Bacterial Over-Expression and Purification of the 3'phosphoadenosine 5'phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling


Abstract
:1. Introduction
2. Results and Discussion
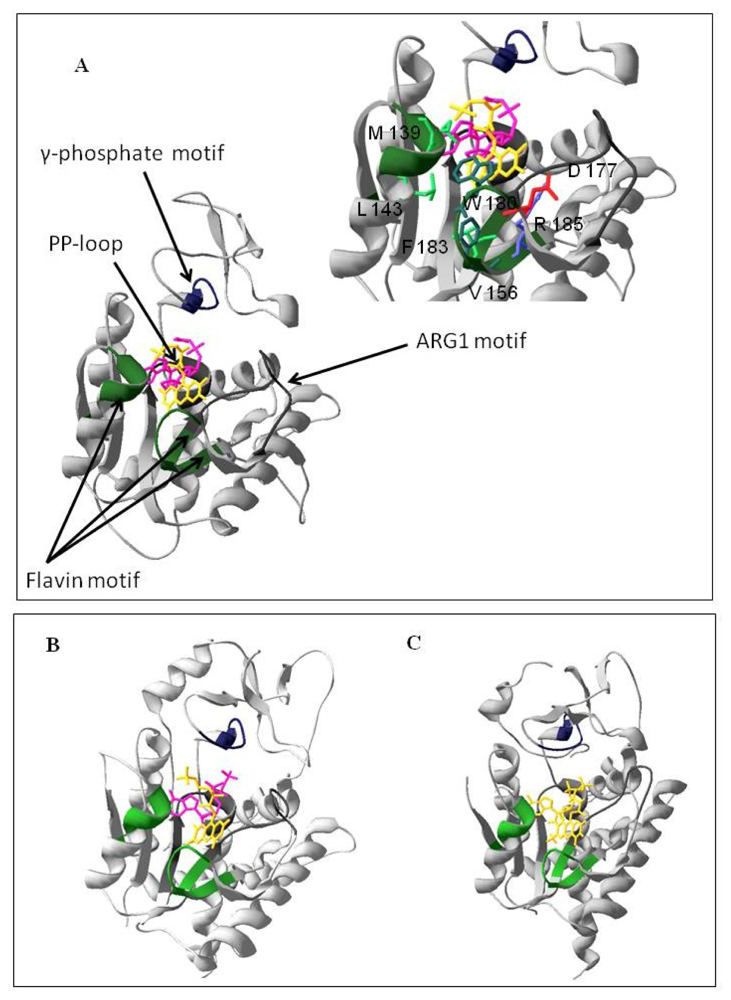
2.1. Homology Modeling of the PAPS Reductase Domain of the hFADS
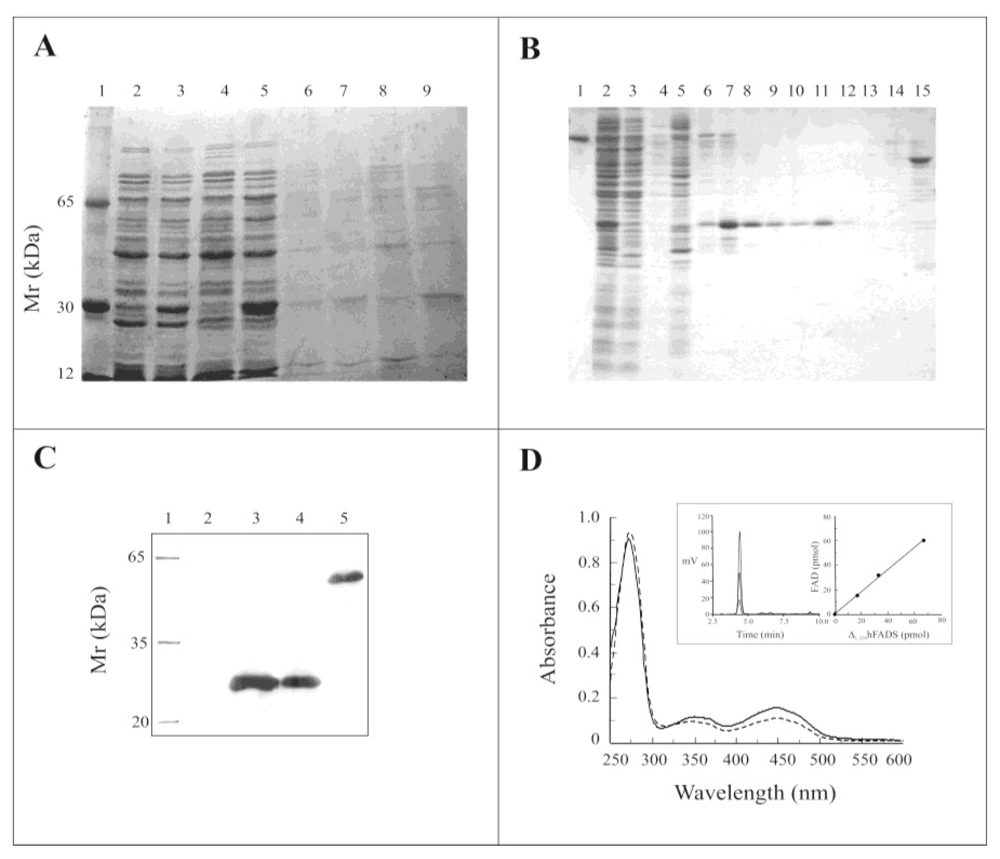
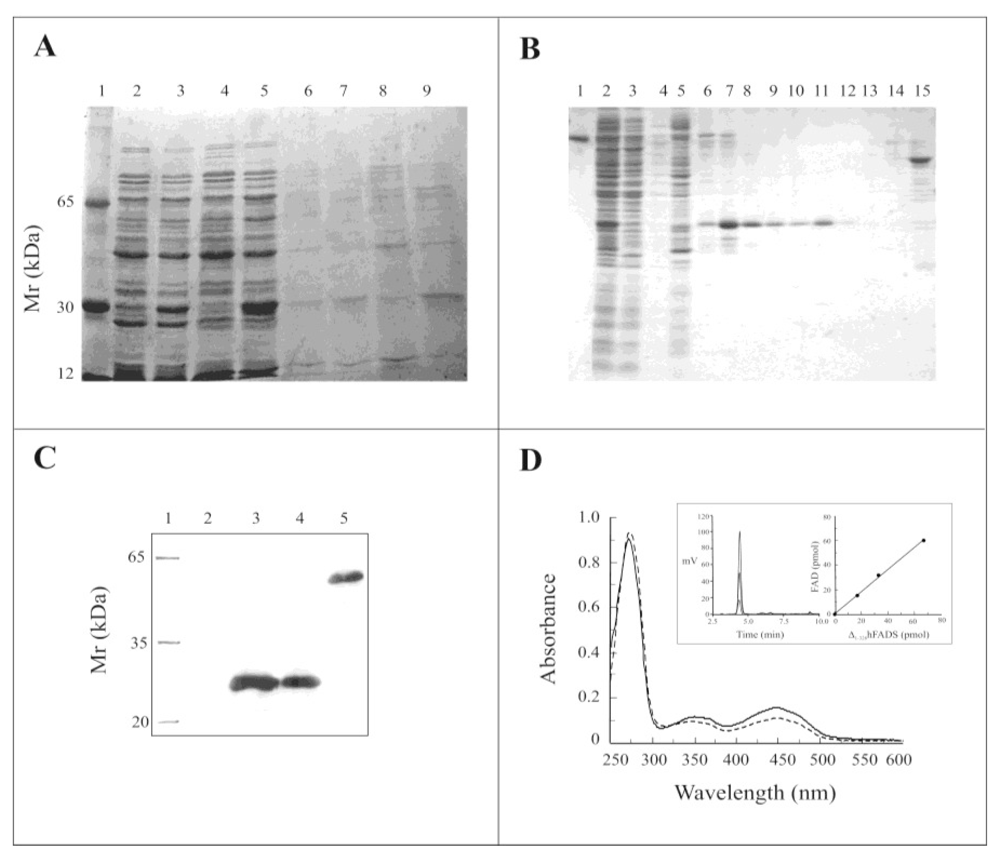
2.2. Expression of the Δ1-328-hFADS and Purification as a FAD-Binding Domain
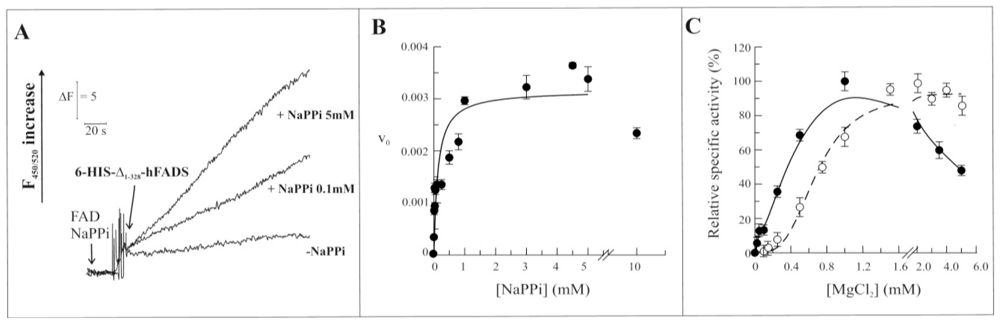
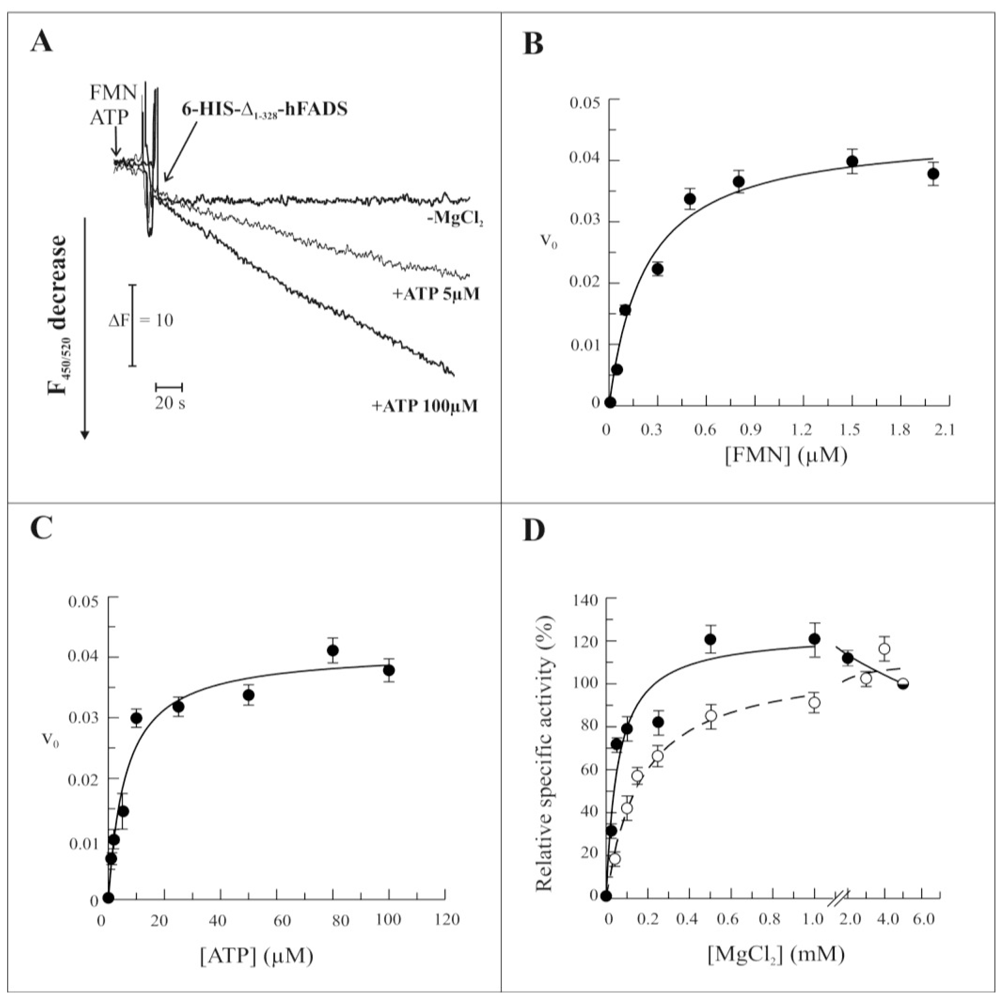
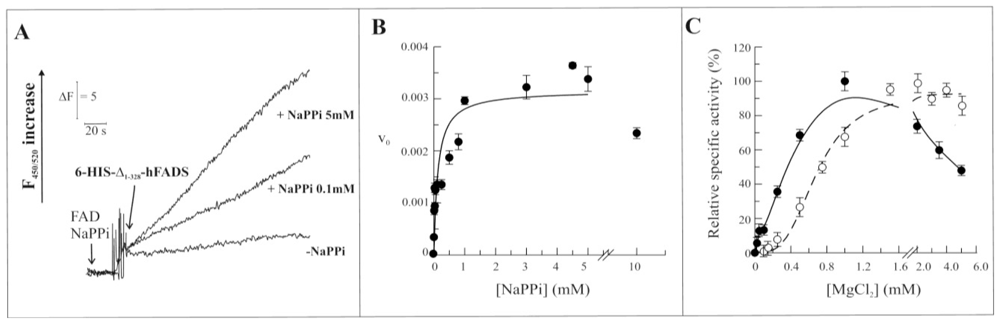
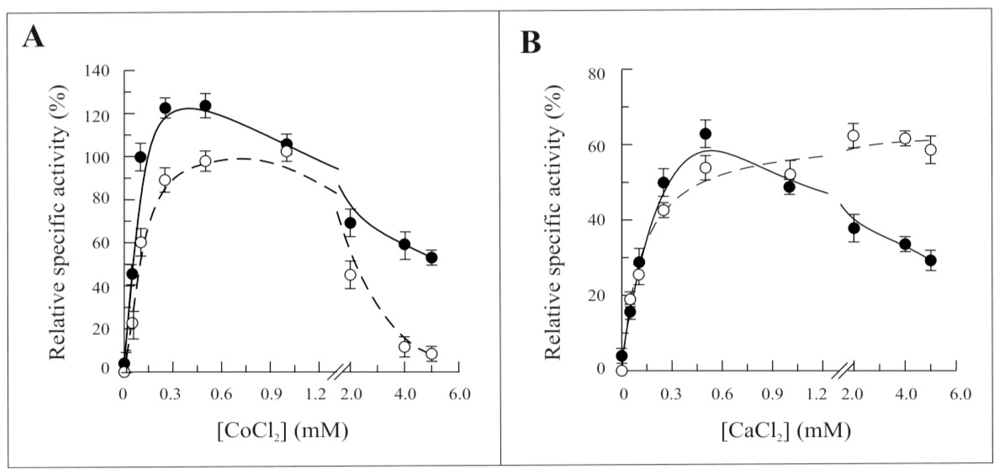
2.3. Biochemical and Functional Characterization of 6-His-Δ1-328-hFADS
2.4. Discussion
3. Experimental Section
3.1. Materials
3.2. Identification of PAPS Reductase Domain of the Human FAD Synthase
3.3. Homology Modeling of the Human PAPS Reductase Domain
3.4. Cloning of PAPS Reductase Domain of the Human FAD Synthase
3.5. Expression of the PAPS Reductase Domain of the Human FAD Synthase in E. coli
3.6. Purification of Recombinant 6-His-Δ1-328-hFADS of the Human FAD Synthase
3.7. Protein Concentration and FAD/Protein Monomer Ratio Measurements
3.8. Measurements of FAD Synthesis Rate
3.9. Measurements of FAD Cleavage Rate
3.10. Other Assays
3.11. Kinetic Data Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
Abbreviations
| Rf | riboflavin |
| RFK | riboflavin kinase |
| FADS | FAD synthase or synthetase |
