Epigenetic Deregulation of MicroRNAs in Rhabdomyosarcoma and Neuroblastoma and Translational Perspectives


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
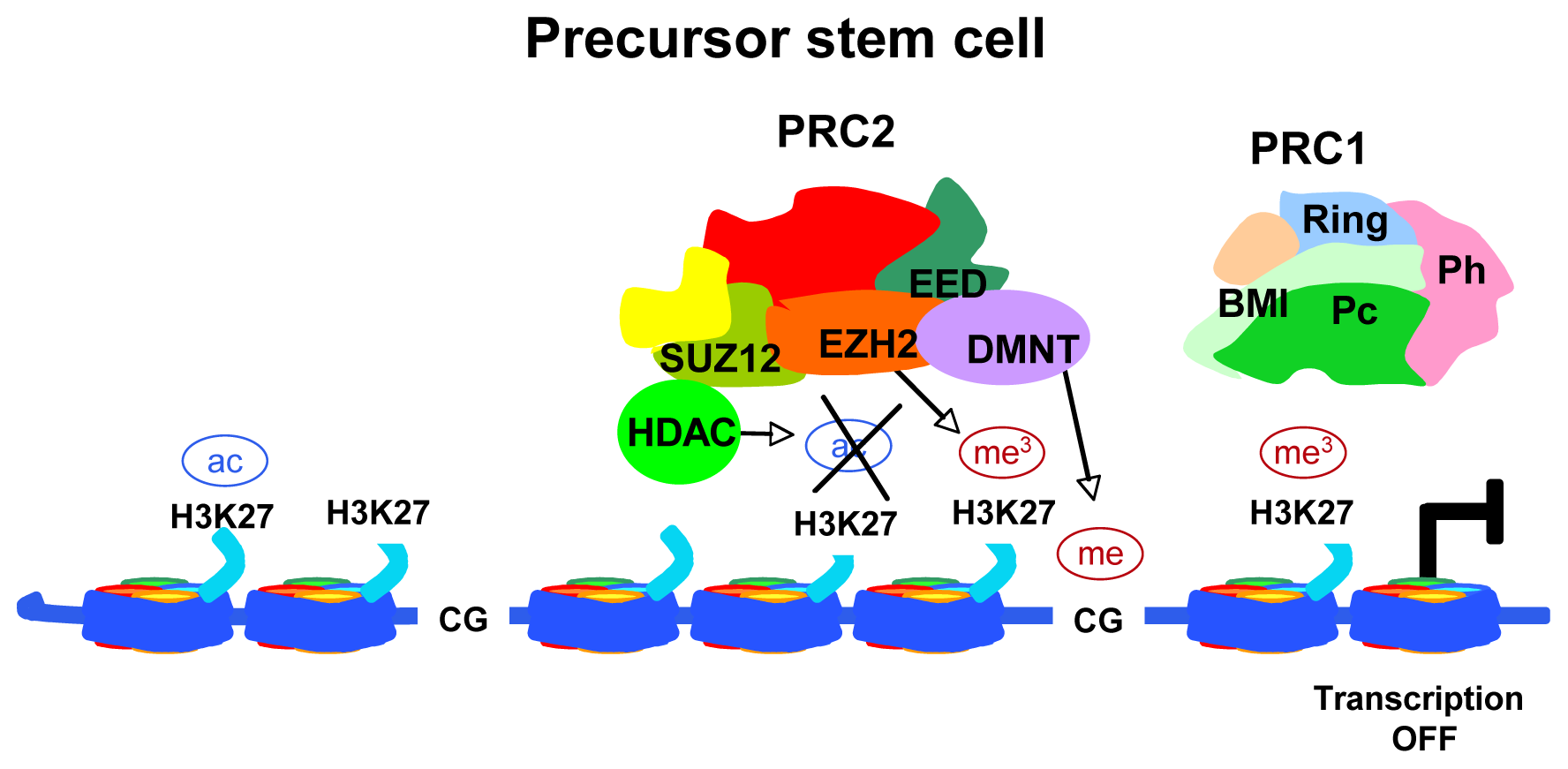
2. What the Term “Epigenetics” Means
3. The Small Non-Coding RNAs, “microRNAs”
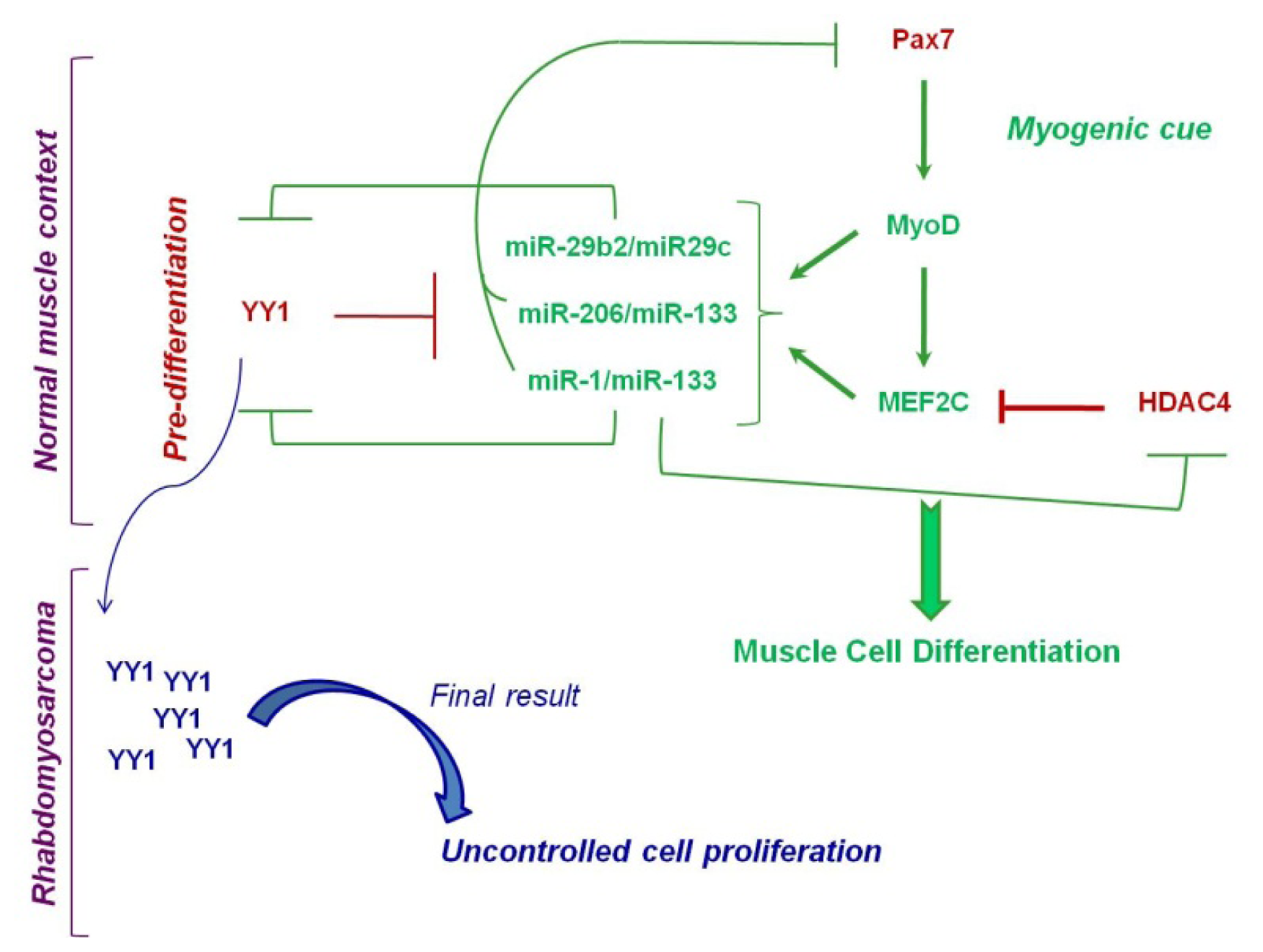
4. Rhabdomyosarcoma
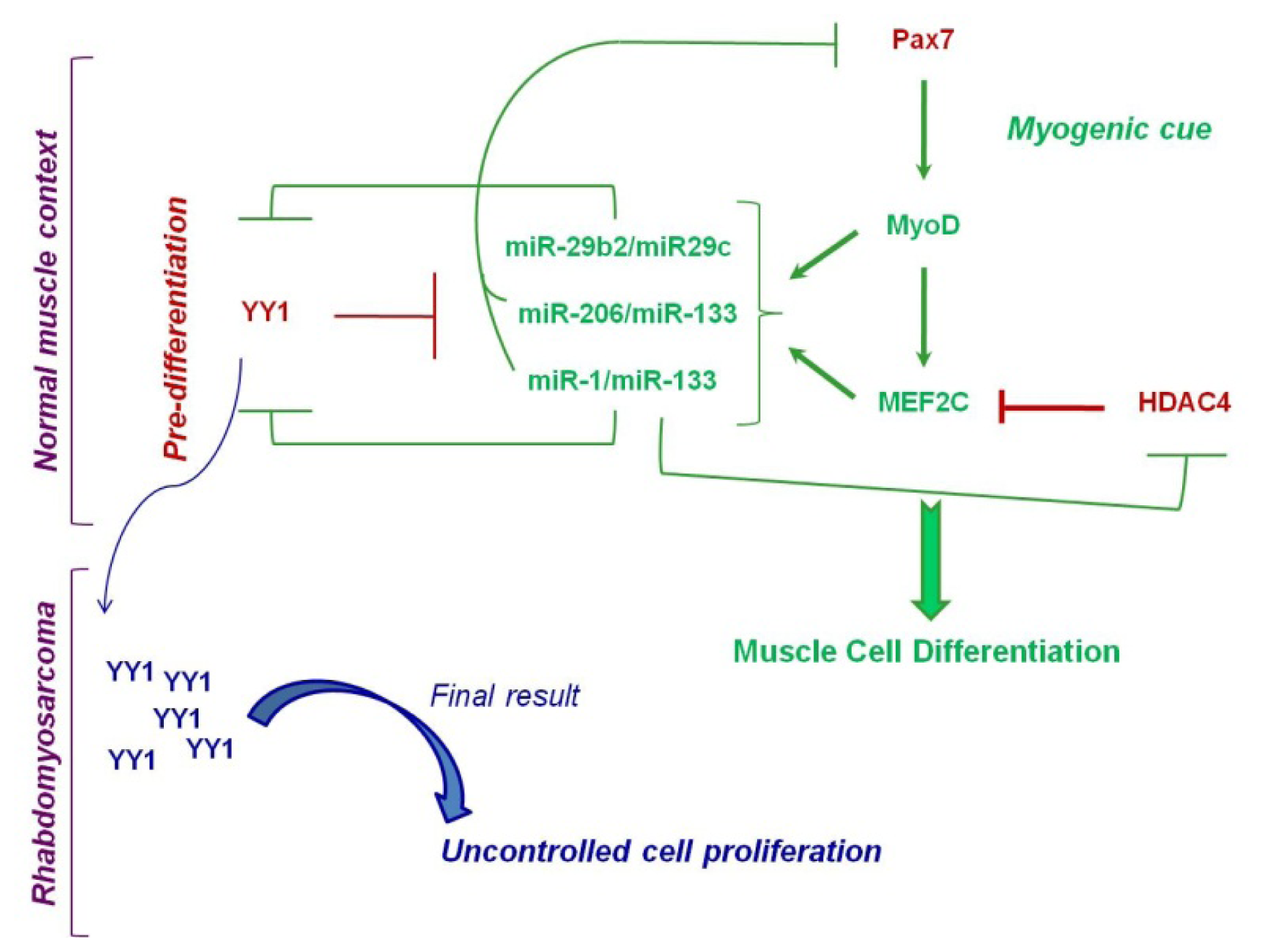
4.1. Epigenetically Deregulated miRNAs in Rhabdomyosarcoma
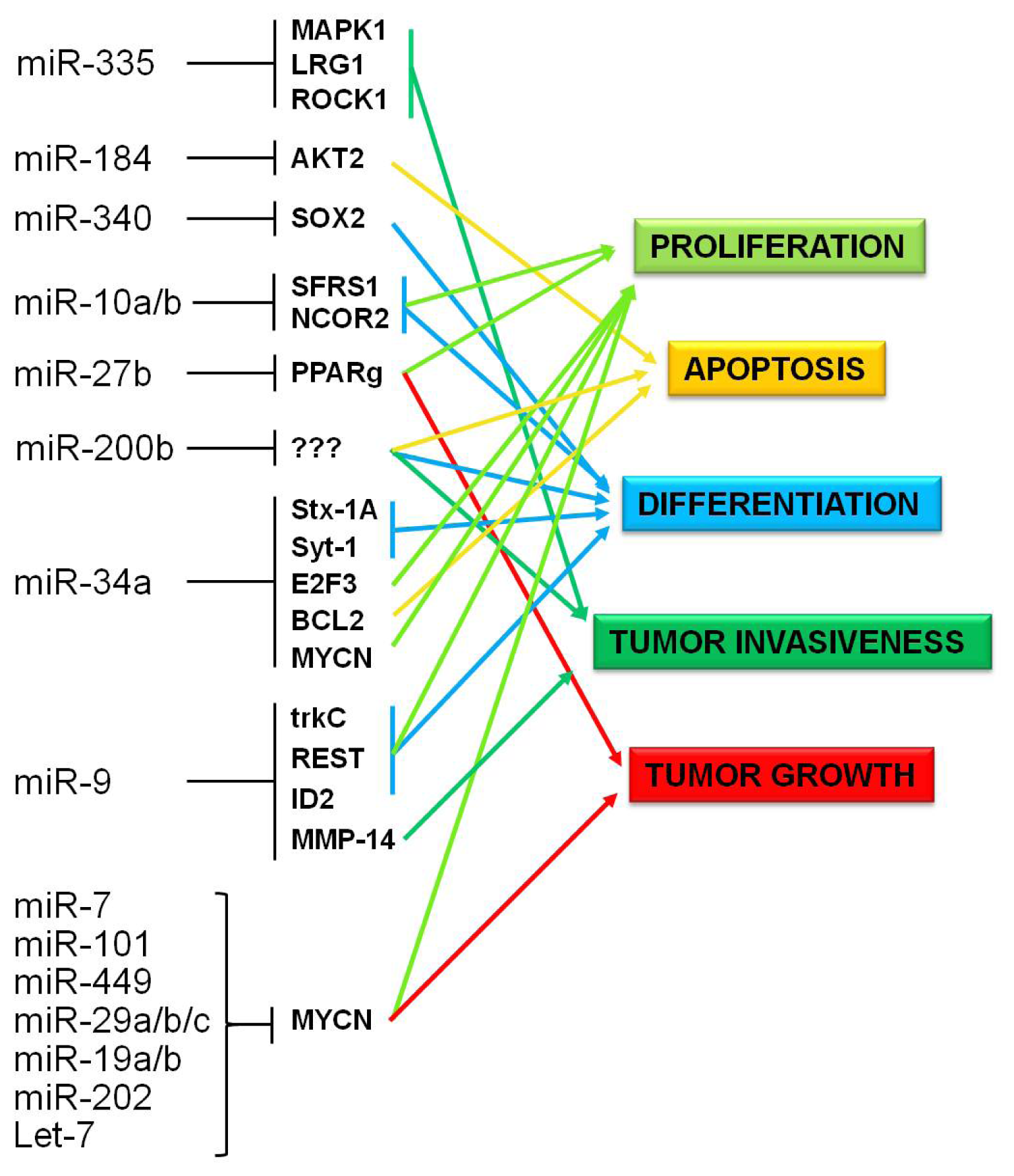
5. Neuroblastoma
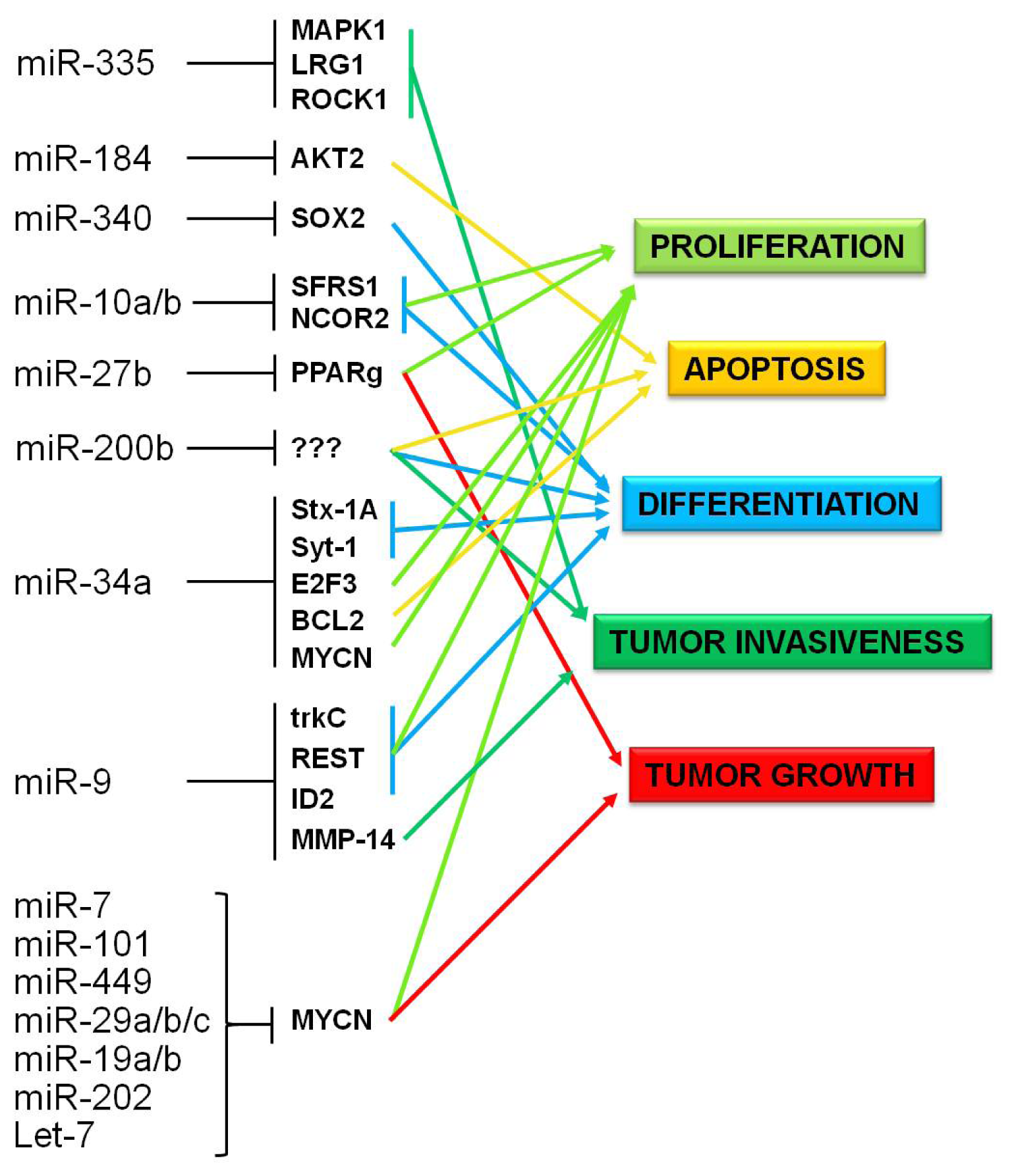
5.1. Epigenetically Deregulated miRNAs in Neuroblastoma
6. Translational Perspectives and Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Iorio, M.V.; Croce, C.M. MicroRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar]
- Crippa, S.; Cassano, M.; Sampaolesi, M. Role of miRNAs in muscle stem cell biology: proliferation, differentiation and death. Curr. Pharmaceut. Des 2012, 18, 1718–1729. [Google Scholar]
- Mondol, V.; Pasquinelli, A.E. Let’s make it happen: the role of let-7 microRNA in development. Curr. Top. Dev. Biol 2012, 99, 1–30. [Google Scholar]
- Mansfield, J.H.; McGlinn, E. Evolution, expression, and developmental function of Hox-embedded miRNAs. Curr. Top. Dev. Biol 2012, 99, 31–57. [Google Scholar]
- Sokol, N.S. The role of microRNAs in muscle development. Curr. Top. Dev. Biol 2012, 99, 59–78. [Google Scholar]
- Cochella, L.; Hobert, O. Diverse functions of microRNAs in nervous system development. Curr. Top. Dev. Biol 2012, 99, 115–143. [Google Scholar]
- O’Connell, R.M.; Baltimore, D. MicroRNAs and hematopoietic cell development. Curr. Top. Dev. Biol 2012, 99, 145–174. [Google Scholar]
- Boettger, T.; Braun, T. A new level of complexity: The role of microRNAs in cardiovascular development. Circ. Res 2012, 110, 1000–1013. [Google Scholar]
- Bell, J.T.; Spector, T.D. A twin approach to unraveling epigenetics. Trends Genet 2011, 27, 116–125. [Google Scholar]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol 2005, 2, S4–S11. [Google Scholar]
- Martin-Subero, J.I. How epigenomics brings phenotype into being. Pediatr. Endocrinol. Rev 2011, 9, 506–510. [Google Scholar]
- Richly, H.; Aloia, L.; Di Croce, L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death. Dis 2011, 2, e204. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev 2011, 12, 861–874. [Google Scholar]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar]
- Duursma, A.M.; Kedde, M.; Schrier, M.; le Sage, C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004, 23, 4051–4060. [Google Scholar]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol 2006, 13, 1097–1101. [Google Scholar]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol 2009, 11, 228–334. [Google Scholar]
- Butcher, J.; Abdou, H.; Morin, K.; Liu, Y. Micromanaging oligodendrocyte differentiation by noncoding RNA: toward a better understanding of the lineage commitment process. J. Neurosci 2009, 29, 5365–5366. [Google Scholar]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol 2009, 10, 116–125. [Google Scholar]
- Wang, Y.; Russell, I.; Chen, C. MicroRNA and stem cell regulation. Curr. Opin. Mol. Ther 2009, 11, 292–298. [Google Scholar]
- Mallanna, S.K.; Rizzino, A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev. Biol 2010, 344, 16–25. [Google Scholar]
- Laurent, L.C. MicroRNAs in embryonic stem cells and early embryonic development. J. Cell Mol. Med 2008, 12, 2181–2188. [Google Scholar]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar]
