Biotransformations Utilizing β-Oxidation Cycle Reactions in the Synthesis of Natural Compounds and Medicines

Abstract
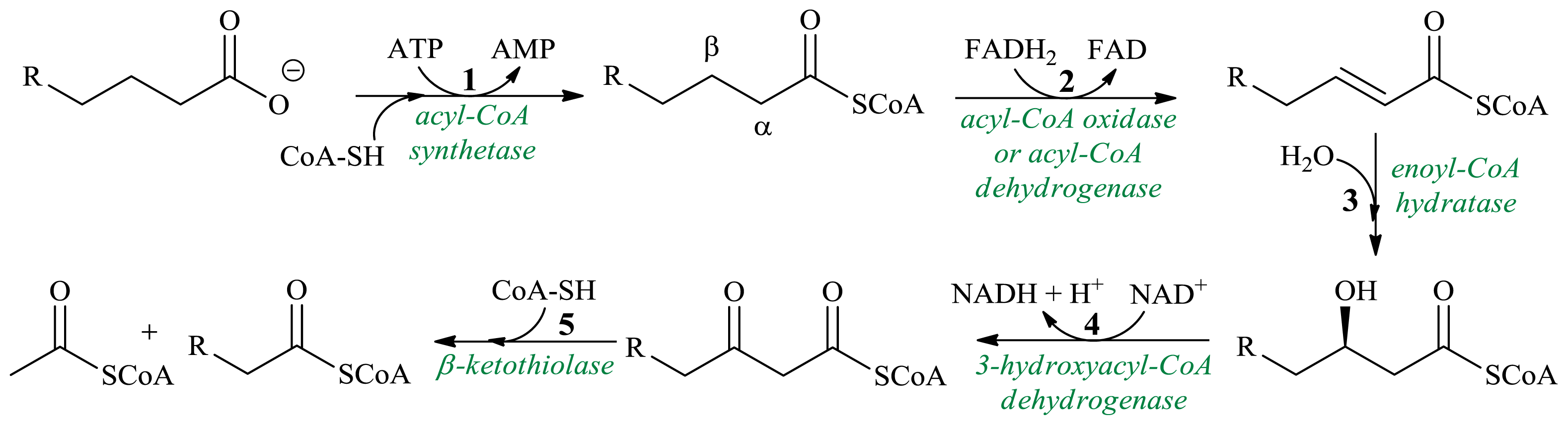
:1. Introduction
