The Implications of Cancer Stem Cells for Cancer Therapy



Abstract
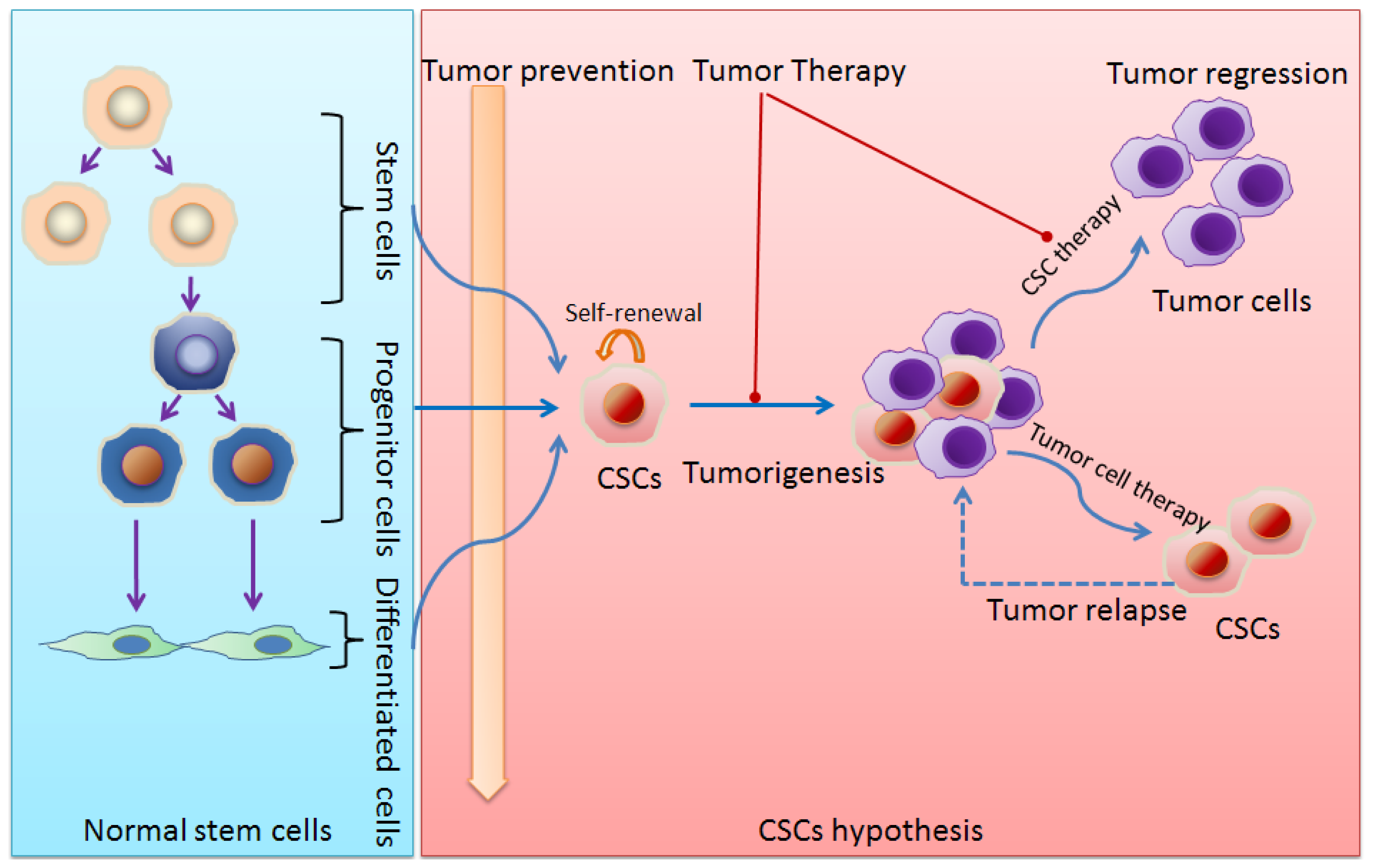
:1. Stem Cells and Cancer Stem Cells (CSCs)
2. Evidence of CSCs
3. CSCs and Cancer Progenitor Cells
4. Origins of CSCs
4.1. From Stem Cells
4.2. From Progenitor Cells
4.3. Other Possible Sources
5. Molecular Mechanisms Controlling CSCs
5.1. Notch Signaling Pathway
5.2. Wnt/β-Catenin Signaling Pathway
5.3. Other Signaling Pathways Implicated in CSCs
6. Implications for Cancer Treatment
7. Conclusions
Acknowledgements
- Competing InterestsThe authors declare no conflict of interest.