| FMNAT | FMN adenylyl transferase |
| hFADS | human FAD synthase |
| hFADS1 | human FAD synthase isoform 1 |
| hFADS2 | human FAD synthase isoform 2 |
| Δ1-328-hFADS | “truncated” form of hFADS lacking the N-terminal domain |
| PAPS | 3′-phosphoadenosine 5′-phosphosulfate |
| IPTG | isopropyl-thio-β-d-galactoside |
References
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans 2000, 28, 283–296. [Google Scholar]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol 2007, 11, 195–202. [Google Scholar]
- Ghisla, S.; Massey, V. Mechanisms of flavoprotein-catalyzed reactions. Eur. J. Biochem 1989, 181, 1–17. [Google Scholar]
- Santos, M.A.; Jimenez, A.; Revuelta, J.L. Molecular characterization of FMN1, the structural gene for the monofunctional flavokinase of Saccharomyces cerevisiae. J. Biol. Chem 2000, 275, 28618–28624. [Google Scholar]
- Wu, M.; Repetto, B.; Glerum, D.M.; Tzagoloff, A. Cloning and characterization of FAD1, the structural gene for flavin adenine dinucleotide synthetase of Saccharomyces cerevisiae. Mol. Cell. Biol 1995, 15, 264–271. [Google Scholar]
- Efimov, I.; Kuusk, V.; Zhang, X.; McIntire, W.S. Proposed steady-state kinetic mechanism for Corynebacterium ammoniagenes FAD synthetase produced by Escherichia coli. Biochemistry 1998, 37, 9716–9723. [Google Scholar]
- Kearney, E.B.; Goldenberg, J.; Lipsick, J.; Perl, M. Flavokinase and FAD synthetase from Bacillus subtilis specific for reduced flavins. J. Biol. Chem 1979, 254, 9551–9557. [Google Scholar]