- Garzon, R.; Fabbri, M.; Cimmino, A.; Calin, G.A.; Croce, C.M. MicroRNA expression and function in cancer. Trends Mol. Med 2006, 12, 580–587. [Google Scholar]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Biophys. Acta 2010, 1799, 694–701. [Google Scholar]
- Fabbri, M.; Calore, F.; Paone, A.; Galli, R.; Calin, G.A. Epigenetic Regulation of miRNAs in Cancer. Adv. Exp.Med. Bio 2013, 754, 137–148. [Google Scholar]
- Chhabra, R.; Dubey, R.; Saini, N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24–2 cluster and its implication in human diseases. Mol. Cancer 2010, 9, 232. [Google Scholar]
- Di Leva, G.; Croce, C.M. Roles of small RNAs in tumor formation. Trends Mol. Med 2010, 16, 257–267. [Google Scholar]
- Perera, R.J.; Ray, A. Epigenetic regulation of miRNA genes and their role in human melanomas. Epigenomics 2012, 4, 81–90. [Google Scholar]
- Ferretti, E.; De Smaele, E.; Miele, E.; Laneve, P.; Po, A.; Pelloni, M.; Paganelli, A.; Di Marcotullio, L.; Caffarelli, E.; Screpanti, I.; et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J 2008, 27, 2616–2627. [Google Scholar]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar]
- Huang, T.H.; Esteller, M. Chromatin remodeling in mammary gland differentiation and breast tumorigenesis. Cold Spring Harb. Perspect. Biol 2010, 2, a004515. [Google Scholar]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 6. [Google Scholar]
- Taulli, R.; Bersani, F.; Ponzetto, C. Micro-orchestrating differentiation in cancer. Cell Cycle 2010, 9, 918–922. [Google Scholar]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar]
- Fontana, L.; Fiori, M.E.; Albini, S.; Cifaldi, L.; Giovinazzi, S.; Forloni, M.; Boldrini, R.; Donfrancesco, A.; Federici, V.; Giacomini, P.; et al. Antagomir-17–5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PloS One 2008, 3, e2236. [Google Scholar]
- Ciarapica, R.; Russo, G.; Verginelli, F.; Raimondi, L.; Donfrancesco, A.; Rota, R.; Giordano, A. Deregulated expression of miR-26a and Ezh2 in rhabdomyosarcoma. Cell Cycle 2009, 8, 172–175. [Google Scholar]
- Taulli, R.; Bersani, F.; Foglizzo, V.; Linari, A.; Vigna, E.; Ladanyi, M.; Tuschl, T.; Ponzetto, C. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J. Clin. Invest. 2009, 119. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Loeb, D.M.; Thornton, K.; Shokek, O. Pediatric soft tissue sarcomas. Surg. Clin. N. Am 2008, 88, 615–627. [Google Scholar]
- Tapscott, S.J.; Thayer, M.J.; Weintraub, H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 1993, 259, 1450–1453. [Google Scholar]
- De Giovanni, C.; Landuzzi, L.; Nicoletti, G.; Lollini, P.L.; Nanni, P. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol 2009, 5, 1449–1475. [Google Scholar]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Takahira, T.; Takahashi, Y.; Tajiri, T.; Taguchi, T.; Suita, S.; Tsuneyoshi, M. Alterations of RB1 gene in embryonal and alveolar rhabdomyosarcoma: special reference to utility of pRB immunoreactivity in differential diagnosis of rhabdomyosarcoma subtype. J. Cancer Res. Clin. Oncol 2008, 134, 1097–1103. [Google Scholar]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J. Clin. Oncol 2001, 19, 3091–3102. [Google Scholar]
- Sorensen, P.H.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J. Clin. Oncol 2002, 20, 2672–2679. [Google Scholar]
- Wachtel, M.; Dettling, M.; Koscielniak, E.; Stegmaier, S.; Treuner, J.; Simon-Klingenstein, K.; Buhlmann, P.; Niggli, F.K.; Schafer, B.W. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2) (q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res 2004, 64, 5539–5545. [Google Scholar]
