DNA Damage and Repair in Atherosclerosis: Current Insights and Future Perspectives




{kind=link}
{kind=link}
Abstract
:1. Introduction
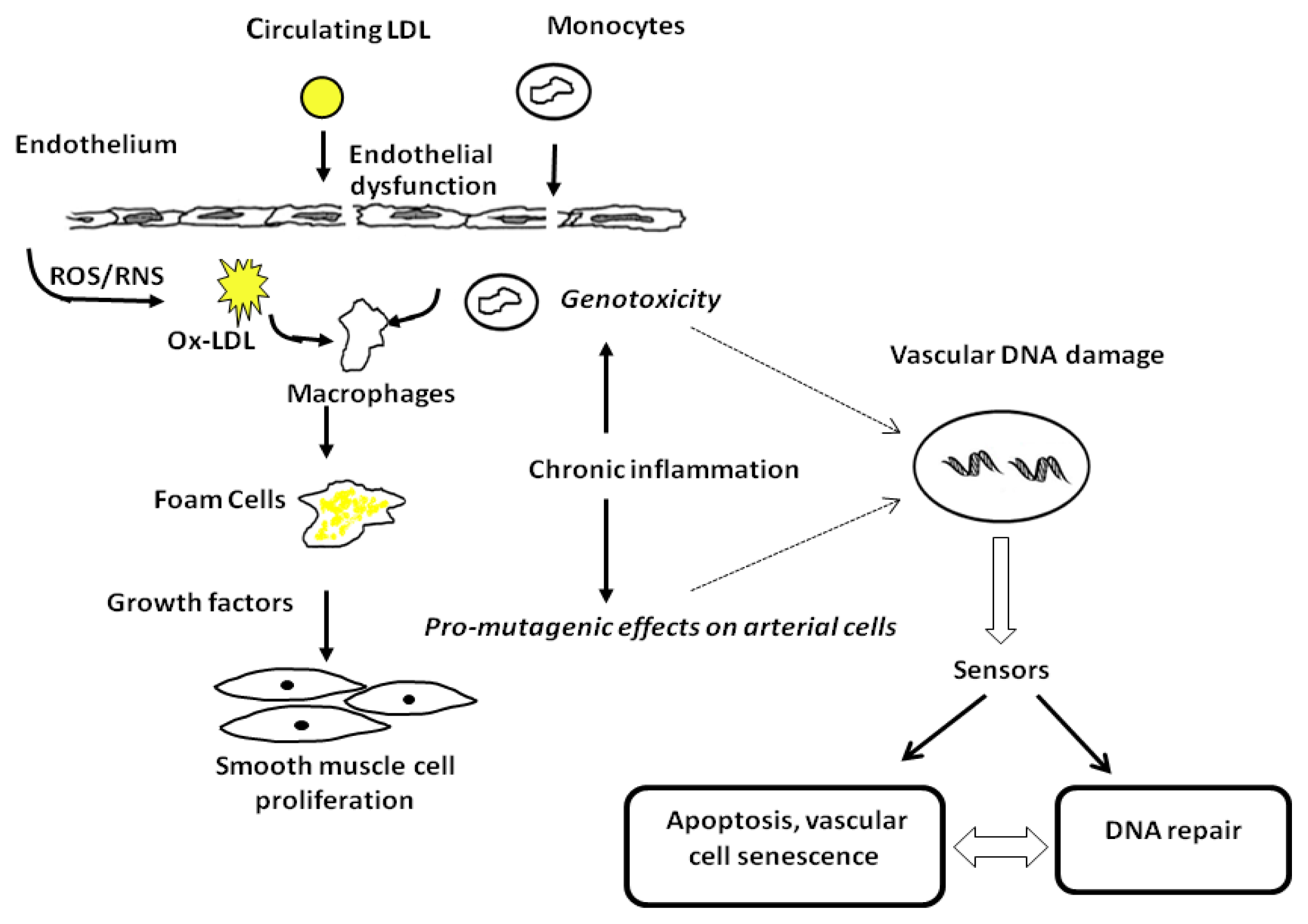
2. Evidence of DNA Damage in Atherosclerosis
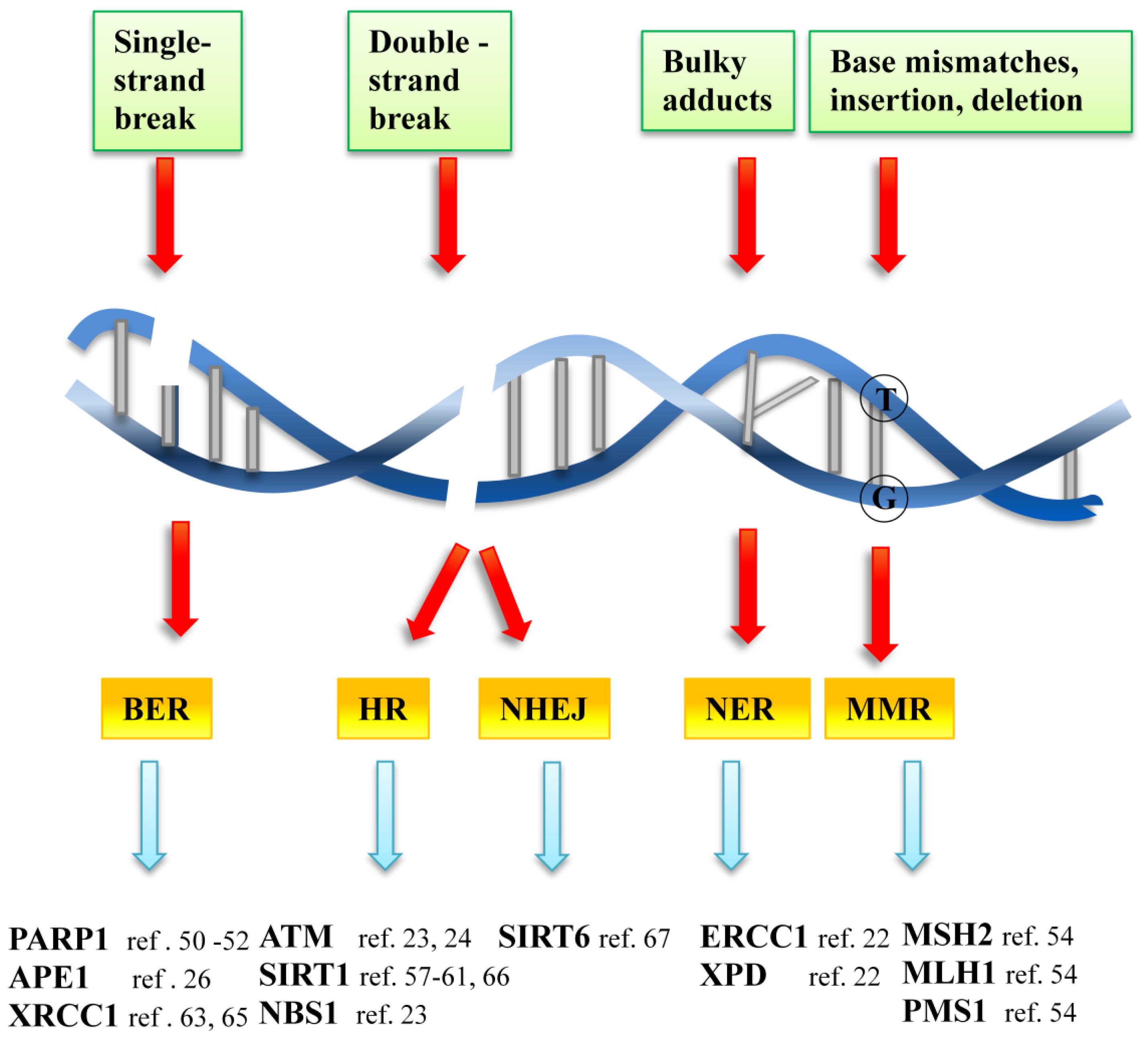
3. DNA Damage Repair Overview
3.1. DNA Repair Systems and Key Components
3.2. Signal Transduction Pathways of the DDR Process
4. DNA Repair in Atherosclerosis
4.1. DNA Repair Proteins in Atherosclerosis
4.2. DNA Damage Signaling Proteins in Atherosclerosis
5. DNA Repair Polymorphisms and Atherosclerosis
6. miRNA, DNA Repair and Atherosclerosis
7. Conclusions and Future Perspectives
References
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart disease and stroke statistics—2008 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008, 117, e25–e146. [Google Scholar]
- Ross, R. Rous-Whipple Award Lecture. Atherosclerosis: A defense mechanism gone awry. Am. J. Pathol 1993, 143, 987–1002. [Google Scholar]
- Andreassi, M.G. Metabolic syndrome, diabetes and atherosclerosis: Influence of gene-environment interaction. Mutat. Res 2009, 667, 35–43. [Google Scholar]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar]
- Andreassi, M.G. DNA damage, vascular senescence and atherosclerosis. J. Mol. Med. (Berl.) 2008, 86, 1033–1043. [Google Scholar]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res 2012, 111, 245–259. [Google Scholar]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res 2008, 102, 201–208. [Google Scholar]
- Izzotti, A.; Cartiglia, C.; Lewtas, J.; De Flora, S. Increased DNA alterations in atherosclerotic lesions of individuals lacking the GSTM1 genotype. FASEB J 2001, 15, 752–757. [Google Scholar]
- Andreassi, M.G.; Barale, R.; Iozzo, P.; Picano, E. The association of micronucleus frequency with obesity, diabetes and cardiovascular disease. Mutagenesis 2011, 26, 77–83. [Google Scholar]
- Kiaris, H.; Hatzistamou, J.; Spandidos, D.A. Instability at the H-ras minisatellite in human atherosclerotic plaques. Atherosclerosis 1996, 125, 47–51. [Google Scholar]
- Hatzistamou, J.; Kiaris, H.; Ergazaki, M.; Spandidos, D.A. Loss of heterozygosity and microsatellite instability in human atherosclerotic plaques. Biochem. Biophys. Res. Commun 1996, 225, 186–190. [Google Scholar]
- Spandidos, D.; Ergazaki, M.; Hatzistamou, J.; Kiaris, H.; Bouros, D.; Tzortzaki, E.; Siafakas, N. Microsatellite instability in patients with chronic obstructive pulmonary disease. Oncol. Rep 1996, 3, 489–491. [Google Scholar]
- McCaffrey, T.A.; Du, B.; Consigli, S.; Szabo, P.; Bray, P.J.; Hartner, L.; Weksler, B.B.; Sanborn, T.A.; Bergman, G.; Bush, H.L., Jr. Genomic instability in the type II TGF-beta1 receptor gene in atherosclerotic and restenotic vascular cells. J. Clin. Invest. 1997, 100, 2182–2188. [Google Scholar]