- Leulliot, N.; Blondeau, K.; Keller, J.; Ulryck, N.; Quevillon-Cheruel, S.; van Tilbeurgh, H. Crystal structure of yeast FAD synthetase (Fad1) in complex with FAD. J. Mol. Biol 2010, 398, 641–646. [Google Scholar]
- Huerta, C.; Borek, D.; Machius, M.; Grishin, N.V.; Zhang, H. Structure and mechanism of a eukaryotic FMN adenylyltransferase. J. Mol. Biol 2009, 389, 388–400. [Google Scholar]
- Gerdes, S.Y.; Scholle, M.D.; D’Souza, M.; Bernal, A.; Baev, M.V.; Farrell, M.; Kurnasov, O.V.; Daugherty, M.D.; Mseeh, F.; Polanuyer, B.M.; et al. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J. Bacteriol 2002, 184, 4555–4572. [Google Scholar]
- Pedrolli, D.B.; Nakanishi, S.; Barile, M.; Mansurova, M.; Carmona, E.C.; Lux, A.; Gartner, W.; Mack, M. The antibiotics roseoflavin and 8-demethyl-8-amino-riboflavin from Streptomyces davawensis are metabolized by human flavokinase and human FAD synthetase. Biochem. Pharmacol 2011, 82, 1853–1859. [Google Scholar]
- Brizio, C.; Galluccio, M.; Wait, R.; Torchetti, E.M.; Bafunno, V.; Accardi, R.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun 2006, 344, 1008–1016. [Google Scholar]
- Galluccio, M.; Brizio, C.; Torchetti, E.M.; Ferranti, P.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase. Protein Expr. Purif 2007, 52, 175–181. [Google Scholar]
