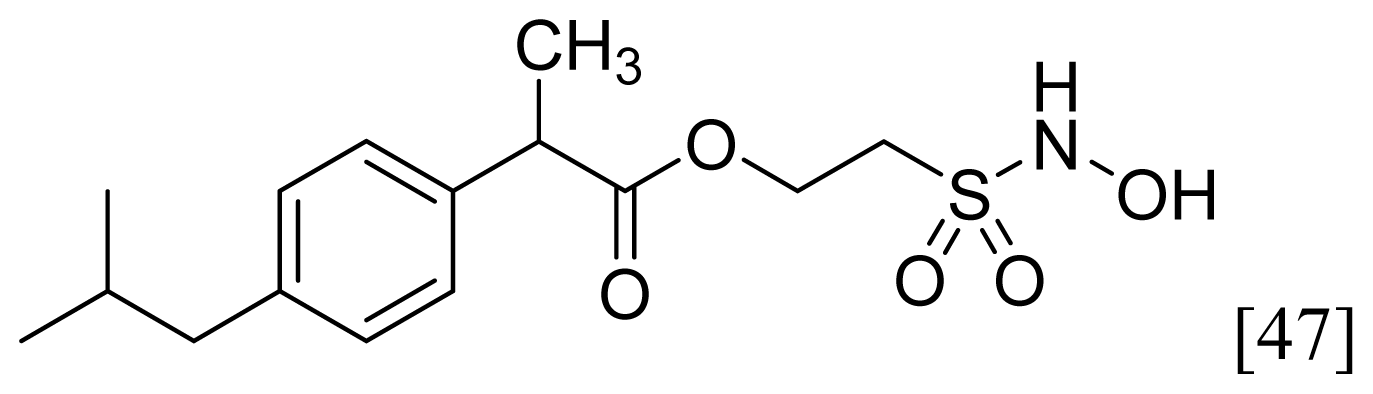 |
| Nitrosothiol Esters |  | 1 | 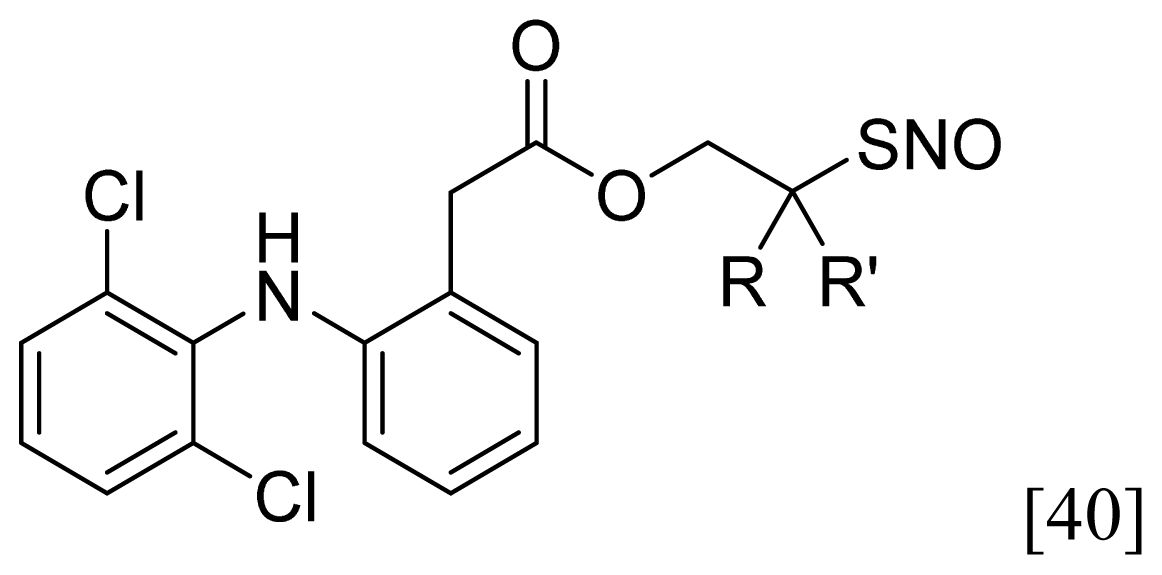 |
| Compound | t1/2, pH 7.4 (h) | % NSAID released in plasma after 2 h | pA2** | % inhibition of Edema Volume | UI |
|---|---|---|---|---|---|
| Ketoprofen a | 87 | 0.882 | |||
| 21a–e a | 16–64 | 58.9–79.6 | 5.28–6.43 | 70–89 | 0.306–0.376 |
| 21f a | ND * | ND * | 7.58 | 86 | 0.299 |
| Flurbiprofen b | 0 | 0.800 | |||
| 22a–e b | 121–505 | 17.9–51.1 | 4.73–5.79 | 79–89 | 0.130–0.230 |
| 22f b | ND * | ND * | 6.31 | 0 | 0.00 |
| Indomethacin c | 45 | 0.55 | |||
| 23a–e c | 34–99 | 49.6–79.9 | 4.49–5.19 | 17–46 | 0.130–0.320 |
| Atropine a,b,c | 8.02 |
| Comd | % reduction in edema (Mouse ear vesicant model) [81] | Acetylcholinesterase Inhibition [72] | Plasma t1/2 (min) | Calculated LogP | |
|---|---|---|---|---|---|
| CEES | TPA | IC50 (μM) | |||
| 24a | 20 | 41 | 1.93 ± 0.64 | 204 | 7.37 |
| 25b | 45 | 70 | 0.83 ± 0.15 | 253 | 6.67 |
| 26a | 91 | 21 | 2.29 ± 0.94 | 468 | 7.87 |
| 27a | 90 | 24 | 0.51 ± 0.02 | 357 | 8.41 |