- Casalone, R.; Granata, P.; Minelli, E.; Portentoso, P.; Giudici, A.; Righi, R.; Castelli, P.; Socrate, A.; Frigerio, B. Cytogenetic analysis reveals clonal proliferation of smooth muscle cells in atherosclerotic plaques. Hum. Genet 1991, 87, 139–143. [Google Scholar]
- Matturri, L.; Cazzullo, A.; Turconi, P.; Lavezzi, A.M. Cytogenetic aspects of cell proliferation in atherosclerotic plaques. Cardiologia 1997, 42, 833–836. [Google Scholar]
- De Flora, S.; Izzotti, A.; Walsh, D.; Degan, P.; Petrilli, G.L.; Lewtas, J. Molecular epidemiology of atherosclerosis. FASEB J 1997, 11, 1021–1031. [Google Scholar]
- Izzotti, A.; Piana, A.; Minniti, G.; Vercelli, M.; Perrone, L.; de Flora, S. Survival of atherosclerotic patients as related to oxidative stress and gene polymorphisms. Mutat. Res 2007, 621, 119–128. [Google Scholar]
- Nair, J.; de Flora, S.; Izzotti, A.; Bartsch, H. Lipid peroxidation-derived etheno-DNA adducts in human atherosclerotic lesions. Mutat. Res 2007, 621, 95–105. [Google Scholar]
- Demirbag, R.; Yilmaz, R.; Gur, M.; Kocyigit, A.; Celik, H.; Guzel, S.; Selek, S. Lymphocyte DNA damage in patients with acute coronary syndrome and its relationship with severity of acute coronary syndrome. Mutat. Res 2005, 578, 298–307. [Google Scholar]
- Federici, C.; Botto, N.; Manfredi, S.; Rizza, A.; del Fiandra, M.; Andreassi, M.G. Relation of increased chromosomal damage to future adverse cardiac events in patients with known coronary artery disease. Am. J. Cardiol 2008, 102, 1296–1300. [Google Scholar]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res 2006, 99, 156–164. [Google Scholar]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar]
- Mahmoudi, M.; Gorenne, I.; Mercer, J.; Figg, N.; Littlewood, T.; Bennett, M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ. Res 2008, 103, 717–725. [Google Scholar]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res 2010, 107, 1021–1031. [Google Scholar]
- Perez-Rivero, G.; Ruiz-Torres, M.P.; Rivas-Elena, J.V.; Jerkic, M.; Diez-Marques, M.L.; Lopez-Novoa, J.M.; Blasco, M.A.; Rodriguez-Puyol, D. Mice deficient in telomerase activity develop hypertension because of an excess of endothelin production. Circulation 2006, 114, 309–317. [Google Scholar]
- Jeon, B.H.; Gupta, G.; Park, Y.C.; Qi, B.; Haile, A.; Khanday, F.A.; Liu, Y.X.; Kim, J.M.; Ozaki, M.; White, A.R.; et al. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ. Res 2004, 95, 902–910. [Google Scholar]
- Friedberg, E.C.W.; Siede, G.C.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006; pp. 4–5. [Google Scholar]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet 2009, 10, 756–768. [Google Scholar]