- Missiaglia, E.; Shepherd, C.J.; Patel, S.; Thway, K.; Pierron, G.; Pritchard-Jones, K.; Renard, M.; Sciot, R.; Rao, P.; Oberlin, O.; et al. MicroRNA-206 expression levels correlate with clinical behaviour of rhabdomyosarcomas. Br. J. Cancer 2010, 102, 1769–1777. [Google Scholar]
- Williamson, D.; Missiaglia, E.; de Reynies, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; Thway, K.; Orbach, D.; Lae, M.; Freneaux, P.; et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol 2010, 28, 2151–2158. [Google Scholar]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.; et al. Molecular classification of rhabdomyosarcoma-genotypic and phenotypic determinants of diagnosis: A report from the Children’s Oncology Group. Am. J. Pathol 2009, 174, 550–564. [Google Scholar]
- Lae, M.; Ahn, E.H.; Mercado, G.E.; Chuai, S.; Edgar, M.; Pawel, B.R.; Olshen, A.; Barr, F.G.; Ladanyi, M. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J. Pathol 2007, 212, 143–151. [Google Scholar]
- Sumegi, J.; Streblow, R.; Frayer, R.W.; Dal Cin, P.; Rosenberg, A.; Meloni-Ehrig, A.; Bridge, J.A. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 2010, 49, 224–236. [Google Scholar]
- Lagha, M.; Sato, T.; Regnault, B.; Cumano, A.; Zuniga, A.; Licht, J.; Relaix, F.; Buckingham, M. Transcriptome analyses based on genetic screens for Pax3 myogenic targets in the mouse embryo. BMC Genom 2010, 11, 696. [Google Scholar]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar]
- Rota, R.; Ciarapica, R.; Giordano, A.; Miele, L.; Locatelli, F. MicroRNAs in rhabdomyosarcoma: pathogenetic implications and translational potentiality. Mol. Cancer 2011, 10, 120. [Google Scholar] [Green Version]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar]
- Subramanian, S.; Kartha, R.V. MicroRNA-mediated gene regulations in human sarcomas. Cell Mol. Life Sci 2012, 69, 3571–3585. [Google Scholar]
- Gagan, J.; Dey, B.K.; Dutta, A. MicroRNAs regulate and provide robustness to the myogenic transcriptional network. Curr. Opin. Pharmacol 2012, 12, 383–388. [Google Scholar]
- Lu, L.; Zhou, L.; Chen, E.Z.; Sun, K.; Jiang, P.; Wang, L.; Su, X.; Sun, H.; Wang, H. A Novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS One 2012, 7, e27596. [Google Scholar]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar]
- Dey, B.K.; Gagan, J.; Dutta, A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol. Cell. Biol 2010, 31, 203–214. [Google Scholar]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet 2006, 38, 228–233. [Google Scholar]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar]
- Hecker, R.M.; Amstutz, R.A.; Wachtel, M.; Walter, D.; Niggli, F.K.; Schafer, B.W. p21 Downregulation is an important component of PAX3/FKHR oncogenicity and its reactivation by HDAC inhibitors enhances combination treatment. Oncogene 2010, 29, 3942–3952. [Google Scholar] [Green Version]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Monkemeyer, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor effects of a combined 5-aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res 2009, 69, 887–895. [Google Scholar]
- Li, Z.; Hassan, M.Q.; Jafferji, M.; Aqeilan, R.I.; Garzon, R.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem 2009, 284, 15676–15684. [Google Scholar]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar]
- Huang, M.B.; Xu, H.; Xie, S.J.; Zhou, H.; Qu, L.H. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS One 2011, 6, e29173. [Google Scholar]
- Olson, E.N.; Spizz, G.; Tainsky, M.A. The oncogenic forms of N-ras or H-ras prevent skeletal myoblast differentiation. Mol. Cell. Biol 1987, 7, 2104–2111. [Google Scholar]