References
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Baker, D.E.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol 2007, 25, 207–215. [Google Scholar]
- Harrison, D.E.; Zhong, R.K. The same exhaustible multilineage precursor produces both myeloid and lymphoid cells as early as 3–4 weeks after marrow transplantation. Proc. Natl. Acad. Sci. USA 1992, 89, 10134–10138. [Google Scholar]
- Morrison, S.J.; Shah, N.M.; Anderson, D.J. Regulatory mechanisms in stem cell biology. Cell 1997, 88, 287–298. [Google Scholar]
- Iwasaki, H.; Suda, T. Cancer stem cells and their niche. Cancer Sci 2009, 100, 1166–1172. [Google Scholar]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med 2011, 17, 313–319. [Google Scholar]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar]
- Shipitsin, M.; Polyak, K. The cancer stem cell hypothesis: In search of definitions, markers, and relevance. Lab. Invest 2008, 88, 459–463. [Google Scholar]
- Yang, Y.M.; Chang, J.W. Current status and issues in cancer stem cell study. Cancer Invest 2008, 26, 741–755. [Google Scholar]
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res 2007, 17, 3–14. [Google Scholar]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar]
- Besancon, R.; Valsesia-Wittmann, S.; Puisieux, A.; Caron de Fromentel, C.; Maguer-Satta, V. Cancer stem cells: The emerging challenge of drug targeting. Curr. Med. Chem 2009, 16, 394–416. [Google Scholar]
- Kamstrup, M.R.; Gniadecki, R.; Skovgaard, G.L. Putative cancer stem cells in cutaneous malignancies. Exp. Dermatol 2007, 16, 297–301. [Google Scholar]
- Leitch, J.; Klein, G.; Tee, R.; Murdock, C.; Teo, W.S. Neurally mediated syncope and atrial fibrillation. N. Engl. J. Med 1991, 324, 495–496. [Google Scholar]
- Guo, W.; Lasky, J.L., 3rd; Wu, H. Cancer stem cells. Pediatr. Res. 2006, 59, 59R–64R. [Google Scholar]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting notch to target cancer stem cells. Clin. Cancer Res 2010, 16, 3141–3152. [Google Scholar]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side population is enriched in tumorigenic, stem-like cancer cells, whereas abcg2+ and abcg2− cancer cells are similarly tumorigenic. Cancer Res 2005, 65, 6207–6219. [Google Scholar]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar]
- Yuan, Y.; Shen, H.; Franklin, D.S.; Scadden, D.T.; Cheng, T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early g1-phase inhibitor, p18ink4c. Nat. Cell Biol 2004, 6, 436–442. [Google Scholar]
- Mackillop, W.J.; Ciampi, A.; Till, J.E.; Buick, R.N. A stem cell model of human tumor growth: Implications for tumor cell clonogenic assays. J. Natl. Cancer Inst 1983, 70, 9–16. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Blair, A.; Hogge, D.E.; Ailles, L.E.; Lansdorp, P.M.; Sutherland, H.J. Lack of expression of thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 1997, 89, 3104–3112. [Google Scholar]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol 2007, 23, 675–699. [Google Scholar]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res 2007, 67, 4827–4833. [Google Scholar]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death. Differ 2008, 15, 504–514. [Google Scholar]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res 2007, 67, 1030–1037. [Google Scholar]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (sc) and tracks sc overpopulation during colon tumorigenesis. Cancer Res 2009, 69, 3382–3389. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar]
- Schatton, T.; Frank, M.H. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res 2008, 21, 39–55. [Google Scholar]
- Suva, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suva, D.; Clement, V.; Provero, P.; Cironi, L.; et al. Identification of cancer stem cells in ewing’s sarcoma. Cancer Res 2009, 69, 1776–1781. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Gibbs, C.P.; Kukekov, V.G.; Reith, J.D.; Tchigrinova, O.; Suslov, O.N.; Scott, E.W.; Ghivizzani, S.C.; Ignatova, T.N.; Steindler, D.A. Stem-like cells in bone sarcomas: Implications for tumorigenesis. Neoplasia 2005, 7, 967–976. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Lang, S.H.; Frame, F.M.; Collins, A.T. Prostate cancer stem cells. J. Pathol 2009, 217, 299–306. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar]