- Krokan, H.E.; Standal, R.; Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J 1997, 325, 1–16. [Google Scholar]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012, 26, 417–432. [Google Scholar]
- Kalisch, T.; Ame, J.C.; Dantzer, F.; Schreiber, V. New readers and interpretations of poly(ADP-ribosyl)ation. Trends Biochem. Sci 2012, 37, 381–390. [Google Scholar]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev 2002, 54, 375–429. [Google Scholar]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet 2010, 44, 113–139. [Google Scholar]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol 2011, 12, 90–103. [Google Scholar]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol 2003, 4, 712–720. [Google Scholar]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar]
- Chanoux, R.A.; Yin, B.; Urtishak, K.A.; Asare, A.; Bassing, C.H.; Brown, E.J. ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem 2009, 284, 5994–6003. [Google Scholar]
- Huen, M.S.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res 2008, 18, 8–16. [Google Scholar]
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov 2008, 3, 156–164. [Google Scholar]
- Zeng, L.; Chen, R.; Liang, F.; Tsuchiya, H.; Murai, H.; Nakahashi, T.; Iwai, K.; Takahashi, T.; Kanda, T.; Morimoto, S. Silent information regulator, Sirtuin 1, and age-related diseases. Geriatr. Gerontol. Int 2009, 9, 7–15. [Google Scholar]
- Gorospe, M.; de Cabo, R. AsSIRTing the DNA damage response. Trends Cell Biol 2008, 18, 77–83. [Google Scholar]
- Yuan, Z.; Seto, E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle 2007, 6, 2869–2871. [Google Scholar]
- Mao, Z.; Hine, C.; Tian, X.; van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar]
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.D.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L.; et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009, 1, 109–121. [Google Scholar]
- Kaidi, A.; Weinert, B.T.; Choudhary, C.; Jackson, S.P. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010, 329, 1348–1353. [Google Scholar]
- Shukla, P.C.; Singh, K.K.; Quan, A.; Al-Omran, M.; Teoh, H.; Lovren, F.; Cao, L.; Rovira, I.I.; Pan, Y.; Brezden-Masley, C.; et al. BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Okuno, Y.; Nakamura-Ishizu, A.; Otsu, K.; Suda, T.; Kubota, Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med 2012, 18, 1208–1216. [Google Scholar]
- Pacher, P.; Szabo, C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc. Drug Rev 2007, 25, 235–260. [Google Scholar]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106, 927–932. [Google Scholar]