- Liu, J.; Luo, X.J.; Xiong, A.W.; Zhang, Z.D.; Yue, S.; Zhu, M.S.; Cheng, S.Y. MicroRNA-214 promotes myogenic differentiation by facilitating exit from mitosis via down-regulation of proto-oncogene N-ras. J. Biol. Chem 2010, 285, 26599–26607. [Google Scholar]
- Wang, H.; Hertlein, E.; Bakkar, N.; Sun, H.; Acharyya, S.; Wang, J.; Carathers, M.; Davuluri, R.; Guttridge, D.C. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell. Biol 2007, 27, 4374–4387. [Google Scholar]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 2004, 18, 2627–2638. [Google Scholar]
- Subramanian, S.; Lui, W.O.; Lee, C.H.; Espinosa, I.; Nielsen, T.O.; Heinrich, M.C.; Corless, C.L.; Fire, A.Z.; van de Rijn, M. MicroRNA expression signature of human sarcomas. Oncogene 2008, 27, 2015–2026. [Google Scholar]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar]
- Amodio, N.; Leotta, M.; Bellizzi, D.; Di Martino, M.T.; D’Aquila, P.; Lionetti, M.; Fabiani, F.; Leone, E.; Gulla, A.M.; Passarino, G.; et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012, 3, 1246–1258. [Google Scholar]
- Hoehner, J.C.; Gestblom, C.; Hedborg, F.; Sandstedt, B.; Olsen, L.; Pahlman, S. A developmental model of neuroblastoma: differentiating stroma-poor tumors’ progress along an extra-adrenal chromaffin lineage. Lab. Investig 1996, 75, 659–675. [Google Scholar]
- Cooper, M.J.; Hutchins, G.M.; Cohen, P.S.; Helman, L.J.; Mennie, R.J.; Israel, M.A. Human neuroblastoma tumor cell lines correspond to the arrested differentiation of chromaffin adrenal medullary neuroblasts. Cell Growth Differ 1990, 1, 149–159. [Google Scholar]
- Gaetano, C.; Matsumoto, K.; Thiele, C.J. In vitro activation of distinct molecular and cellular phenotypes after induction of differentiation in a human neuroblastoma cell line. Cancer Res 1992, 52, 4402–4407. [Google Scholar]
- Brodeur, G.M. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar]
- Van Roy, N.; Laureys, G.; Cheng, N.C.; Willem, P.; Opdenakker, G.; Versteeg, R.; Speleman, F. 1;17 translocations and other chromosome 17 rearrangements in human primary neuroblastoma tumors and cell lines. Genes. Chromosomes Cancer 1994, 10, 103–114. [Google Scholar]
- Fong, C.T.; Dracopoli, N.C.; White, P.S.; Merrill, P.T.; Griffith, R.C.; Housman, D.E.; Brodeur, G.M. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: correlation with N-myc amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 3753–3757. [Google Scholar]
- Janoueix-Lerosey, I.; Novikov, E.; Monteiro, M.; Gruel, N.; Schleiermacher, G.; Loriod, B.; Nguyen, C.; Delattre, O. Gene expression profiling of 1p35–36 genes in neuroblastoma. Oncogene 2004, 23, 5912–5922. [Google Scholar]
- Fujita, T.; Igarashi, J.; Okawa, E.R.; Gotoh, T.; Manne, J.; Kolla, V.; Kim, J.; Zhao, H.; Pawel, B.R.; London, W.B.; et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J. Natl. Cancer Inst. 2008, 100, 940–949. [Google Scholar]
- Saito-Ohara, F.; Imoto, I.; Inoue, J.; Hosoi, H.; Nakagawara, A.; Sugimoto, T.; Inazawa, J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res 2003, 63, 1876–1883. [Google Scholar]
- Van Noesel, M.M.; Versteeg, R. Pediatric neuroblastomas: Genetic and epigenetic “danse macabre”. Gene 2004, 325, 1–15. [Google Scholar]
- Oppenheimer, O.; Alaminos, M.; Gerald, W.L. Genomic medicine and neuroblastoma. Expert. Rev. Mol. Diagn 2003, 3, 39–54. [Google Scholar]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., II; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar]
- Osajima-Hakomori, Y.; Miyake, I.; Ohira, M.; Nakagawara, A.; Nakagawa, A.; Sakai, R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am. J. Pathol 2005, 167, 213–222. [Google Scholar]