- Douville, J.; Beaulieu, R.; Balicki, D. Aldh1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev 2009, 18, 17–25. [Google Scholar]
- Corti, S.; Locatelli, F.; Papadimitriou, D.; Donadoni, C.; Salani, S.; Del Bo, R.; Strazzer, S.; Bresolin, N.; Comi, G.P. Identification of a primitive brain-derived neural stem cell population based on aldehyde dehydrogenase activity. Stem Cells 2006, 24, 975–985. [Google Scholar]
- Ma, S.; Chan, K.W.; Lee, T.K.; Tang, K.H.; Wo, J.Y.; Zheng, B.J.; Guan, X.Y. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol. Cancer Res 2008, 6, 1146–1153. [Google Scholar]
- Gunaratne, P.H. Embryonic stem cell micrornas: Defining factors in induced pluripotent (ips) and cancer (csc) stem cells? Curr. Stem Cell Res. Ther 2009, 4, 168–177. [Google Scholar]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In vitro reprogramming of fibroblasts into a pluripotent es-cell-like state. Nature 2007, 448, 318–324. [Google Scholar]
- Zhang, Y.; Peng, J.; Zhang, H.; Zhu, Y.; Wan, L.; Chen, J.; Chen, X.; Lin, R.; Li, H.; Mao, X.; et al. Notch1 signaling is activated in cells expressing embryonic stem cell proteins in human primary nasopharyngeal carcinoma. J. Otolaryngol. Head Neck Surg 2010, 39, 157–166. [Google Scholar]
- Zhang, H.B.; Ren, C.P.; Yang, X.Y.; Wang, L.; Li, H.; Zhao, M.; Yang, H.; Yao, K.T. Identification of label-retaining cells in nasopharyngeal epithelia and nasopharyngeal carcinoma tissues. Histochem. Cell Biol 2007, 127, 347–354. [Google Scholar]
- Cheng, A.L.; Huang, W.G.; Chen, Z.C.; Peng, F.; Zhang, P.F.; Li, M.Y.; Li, F.; Li, J.L.; Li, C.; Yi, H.; et al. Identification of novel nasopharyngeal carcinoma biomarkers by laser capture microdissection and proteomic analysis. Clin. Cancer Res 2008, 14, 435–445. [Google Scholar]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res 2012, 72, 576–580. [Google Scholar]
- Passegue, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis cml. N. Engl. J. Med 2004, 351, 657–667. [Google Scholar]
- Manz, M.G.; Miyamoto, T.; Akashi, K.; Weissman, I.L. Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. USA 2002, 99, 11872–11877. [Google Scholar]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar]
- Lowenberg, B.; Touw, I.P. Hematopoietic growth factors and their receptors in acute leukemia. Blood 1993, 81, 281–292. [Google Scholar]
- Griffin, J.D.; Lowenberg, B. Clonogenic cells in acute myeloblastic leukemia. Blood 1986, 68, 1185–1195. [Google Scholar]
- McCulloch, E.A.; Izaguirre, C.A.; Chang, L.J.; Smith, L.J. Renewal and determination in leukemic blast populations. J. Cell Physiol. Suppl 1982, 1, 103–111. [Google Scholar]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to sti571 in vitro. Blood 2002, 99, 319–325. [Google Scholar]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (bms-354825) targets an earlier progenitor population than imatinib in primary cml but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar]
- Jorgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ cml cells. Blood 2007, 109, 4016–4019. [Google Scholar]
- Testa, U. Leukemia stem cells. Ann. Hematol 2011, 90, 245–271. [Google Scholar]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst 2008, 100, 672–679. [Google Scholar]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar]
- Jiang, X.; Zhao, Y.; Chan, W.Y.; Pang, E.; Eaves, A.; Eaves, C. Leukemic stem cells of chronic phase cml patients consistently display very high bcr-abl transcript levels and reduced responsiveness to imatinib mesylate in addition to generating a rare subset that produce imatinib mesylate-resistant differentiated progeny. Blood 2004, 104, 711–722. [Google Scholar]