- Benko, R.; Pacher, P.; Vaslin, A.; Kollai, M.; Szabo, C. Restoration of the endothelial function in the aortic rings of apolipoprotein E deficient mice by pharmacological inhibition of the nuclear enzyme poly(ADP-ribose) polymerase. Life Sci 2004, 75, 1255–1261. [Google Scholar]
- Radovits, T.; Lin, L.N.; Zotkina, J.; Gero, D.; Szabo, C.; Karck, M.; Szabo, G. Poly(ADP-ribose) polymerase inhibition improves endothelial dysfunction induced by reactive oxidant hydrogen peroxide in vitro. Eur. J. Pharmacol 2007, 564, 158–166. [Google Scholar]
- Pernice, F.; Floccari, F.; Caccamo, C.; Belghity, N.; Mantuano, S.; Pacile, M.E.; Romeo, A.; Nostro, L.; Barilla, A.; Crasci, E.; et al. Chromosomal damage and atherosclerosis. A protective effect from simvastatin. Eur. J. Pharmacol 2006, 532, 223–229. [Google Scholar]
- Flouris, G.A.; Arvanitis, D.A.; Parissis, J.T.; Arvanitis, D.L.; Spandidos, D.A. Loss of heterozygosity in DNA mismatch repair genes in human atherosclerotic plaques. Mol. Cell Biol. Res. Commun 2000, 4, 62–65. [Google Scholar]
- Miniati, P.; Sourvinos, G.; Michalodimitrakis, M.; Spandidos, D.A. Loss of heterozygosity on chromosomes 1, 2, 8, 9 and 17 in cerebral atherosclerotic plaques. Int. J. Biol. Markers 2001, 16, 167–171. [Google Scholar]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res 2007, 100, 1512–1521. [Google Scholar]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res 2008, 80, 191–199. [Google Scholar]
- Tokunaga, O.; Fan, J.L.; Watanabe, T. Atherosclerosis- and age-related multinucleated variant endothelial cells in primary culture from human aorta. Am. J. Pathol 1989, 135, 967–976. [Google Scholar]
- Aviv, H.; Khan, M.Y.; Skurnick, J.; Okuda, K.; Kimura, M.; Gardner, J.; Priolo, L.; Aviv, A. Age dependent aneuploidy and telomere length of the human vascular endothelium. Atherosclerosis 2001, 159, 281–287. [Google Scholar]
- Borradaile, N.M.; Pickering, J.G. Polyploidy impairs human aortic endothelial cell function and is prevented by nicotinamide phosphoribosyltransferase. Am. J. Physiol. Cell Physiol 2010, 298, C66–C74. [Google Scholar]
- Bazo, A.P.; Salvadori, D., Jr; Salvadori, R.A.; Sodre, L.P.; da Silva, G.N.; de Camargo, E.A.; Ribeiro, L.R.; Salvadori, D.M. DNA repair gene polymorphism is associated with the genetic basis of atherosclerotic coronary artery disease. Cardiovasc. Pathol. 2011, 20, e9–e15. [Google Scholar]
- Guven, M.; Guven, G.S.; Oz, E.; Ozaydin, A.; Batar, B.; Ulutin, T.; Hacihanefioglu, S.; Domanic, N. DNA repair gene XRCC1 and XPD polymorphisms and their association with coronary artery disease risks and micronucleus frequency. Heart Vessels 2007, 22, 355–360. [Google Scholar]