- Lim, M.S.; Carlson, M.L.; Crockett, D.K.; Fillmore, G.C.; Abbott, D.R.; Elenitoba-Johnson, O.F.; Tripp, S.R.; Rassidakis, G.Z.; Medeiros, L.J.; Szankasi, P.; et al. The proteomic signature of NPM/ALK reveals deregulation of multiple cellular pathways. Blood 2009, 114, 1585–1595. [Google Scholar]
- Decock, A.; Ongenaert, M.; Vandesompele, J.; Speleman, F. Neuroblastoma epigenetics: From candidate gene approaches to genome-wide screenings. Epigenetics 2011, 6, 962–790. [Google Scholar]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med 2000, 6, 529–535. [Google Scholar]
- Astuti, D.; Agathanggelou, A.; Honorio, S.; Dallol, A.; Martinsson, T.; Kogner, P.; Cummins, C.; Neumann, H.P.; Voutilainen, R.; Dahia, P.; et al. RASSF1A promoter region CpG island hypermethylation in phaeochromocytomas and neuroblastoma tumours. Oncogene 2001, 20, 7573–7577. [Google Scholar]
- Murphy, D.M.; Buckley, P.G.; Bryan, K.; Das, S.; Alcock, L.; Foley, N.H.; Prenter, S.; Bray, I.; Watters, K.M.; Higgins, D.; et al. Global MYCN transcription factor binding analysis in neuroblastoma reveals association with distinct E-box motifs and regions of DNA hypermethylation. PLoS One 2009, 4, e8154. [Google Scholar]
- Buckley, P.G.; Alcock, L.; Bryan, K.; Bray, I.; Schulte, J.H.; Schramm, A.; Eggert, A.; Mestdagh, P.; De Preter, K.; Vandesompele, J.; et al. Chromosomal and microRNA expression patterns reveal biologically distinct subgroups of 11q-neuroblastoma. Clin. Cancer Res 2010, 16, 2971–2978. [Google Scholar]
- Caren, H.; Djos, A.; Nethander, M.; Sjoberg, R.M.; Kogner, P.; Enstrom, C.; Nilsson, S.; Martinsson, T. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 2011, 11, 66. [Google Scholar] [Green Version]
- Chavali, P.L.; Funa, K.; Chavali, S. Cis-regulation of microRNA expression by scaffold/matrix-attachment regions. Nucleic Acids Res 2011, 39, 6908–6918. [Google Scholar]
- Angrisano, T.; Sacchetti, S.; Natale, F.; Cerrato, A.; Pero, R.; Keller, S.; Peluso, S.; Perillo, B.; Avvedimento, V.E.; Fusco, A.; et al. Chromatin and DNA methylation dynamics during retinoic acid-induced RET gene transcriptional activation in neuroblastoma cells. Nucleic Acids Res 2011, 39, 1993–2006. [Google Scholar]
- Das, S.; Foley, N.; Bryan, K.; Watters, K.M.; Bray, I.; Murphy, D.M.; Buckley, P.G.; Stallings, R.L. MicroRNA mediates DNA demethylation events triggered by retinoic acid during neuroblastoma cell differentiation. Cancer Res 2010, 70, 7874–7881. [Google Scholar]
- Das, S.; Bryan, K.; Buckley, P.G.; Piskareva, O.; Bray, I.M.; Foley, N.; Ryan, J.; Lynch, J.; Creevey, L.; Fay, J.; et al. Modulation of neuroblastoma disease pathogenesis by an extensive network of epigenetically regulated microRNAs. Oncogene 2012. [Google Scholar] [CrossRef]
- Stallings, R.L.; Foley, N.H.; Bray, I.M.; Das, S.; Buckley, P.G. MicroRNA and DNA methylation alterations mediating retinoic acid induced neuroblastoma cell differentiation. Semin. Cancer Biol 2011, 21, 283–290. [Google Scholar]
- Meseguer, S.; Mudduluru, G.; Escamilla, J.M.; Allgayer, H.; Barettino, D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J. Biol. Chem 2011, 286, 4150–4164. [Google Scholar]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol 2007, 14, 185–193. [Google Scholar]
- Foley, N.H.; Bray, I.; Watters, K.M.; Das, S.; Bryan, K.; Bernas, T.; Prehn, J.H.; Stallings, R.L. MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell Death Differ 2011, 18, 1089–1098. [Google Scholar]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar]