- Venugopal, C.; Li, N.; Wang, X.; Manoranjan, B.; Hawkins, C.; Gunnarsson, T.; Hollenberg, R.; Klurfan, P.; Murty, N.; Kwiecien, J.; et al. Bmi1 marks intermediate precursors during differentiation of human brain tumor initiating cells. Stem Cell Res 2012, 8, 141–153. [Google Scholar]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar]
- Bonnet, D. Cancer stem cells: Amls show the way. Biochem. Soc. Trans 2005, 33, 1531–1533. [Google Scholar]
- Wang, Y.; Yang, J.; Zheng, H.; Tomasek, G.J.; Zhang, P.; McKeever, P.E.; Lee, E.Y.; Zhu, Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 2009, 15, 514–526. [Google Scholar]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by mll-af9. Nature 2006, 442, 818–822. [Google Scholar]
- Nakano, I.; Kornblum, H.I. Brain tumor stem cells. Pediatr. Res 2006, 59, 54R–58R. [Google Scholar]
- Wagers, A.J.; Weissman, I.L. Plasticity of adult stem cells. Cell 2004, 116, 639–648. [Google Scholar]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar]
- Brabletz, S.; Schmalhofer, O.; Brabletz, T. Gastrointestinal stem cells in development and cancer. J. Pathol 2009, 217, 307–317. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Chiou, S.H.; Wang, M.L.; Chou, Y.T.; Chen, C.J.; Hong, C.F.; Hsieh, W.J.; Chang, H.T.; Chen, Y.S.; Lin, T.W.; Hsu, H.S.; et al. Coexpression of oct4 and nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res 2010, 70, 10433–10444. [Google Scholar]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. Lin28b promotes colon cancer progression and metastasis. Cancer Res 2011, 71, 4260–4268. [Google Scholar]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cazares, H.; Eberhart, C.G.; et al. C-met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent es-cell-like state. RNA 2008, 14, 2115–2124. [Google Scholar]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L.; et al. Targeting the nf-kappab signaling pathway in notch1-induced t-cell leukemia. Nat. Med 2007, 13, 70–77. [Google Scholar]
- McCleary-Wheeler, A.L.; McWilliams, R.; Fernandez-Zapico, M.E. Aberrant signaling pathways in pancreatic cancer: A two compartment view. Mol. Carcinog 2012, 51, 25–39. [Google Scholar]
- Hu, Y.Y.; Zheng, M.H.; Zhang, R.; Liang, Y.M.; Han, H. Notch signaling pathway and cancer metastasis. Adv. Exp. Med. Biol 2012, 727, 186–198. [Google Scholar]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar]
- Stockhausen, M.T.; Kristoffersen, K.; Poulsen, H.S. The functional role of notch signaling in human gliomas. Neuro. Oncol 2010, 12, 199–211. [Google Scholar]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar]
- Huelsken, J.; Behrens, J. The wnt signalling pathway. J. Cell Sci 2002, 115, 3977–3978. [Google Scholar]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin d1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-myc as a target of the apc pathway. Science 1998, 281, 1509–1512. [Google Scholar]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/tcf-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar]
- Rich, J.N. Cancer stem cells in radiation resistance. Cancer Res 2007, 67, 8980–8984. [Google Scholar]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol 2012, 4, a0080052. [Google Scholar]
- Ayyanan, A.; Civenni, G.; Ciarloni, L.; Morel, C.; Mueller, N.; Lefort, K.; Mandinova, A.; Raffoul, W.; Fiche, M.; Dotto, G.P.; et al. Increased wnt signaling triggers oncogenic conversion of human breast epithelial cells by a notch-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 3799–3804. [Google Scholar]
- Shiras, A.; Chettiar, S.T.; Shepal, V.; Rajendran, G.; Prasad, G.R.; Shastry, P. Spontaneous transformation of human adult nontumorigenic stem cells to cancer stem cells is driven by genomic instability in a human model of glioblastoma. Stem Cells 2007, 25, 1478–1489. [Google Scholar]
- Zhang, T.; Otevrel, T.; Gao, Z.; Ehrlich, S.M.; Fields, J.Z.; Boman, B.M. Evidence that apc regulates survivin expression: A possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 2001, 61, 8664–8667. [Google Scholar]