- Mahmoudi, M.; Mercer, J.; Bennett, M. DNA damage and repair in atherosclerosis. Cardiovasc. Res 2006, 71, 259–268. [Google Scholar]
- Shyu, H.Y.; Shieh, J.C.; Ji-Ho, L.; Wang, H.W.; Cheng, C.W. Polymorphisms of DNA repair pathway genes and cigarette smoking in relation to susceptibility to large artery atherosclerotic stroke among ethnic Chinese in Taiwan. J. Atheroscler. Thromb 2012, 19, 316–325. [Google Scholar]
- Shimoyama, Y.; Mitsuda, Y.; Tsuruta, Y.; Suzuki, K.; Hamajima, N.; Niwa, T. SIRTUIN 1 gene polymorphisms are associated with cholesterol metabolism and coronary artery calcification in Japanese hemodialysis patients. J. Ren. Nutr 2012, 22, 114–119. [Google Scholar]
- Dong, C.; Della-Morte, D.; Wang, L.; Cabral, D.; Beecham, A.; McClendon, M.S.; Luca, C.C.; Blanton, S.H.; Sacco, R.L.; Rundek, T. Association of the sirtuin and mitochondrial uncoupling protein genes with carotid plaque. PLoS One 2011, 6, e27157. [Google Scholar]
- Samani, N.J.; Schunkert, H. Chromosome 9p21 and cardiovascular disease: the story unfolds. Circ. Cardiovasc. Genet 2008, 1, 81–84. [Google Scholar]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar]
- Crippa, S.; Cassano, M.; Sampaolesi, M. Role of miRNAs in muscle stem cell biology: Proliferation, differentiation and death. Curr. Pharm. Des 2012, 18, 1718–1729. [Google Scholar]
- Karp, X.; Ambros, V. Developmental biology. Encountering microRNAs in cell fate signaling. Science 2005, 310, 1288–1289. [Google Scholar]
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5. [Google Scholar] [CrossRef]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet 2004, 20, 617–624. [Google Scholar]
- Hu, H.; Gatti, R.A. MicroRNAs: New players in the DNA damage response. J. Mol. Cell Biol 2011, 3, 151–158. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Ikeda, S.; Kong, S.W.; Lu, J.; Bisping, E.; Zhang, H.; Allen, P.D.; Golub, T.R.; Pieske, B.; Pu, W.T. Altered microRNA expression in human heart disease. Physiol. Genomics 2007, 31, 367–373. [Google Scholar]
- Hartmann, D.; Thum, T. MicroRNAs and vascular (dys)function. Vascul. Pharmacol 2011, 55, 92–105. [Google Scholar]
- Thum, T. MicroRNA therapeutics in cardiovascular medicine. EMBO Mol. Med 2012, 4, 3–14. [Google Scholar]
- Hammond, S.M. RNAi, microRNAs, and human disease. Cancer Chemother. Pharmacol 2006, 58, s63–s68. [Google Scholar]
- Mann, D.L. MicroRNAs and the failing heart. N. Eng. J. Med 2007, 356, 2644–2645. [Google Scholar]
- Fichtlscherer, S.; De Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Roxe, T.; Muller-Ardogan, M.; et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res 2010, 107, 677–684. [Google Scholar]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res 2010, 107, 810–817. [Google Scholar]