- Torchetti, E.M.; Brizio, C.; Colella, M.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Roberti, M.; Barile, M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 2010, 10, 263–273. [Google Scholar]
- Torchetti, E.M.; Bonomi, F.; Galluccio, M.; Gianazza, E.; Giancaspero, T.A.; Iametti, S.; Indiveri, C.; Barile, M. Human FAD synthase (isoform 2): A component of the machinery that delivers FAD to apo-flavoproteins. FEBS J 2011, 278, 4434–4449. [Google Scholar]
- Barile, M.; Giancaspero, T.A.; Brizio, C.; Panebianco, C.; Indiveri, C.; Galluccio, M.; Vergani, L.; Eberini, I.; Gianazza, E. Biosynthesis of flavin cofactors in man: Implications in health and disease. Curr. Pharmaceut. Des. 2012, in press. [Google Scholar]
- Leimkuhler, S.; Wuebbens, M.M.; Rajagopalan, K.V. The History of the Discovery of the Molybdenum Cofactor and Novel Aspects of its Biosynthesis in Bacteria. Coord. Chem. Rev 2011, 255, 1129–1144. [Google Scholar]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010, 66, 12–21. [Google Scholar]
- Schrecker, A.W.; Kornberg, A. Reversible enzymatic synthesis of flavin-adenine dinucleotide. J. Biol. Chem 1950, 182, 795–803. [Google Scholar]
- Bevers, L.E.; Hagedoorn, P.L.; Santamaria-Araujo, J.A.; Magalon, A.; Hagen, W.R.; Schwarz, G. Function of MoaB proteins in the biosynthesis of the molybdenum and tungsten cofactors. Biochemistry 2008, 47, 949–956. [Google Scholar]