- Kim, P.J.; Plescia, J.; Clevers, H.; Fearon, E.R.; Altieri, D.C. Survivin and molecular pathogenesis of colorectal cancer. Lancet 2003, 362, 205–209. [Google Scholar]
- Bienz, M.; Clevers, H. Linking colorectal cancer to wnt signaling. Cell 2000, 103, 311–320. [Google Scholar]
- Polakis, P. Wnt signaling and cancer. Genes Dev 2000, 14, 1837–1851. [Google Scholar]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and cml stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar]
- Vermeulen, L.; de Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol 2010, 12, 468–476. [Google Scholar]
- Chen, M.S.; Woodward, W.A.; Behbod, F.; Peddibhotla, S.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. Wnt/beta-catenin mediates radiation resistance of sca1+ progenitors in an immortalized mammary gland cell line. J. Cell Sci 2007, 120, 468–477. [Google Scholar]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. Wnt/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the pten/mtor/stat3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008, 22, 436–448. [Google Scholar]
- Dvorak, P.; Dvorakova, D.; Hampl, A. Fibroblast growth factor signaling in embryonic and cancer stem cells. FEBS Lett 2006, 580, 2869–2874. [Google Scholar]
- Dirks, P.B. Brain tumor stem cells: Bringing order to the chaos of brain cancer. J. Clin. Oncol 2008, 26, 2916–2924. [Google Scholar]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One 2011, 6, e17687. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. Hedgehog-gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol 2007, 17, 165–172. [Google Scholar]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. Her2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. Pten activation contributes to tumor inhibition by trastuzumab, and loss of pten predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bfgf and egf more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through l1cam suppresses glioma growth. Cancer Res 2008, 68, 6043–6048. [Google Scholar]
- Cao, Y.; Lathia, J.D.; Eyler, C.E.; Wu, Q.; Li, Z.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Erythropoietin receptor signaling through stat3 is required for glioma stem cell maintenance. Genes Cancer 2010, 1, 50–61. [Google Scholar]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. Stat3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar]
- Lage, H. An overview of cancer multidrug resistance: A still unsolved problem. Cell Mol. Life Sci 2008, 65, 3145–3167. [Google Scholar]
- Rangwala, F.; Omenetti, A.; Diehl, A.M. Cancer stem cells: Repair gone awry? J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Kvinlaug, B.T.; Huntly, B.J. Targeting cancer stem cells. Expert Opin. Ther. Targets 2007, 11, 915–927. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar]
- Azzi, S.; Bruno, S.; Giron-Michel, J.; Clay, D.; Devocelle, A.; Croce, M.; Ferrini, S.; Chouaib, S.; Vazquez, A.; Charpentier, B.; et al. Differentiation therapy: Targeting human renal cancer stem cells with interleukin 15. J. Natl. Cancer Inst 2011, 103, 1884–1898. [Google Scholar]
- Pham, P.V.; Phan, N.L.; Nguyen, N.T.; Truong, N.H.; Duong, T.T.; Le, D.V.; Truong, K.D.; Phan, N.K. Differentiation of breast cancer stem cells by knockdown of CD44: Promising differentiation therapy. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef]
- Cole, M.F.; Johnstone, S.E.; Newman, J.J.; Kagey, M.H.; Young, R.A. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev 2008, 22, 746–755. [Google Scholar]
- Wei, G.; Ku, S.; Ma, G.K.; Saito, S.; Tang, A.A.; Zhang, J.; Mao, J.H.; Appella, E.; Balmain, A.; Huang, E.J. Hipk2 represses beta-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 13040–13045. [Google Scholar]
- Singh, A.; Settleman, J. Emt, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar]