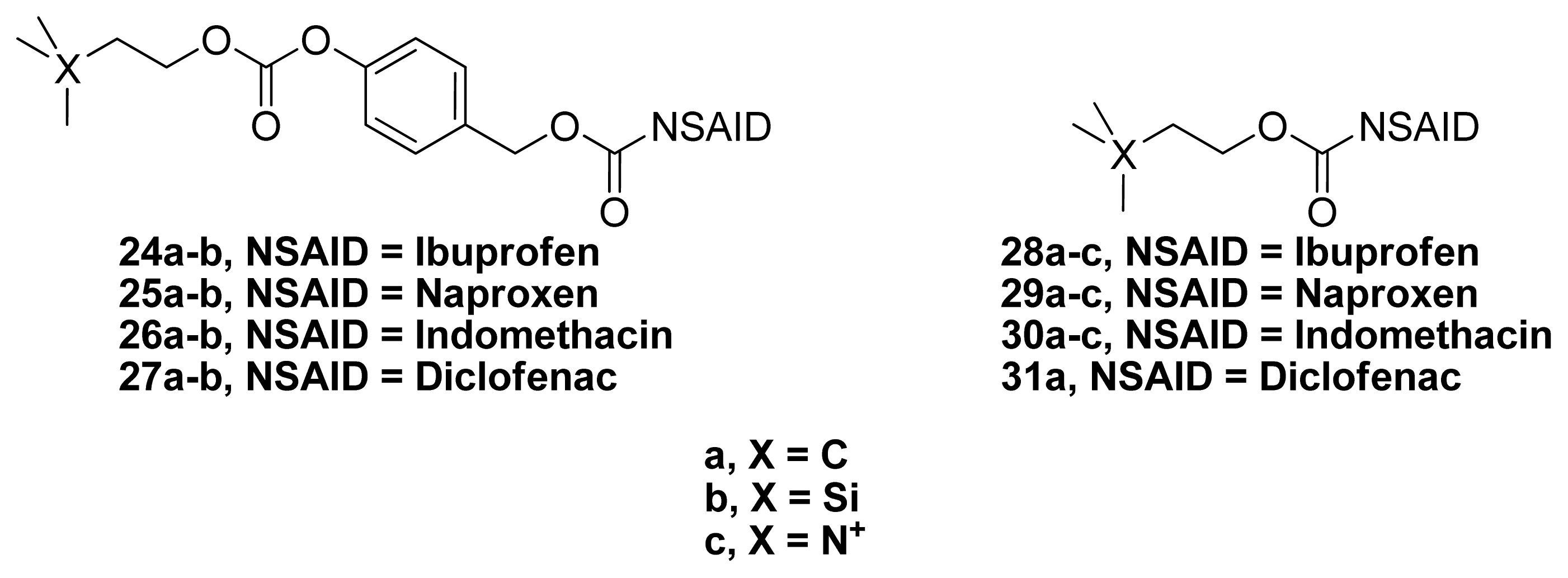
| 31a | 113 | 29 | 2.69 ± 0.15 | 111 | 7.52 |
| Tacrine | - | - | 0.055 ± 0.005 | - | - |
| DIC | 17 | 58 | - | - | - |
| IND | 46 | 55 | - | - | - |
| IBU | -15 | -33 | - | - | - |
| NAP | NS | * | 104 | - | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qandil, A.M. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. Int. J. Mol. Sci. 2012, 13, 17244-17274. https://doi.org/10.3390/ijms131217244
Qandil AM. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. International Journal of Molecular Sciences. 2012; 13(12):17244-17274. https://doi.org/10.3390/ijms131217244
Chicago/Turabian StyleQandil, Amjad M. 2012. "Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review" International Journal of Molecular Sciences 13, no. 12: 17244-17274. https://doi.org/10.3390/ijms131217244
APA StyleQandil, A. M. (2012). Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. International Journal of Molecular Sciences, 13(12), 17244-17274. https://doi.org/10.3390/ijms131217244