- Kuper, J.; Llamas, A.; Hecht, H.J.; Mendel, R.R.; Schwarz, G. Structure of the molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature 2004, 430, 803–806. [Google Scholar]
- Wittle, A.E.; Kamdar, K.P.; Finnerty, V. The Drosophila cinnamon gene is functionally homologous to Arabidopsis cnx1 and has a similar expression pattern to the mammalian gephyrin gene. Mol. Gen. Genet 1999, 261, 672–680. [Google Scholar]
- Nawrotzki, R.; Islinger, M.; Vogel, I.; Volkl, A.; Kirsch, J. Expression and subcellular distribution of gephyrin in non-neuronal tissues and cells. Histochem. Cell Biol 2012, 137, 471–482. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Barile, M.; Brizio, C.; de Virgilio, C.; Delfine, S.; Quagliariello, E.; Passarella, S. Flavin adenine dinucleotide and flavin mononucleotide metabolism in rat liver-the occurrence of FAD pyrophosphatase and FMN phosphohydrolase in isolated mitochondria. Eur. J. Biochem 1997, 249, 777–785. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Brizio, C.; Brandsch, R.; Bufano, D.; Pochini, L.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, functional characterization and refolding of rat dimethylglycine dehydrogenase. Protein Expr. Purif 2004, 37, 434–442. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6-His-Δ1-328-hFADS (this study) | 6-His-hFADS2 [13] | |
|---|---|---|
| Forward reaction | ||
| kcat (s−1) | 0.042 ± 0.001 | 0.069 ± 0.011 |
| Km FMN (μM) | 0.24 ± 0.06 | 0.35 ± 0.08 |