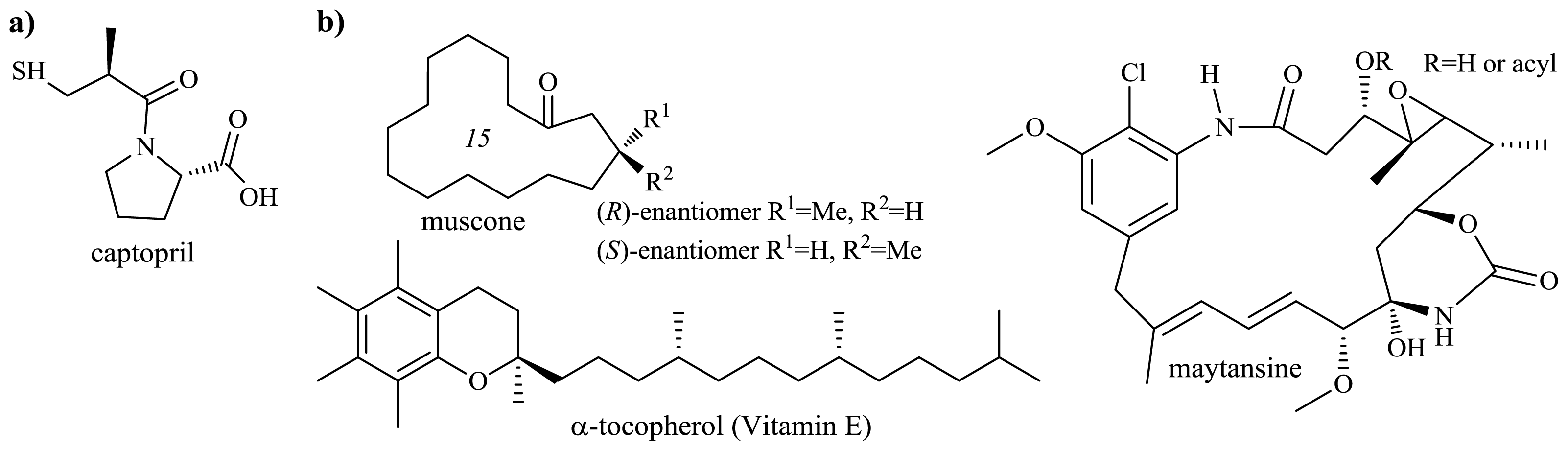
2. Production of Optically Active Compounds
2.1. Syntheses Employing Chiral