- Buechner, J.; Tomte, E.; Haug, B.H.; Henriksen, J.R.; Lokke, C.; Flaegstad, T.; Einvik, C. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br. J. Cancer 2011, 105, 296–303. [Google Scholar]
- Molenaar, J.J.; Domingo-Fernandez, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet 2012, 44, 1199–1206. [Google Scholar]
- Lynch, J.; Fay, J.; Meehan, M.; Bryan, K.; Watters, K.M.; Murphy, D.M.; Stallings, R.L. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-beta signalling pathway. Carcinogenesis 2012, 33, 976–985. [Google Scholar]
- Foley, N.H.; Bray, I.M.; Tivnan, A.; Bryan, K.; Murphy, D.M.; Buckley, P.G.; Ryan, J.; O’Meara, A.; O’Sullivan, M.; Stallings, R.L. MicroRNA-184 inhibits neuroblastoma cell survival through targeting the serine/threonine kinase AKT2. Mol. Cancer 2010, 9, 83. [Google Scholar]
- Tivnan, A.; Foley, N.H.; Tracey, L.; Davidoff, A.M.; Stallings, R.L. MicroRNA-184-mediated inhibition of tumour growth in an orthotopic murine model of neuroblastoma. Anticancer Res 2010, 30, 4391–4395. [Google Scholar]
- Bray, I.; Tivnan, A.; Bryan, K.; Foley, N.H.; Watters, K.M.; Tracey, L.; Davidoff, A.M.; Stallings, R.L. MicroRNA-542–5p as a novel tumor suppressor in neuroblastoma. Cancer Lett 2011, 303, 56–64. [Google Scholar]
- Schulte, J.H.; Marschall, T.; Martin, M.; Rosenstiel, P.; Mestdagh, P.; Schlierf, S.; Thor, T.; Vandesompele, J.; Eggert, A.; Schreiber, S.; et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res 2010, 38, 5919–5928. [Google Scholar]
- Schulte, J.H.; Schowe, B.; Mestdagh, P.; Kaderali, L.; Kalaghatgi, P.; Schlierf, S.; Vermeulen, J.; Brockmeyer, B.; Pajtler, K.; Thor, T.; et al. Accurate prediction of neuroblastoma outcome based on miRNA expression profiles. Int. J. Cancer 2010, 127, 2374–2385. [Google Scholar]
- Laneve, P.; Di Marcotullio, L.; Gioia, U.; Fiori, M.E.; Ferretti, E.; Gulino, A.; Bozzoni, I.; Caffarelli, E. The interplay between microRNAs and the neurotrophin receptor tropomyosin-related kinase C controls proliferation of human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 2007, 104, 7957–7962. [Google Scholar]
- Laneve, P.; Gioia, U.; Andriotto, A.; Moretti, F.; Bozzoni, I.; Caffarelli, E. A minicircuitry involving REST and CREB controls miR-9–2 expression during human neuronal differentiation. Nucleic Acids Res 2010, 38, 6895–6905. [Google Scholar]
- Annibali, D.; Gioia, U.; Savino, M.; Laneve, P.; Caffarelli, E.; Nasi, S. A new module in neural differentiation control: two microRNAs upregulated by retinoic acid, miR-9 and -103, target the differentiation inhibitor ID2. PLoS One 2012, 7, e40269. [Google Scholar]
- Zhang, H.; Qi, M.; Li, S.; Qi, T.; Mei, H.; Huang, K.; Zheng, L.; Tong, Q. MicroRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol. Cancer Therapeut 2012, 11, 1454–1466. [Google Scholar]
- Lee, J.J.; Drakaki, A.; Iliopoulos, D.; Struhl, K. MiR-27b targets PPARgamma to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene 2012, 31, 3818–3825. [Google Scholar]
- Cole, K.A.; Attiyeh, E.F.; Mosse, Y.P.; Laquaglia, M.J.; Diskin, S.J.; Brodeur, G.M.; Maris, J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res 2008, 6, 735–742. [Google Scholar]
- Ragusa, M.; Majorana, A.; Banelli, B.; Barbagallo, D.; Statello, L.; Casciano, I.; Guglielmino, M.R.; Duro, L.R.; Scalia, M.; Magro, G.; et al. MIR152, MIR200B, and MIR338, human positional and functional neuroblastoma candidates, are involved in neuroblast differentiation and apoptosis. J. Mol. Med 2010, 88, 1041–1053. [Google Scholar]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Korner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar]