| Km ATP (μM) | 6.23 ± 1.5 | 15.3 ± 2.2 |
| Ki GTP (mM) | 7.5 (uncompetitive) | 1.8 (uncompetitive) |
| Mg2+50 (mM) | 0.05 ± 0.02 | 0.15 ±0.02 |
| Co2+50 (mM) | 0.08 ± 0.03 | 0.12 ± 0.02 |
| Ca2+50 (mM) | 0.10 ± 0.01 | 0.12 ±0.03 |
| Reverse reaction | ||
| kcat (s−1) | 0.0037 ± 0.0009 | 0.0052 ± 0.0001 |
| Km NaPPI (μM) | 92.0 ± 0.5 | 82.1 ± 2.2 |
| Km FAD (μM) | n.d. | <0.1 |
| Mg2+50 (mM) | 0.20 ± 0.03 | 0.60 ± 0.08 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miccolis, A.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Barile, M. Bacterial Over-Expression and Purification of the 3'phosphoadenosine 5'phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling. Int. J. Mol. Sci. 2012, 13, 16880-16898. https://doi.org/10.3390/ijms131216880
Miccolis A, Galluccio M, Giancaspero TA, Indiveri C, Barile M. Bacterial Over-Expression and Purification of the 3'phosphoadenosine 5'phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling. International Journal of Molecular Sciences. 2012; 13(12):16880-16898. https://doi.org/10.3390/ijms131216880
Chicago/Turabian StyleMiccolis, Angelica, Michele Galluccio, Teresa Anna Giancaspero, Cesare Indiveri, and Maria Barile. 2012. "Bacterial Over-Expression and Purification of the 3'phosphoadenosine 5'phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling" International Journal of Molecular Sciences 13, no. 12: 16880-16898. https://doi.org/10.3390/ijms131216880