Stereogenic Centers of a Natural Product
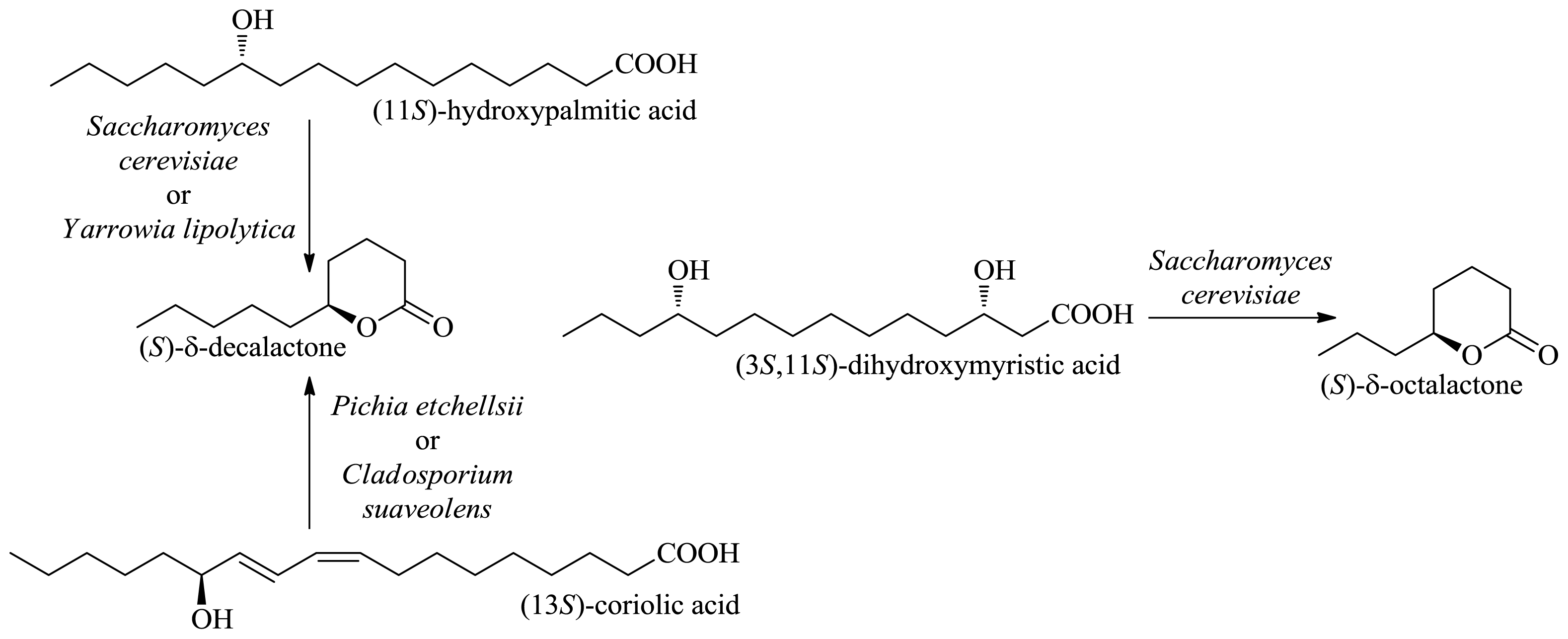