- Alison, M.R.; Lim, S.M.; Nicholson, L.J. Cancer stem cells: Problems for therapy? J. Pathol 2011, 223, 147–161. [Google Scholar]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; Di Fiore, P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010, 140, 62–73. [Google Scholar]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via il6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar]
- Cho, W.C. Targeting signaling pathways in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 1–3. [Google Scholar]
- Zimmerman, A.L.; Wu, S. MicroRNAs, cancer and cancer stem cells. Cancer Lett 2011, 300, 10–19. [Google Scholar]
- Yu, Z.R.; Pestell, R.G. MicroRNAs and cancer stem cells. In MicroRNAs in Cancer Translational Research; Cho, W.C.S., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp. 373–388. [Google Scholar]
- Riccioni, R.; Dupuis, M.L.; Bernabei, M.; Petrucci, E.; Pasquini, L.; Mariani, G.; Cianfriglia, M.; Testa, U. The cancer stem cell selective inhibitor salinomycin is a p-glycoprotein inhibitor. Blood Cells Mol. Dis 2010, 45, 86–92. [Google Scholar]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar]
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of aml. Nat. Biotechnol 2010, 28, 275–280. [Google Scholar]
- Kang, M.K.; Hur, B.I.; Ko, M.H.; Kim, C.H.; Cha, S.H.; Kang, S.K. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef]
- Dingli, D.; Michor, F. Successful therapy must eradicate cancer stem cells. Stem Cells 2006, 24, 2603–2610. [Google Scholar]
- Fuchs, E. The tortoise and the hair: Slow-cycling cells in the stem cell race. Cell 2009, 137, 811–819. [Google Scholar]
- Essers, M.A.; Trumpp, A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol 2010, 4, 443–450. [Google Scholar]
- Tan, B.; Piwnica-Worms, D.; Ratner, L. Multidrug resistance transporters and modulation. Curr. Opin. Oncol 2000, 12, 450–458. [Google Scholar]
- Raguz, S.; Yague, E. Resistance to chemotherapy: New treatments and novel insights into an old problem. Br. J. Cancer 2008, 99, 387–391. [Google Scholar]
- Wang, J.; Guo, L.P.; Chen, L.Z.; Zeng, Y.X.; Lu, S.H. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res 2007, 67, 3716–3724. [Google Scholar]
- Liang, Y.; Zhong, Z.; Huang, Y.; Deng, W.; Cao, J.; Tsao, G.; Liu, Q.; Pei, D.; Kang, T.; Zeng, Y.X. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J. Biol. Chem 2010, 285, 4931–4940. [Google Scholar]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol 2008, 26, 2839–2845. [Google Scholar]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar]


{kind=link}
| Cancer Types | Cell Surface Markers | Reference |
|---|---|---|
| Lung cancer | CD24+, CD44+, CD133+ | [27,28] |
| Hepatic carcinoma | CD90+, CD45−, (CD44+) | [29] |
| Ovarian cancer | CD44+, CD117+ | [30] |
| Colon cancer | CD133+, EpCAM+, CD44+, CD166+, ALDH1+ | [31,33–35] |
| Pancreatic cancer | CD44+, CD24+, ESA+, CD133+ | [32] |
| Melanoma | ABCB5+ | [36] |
| Ewing’s sarcoma | CD133+ | [37] |
| Glioma | CD133+ | [38] |
| Sarcomas | CD105+, CD44+, Stro1+ | [39] |
| Breast cancer | CD44+CD24−/low | [40] |
| Prostate cancer | Sca1+, CD133+, CD44+ | [41] |
| Head & neck squamous cell carcinoma | CD44+ | [26] |
|
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, W.; Peng, J.; Zhang, Y.; Cho, W.C.S.; Jin, K. The Implications of Cancer Stem Cells for Cancer Therapy. Int. J. Mol. Sci. 2012, 13, 16636-16657. https://doi.org/10.3390/ijms131216636
Jiang W, Peng J, Zhang Y, Cho WCS, Jin K. The Implications of Cancer Stem Cells for Cancer Therapy. International Journal of Molecular Sciences. 2012; 13(12):16636-16657. https://doi.org/10.3390/ijms131216636
Chicago/Turabian StyleJiang, Wenjing, Jianhua Peng, Yue Zhang, William C. S. Cho, and Kunlin Jin. 2012. "The Implications of Cancer Stem Cells for Cancer Therapy" International Journal of Molecular Sciences 13, no. 12: 16636-16657. https://doi.org/10.3390/ijms131216636
APA StyleJiang, W., Peng, J., Zhang, Y., Cho, W. C. S., & Jin, K. (2012). The Implications of Cancer Stem Cells for Cancer Therapy. International Journal of Molecular Sciences, 13(12), 16636-16657. https://doi.org/10.3390/ijms131216636