- Wei, J.S.; Song, Y.K.; Durinck, S.; Chen, Q.R.; Cheuk, A.T.; Tsang, P.; Zhang, Q.; Thiele, C.J.; Slack, A.; Shohet, J.; et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008, 27, 5204–5213. [Google Scholar]
- Agostini, M.; Tucci, P.; Steinert, J.R.; Shalom-Feuerstein, R.; Rouleau, M.; Aberdam, D.; Forsythe, I.D.; Young, K.W.; Ventura, A.; Concepcion, C.P.; et al. MicroRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc. Natl. Acad. Sci. USA 2011, 108, 21099–21104. [Google Scholar]
- Gougelet, A.; Perez, J.; Pissaloux, D.; Besse, A.; Duc, A.; Decouvelaere, A.V.; Ranchere-Vince, D.; Blay, J.Y.; Alberti, L. miRNA Profiling: How to Bypass the Current Difficulties in the Diagnosis and Treatment of Sarcomas. Sarcoma 2011, 2011, 460650. [Google Scholar]
- Lin, R.J.; Lin, Y.C.; Chen, J.; Kuo, H.H.; Chen, Y.Y.; Diccianni, M.B.; London, W.B.; Chang, C.H.; Yu, A.L. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010, 70, 7841–7850. [Google Scholar]
- De Preter, K.; Mestdagh, P.; Vermeulen, J.; Zeka, F.; Naranjo, A.; Bray, I.; Castel, V.; Chen, C.; Drozynska, E.; Eggert, A.; et al. MiRNA expression profiling enables risk stratification in archived and fresh neuroblastoma tumor samples. Clin. Cancer Res 2011, 17, 7684–7692. [Google Scholar]
- Cortez, M.A.; Calin, G.A. MicroRNA identification in plasma and serum: A new tool to diagnose and monitor diseases. Expert Opin. Biol. Ther 2009, 9, 703–711. [Google Scholar]
- Miyachi, M.; Tsuchiya, K.; Yoshida, H.; Yagyu, S.; Kikuchi, K.; Misawa, A.; Iehara, T.; Hosoi, H. Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochem. Biophys. Res. Commun 2010, 400, 89–93. [Google Scholar]
- Hogrefe, R.I.; Lebedev, A.V.; Zon, G.; Pirollo, K.F.; Rait, A.; Zhou, Q.; Yu, W.; Chang, E.H. Chemically modified short interfering hybrids (siHYBRIDS): nanoimmunoliposome delivery in vitro and in vivo for RNAi of HER-2. Nucleos. Nucleot. Nucleic Acids 2006, 25, 889–907. [Google Scholar]
- Pirollo, K.F.; Rait, A.; Zhou, Q.; Hwang, S.H.; Dagata, J.A.; Zon, G.; Hogrefe, R.I.; Palchik, G.; Chang, E.H. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res 2007, 67, 2938–2943. [Google Scholar]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar]
- Anand, S.; Majeti, B.K.; Acevedo, L.M.; Murphy, E.A.; Mukthavaram, R.; Scheppke, L.; Huang, M.; Shields, D.J.; Lindquist, J.N.; Lapinski, P.E.; et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med 2010, 16, 909–914. [Google Scholar]
- Su, J.; Baigude, H.; McCarroll, J.; Rana, T.M. Silencing microRNA by interfering nanoparticles in mice. Nucleic Acids Res 2011, 39, e38. [Google Scholar]
- Almeida, M.I.; Reis, R.M.; Calin, G.A. MicroRNA history: discovery, recent applications, and next frontiers. Mutat. Res 2011, 717, 1–8. [Google Scholar]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernandez, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS One 2012, 7, e38129. [Google Scholar]
- Issa, J.P.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar]
- Vigil, C.E.; Martin-Santos, T.; Garcia-Manero, G. Safety and efficacy of azacitidine in myelodysplastic syndromes. Drug Des. Devel. Ther 2010, 4, 221–229. [Google Scholar]
- Candelaria, M.; Herrera, A.; Labardini, J.; Gonzalez-Fierro, A.; Trejo-Becerril, C.; Taja-Chayeb, L.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Arias-Bofill, D.; Vidal, S.; et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann. Hematol 2010, 90, 379–387. [Google Scholar]