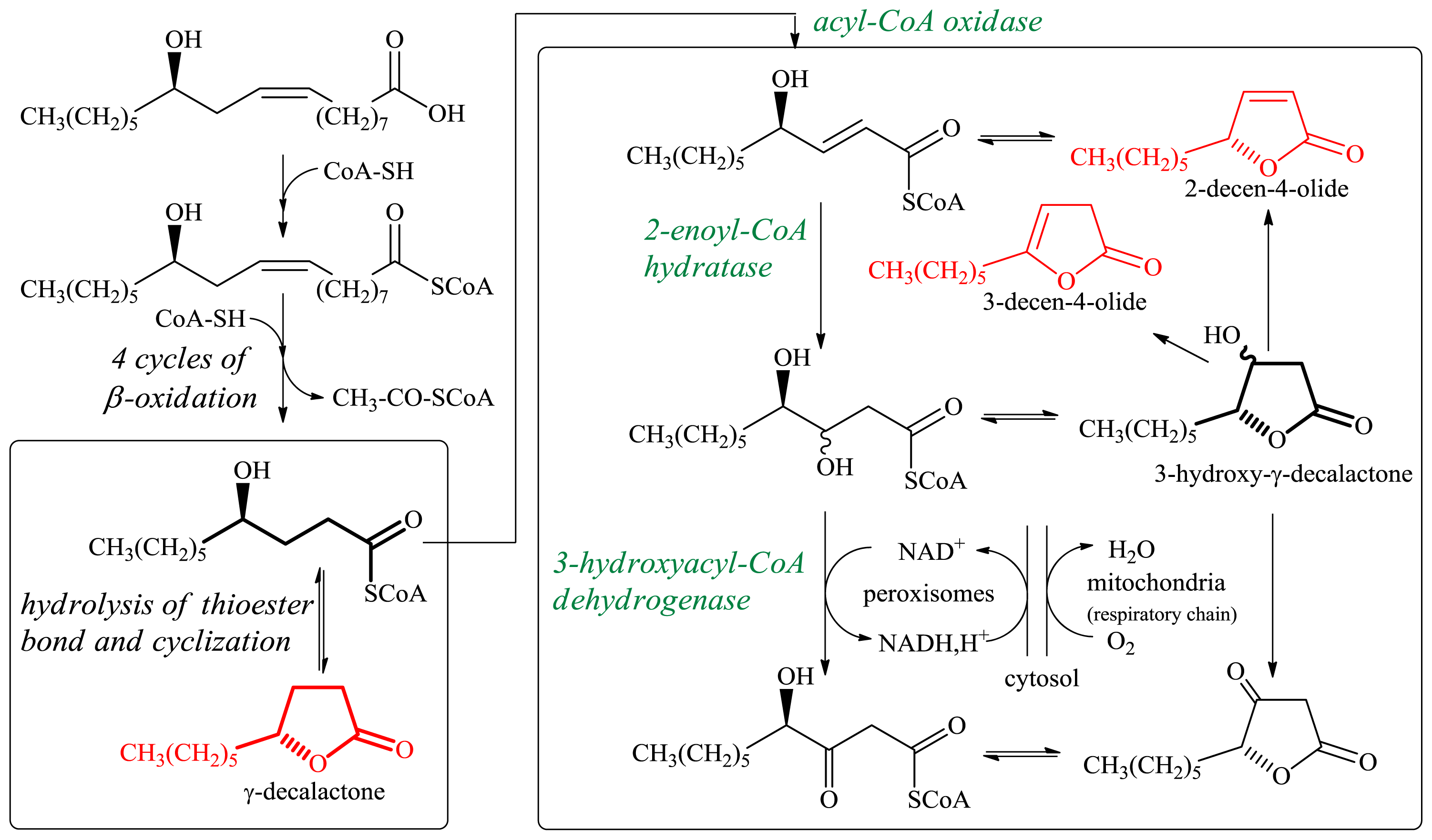
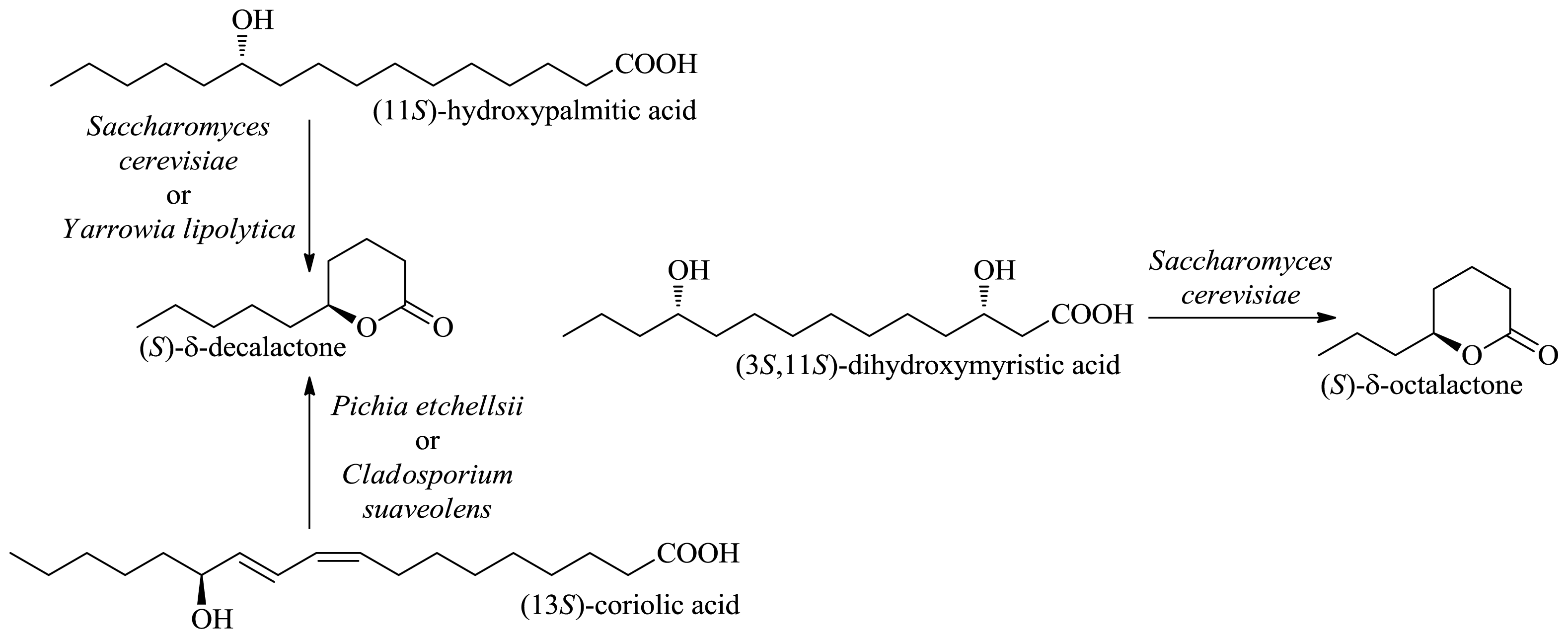
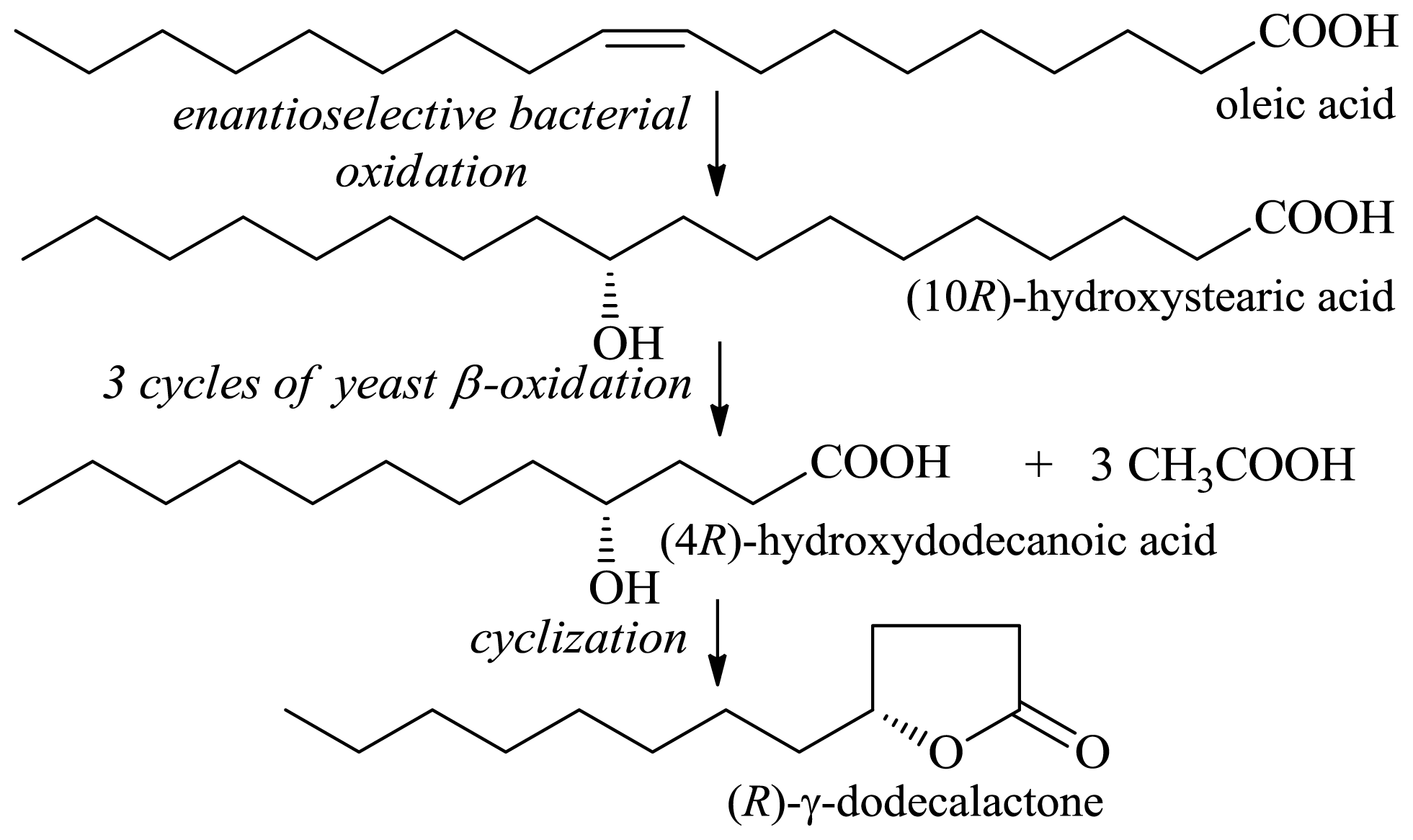