- Raitoharju, E.; Lyytikainen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kahonen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar]
- Wan, G.; Mathur, R.; Hu, X.; Zhang, X.; Lu, X. miRNA response to DNA damage. Trends Biochem. Sci 2011, 36, 478–484. [Google Scholar]
- Simone, N.L.; Soule, B.P.; Ly, D.; Saleh, A.D.; Savage, J.E.; Degraff, W.; Cook, J.; Harris, C.C.; Gius, D.; Mitchell, J.B. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One 2009, 4, e6377. [Google Scholar]
- Yu, Y.; Wang, Y.; Ren, X.; Tsuyada, A.; Li, A.; Liu, L.J.; Wang, S.E. Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor beta contributes to chemoresistance in breast cancer cells. Mol. Cancer Res 2010, 8, 1633–1642. [Google Scholar]
- Lin, Y.; Liu, X.; Cheng, Y.; Yang, J.; Huo, Y.; Zhang, C. Involvement of MicroRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J. Biol. Chem 2009, 284, 7903–7913. [Google Scholar]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun 2010, 398, 735–740. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar]
- Kobayashi, K.; Imanishi, T.; Akasaka, T. Endothelial progenitor cell differentiation and senescence in an angiotensin II-infusion rat model. Hypertens. Res 2006, 29, 449–455. [Google Scholar]
- Imanishi, T.; Kobayashi, K.; Kuroi, A.; Mochizuki, S.; Goto, M.; Yoshida, K.; Akasaka, T. Effects of angiotensin II on NO bioavailability evaluated using a catheter-type NO sensor. Hypertension 2006, 48, 1058–1065. [Google Scholar]
- Oeseburg, H.; Iusuf, D.; van der Harst, P.; van Gilst, W.H.; Henning, R.H.; Roks, A.J. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension 2009, 53, 417–422. [Google Scholar]
- Addabbo, F.; Montagnani, M.; Goligorsky, M.S. Mitochondria and reactive oxygen species. Hypertension 2009, 53, 885–892. [Google Scholar]
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and disease: The road ahead. Front. Pharmacol. 2012, 3. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cervelli, T.; Borghini, A.; Galli, A.; Andreassi, M.G. DNA Damage and Repair in Atherosclerosis: Current Insights and Future Perspectives. Int. J. Mol. Sci. 2012, 13, 16929-16944. https://doi.org/10.3390/ijms131216929
Cervelli T, Borghini A, Galli A, Andreassi MG. DNA Damage and Repair in Atherosclerosis: Current Insights and Future Perspectives. International Journal of Molecular Sciences. 2012; 13(12):16929-16944. https://doi.org/10.3390/ijms131216929
Chicago/Turabian StyleCervelli, Tiziana, Andrea Borghini, Alvaro Galli, and Maria Grazia Andreassi. 2012. "DNA Damage and Repair in Atherosclerosis: Current Insights and Future Perspectives" International Journal of Molecular Sciences 13, no. 12: 16929-16944. https://doi.org/10.3390/ijms131216929