- Fu, S.; Hu, W.; Iyer, R.; Kavanagh, J.J.; Coleman, R.L.; Levenback, C.F.; Sood, A.K.; Wolf, J.K.; Gershenson, D.M.; Markman, M.; et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2010, 117, 1661–1669. [Google Scholar]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011, 1, 598–607. [Google Scholar]
- Abele, R.; Clavel, M.; Dodion, P.; Bruntsch, U.; Gundersen, S.; Smyth, J.; Renard, J.; van Glabbeke, M.; Pinedo, H.M. The EORTC Early Clinical Trials Cooperative Group experience with 5-aza-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur. J. Cancer Clin. Oncol 1987, 23, 1921–1924. [Google Scholar]
- Momparler, R.L.; Bouffard, D.Y.; Momparler, L.F.; Dionne, J.; Belanger, K.; Ayoub, J. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anti-Cancer Drugs 1997, 8, 358–368. [Google Scholar]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar]
- Shoemaker, A.R.; Mitten, M.J.; Adickes, J.; Ackler, S.; Refici, M.; Ferguson, D.; Oleksijew, A.; O’Connor, J.M.; Wang, B.; Frost, D.J.; et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin. Cancer Res 2008, 14, 3268–3277. [Google Scholar]
- Jain, H.V.; Meyer-Hermann, M. The molecular basis of synergism between carboplatin and ABT-737 therapy targeting ovarian carcinomas. Cancer Res 2011, 71, 705–715. [Google Scholar]
- Belinsky, S.A.; Klinge, D.M.; Stidley, C.A.; Issa, J.P.; Herman, J.G.; March, T.H.; Baylin, S.B. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res 2003, 63, 7089–7093. [Google Scholar]
- Gore, S.D.; Baylin, S.; Sugar, E.; Carraway, H.; Miller, C.B.; Carducci, M.; Grever, M.; Galm, O.; Dauses, T.; Karp, J.E.; et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 2006, 66, 6361–6369. [Google Scholar]
- Fandy, T.E.; Herman, J.G.; Kerns, P.; Jiemjit, A.; Sugar, E.A.; Choi, S.H.; Yang, A.S.; Aucott, T.; Dauses, T.; Odchimar-Reissig, R.; et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 2009, 114, 2764–2773. [Google Scholar]
- Suva, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 2009, 69, 9211–9218. [Google Scholar]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007, 21, 1050–1063. [Google Scholar]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar]
- Hayden, A.; Johnson, P.W.; Packham, G.; Crabb, S.J. S-adenosylhomocysteine hydrolase inhibition by 3-deazaneplanocin A analogues induces anti-cancer effects in breast cancer cell lines and synergy with both histone deacetylase and HER2 inhibition. Breast Cancer Res. Treat 2010, 102, 109–119. [Google Scholar]
- Yu, Y.; Zeng, P.; Xiong, J.; Liu, Z.; Berger, S.L.; Merlino, G. Epigenetic drugs can stimulate metastasis through enhanced expression of the pro-metastatic Ezrin gene. PLoS One 2010, 5, e12710. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Romania, P.; Bertaina, A.; Bracaglia, G.; Locatelli, F.; Fruci, D.; Rota, R. Epigenetic Deregulation of MicroRNAs in Rhabdomyosarcoma and Neuroblastoma and Translational Perspectives. Int. J. Mol. Sci. 2012, 13, 16554-16579. https://doi.org/10.3390/ijms131216554
Romania P, Bertaina A, Bracaglia G, Locatelli F, Fruci D, Rota R. Epigenetic Deregulation of MicroRNAs in Rhabdomyosarcoma and Neuroblastoma and Translational Perspectives. International Journal of Molecular Sciences. 2012; 13(12):16554-16579. https://doi.org/10.3390/ijms131216554
Chicago/Turabian StyleRomania, Paolo, Alice Bertaina, Giorgia Bracaglia, Franco Locatelli, Doriana Fruci, and Rossella Rota. 2012. "Epigenetic Deregulation of MicroRNAs in Rhabdomyosarcoma and Neuroblastoma and Translational Perspectives" International Journal of Molecular Sciences 13, no. 12: 16554-16579. https://doi.org/10.3390/ijms131216554