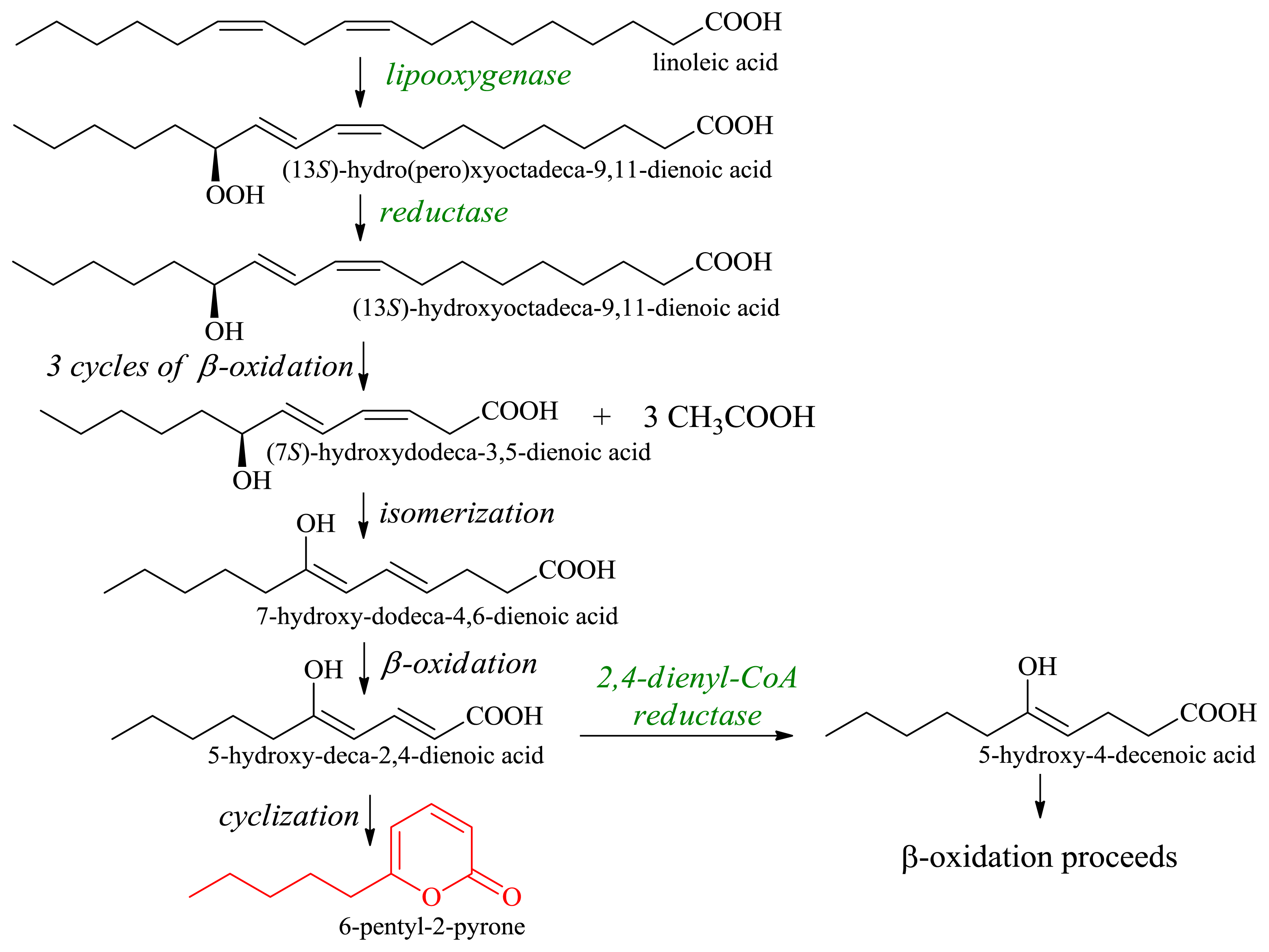
2.1.1. Production of Flavor-Active Lactones
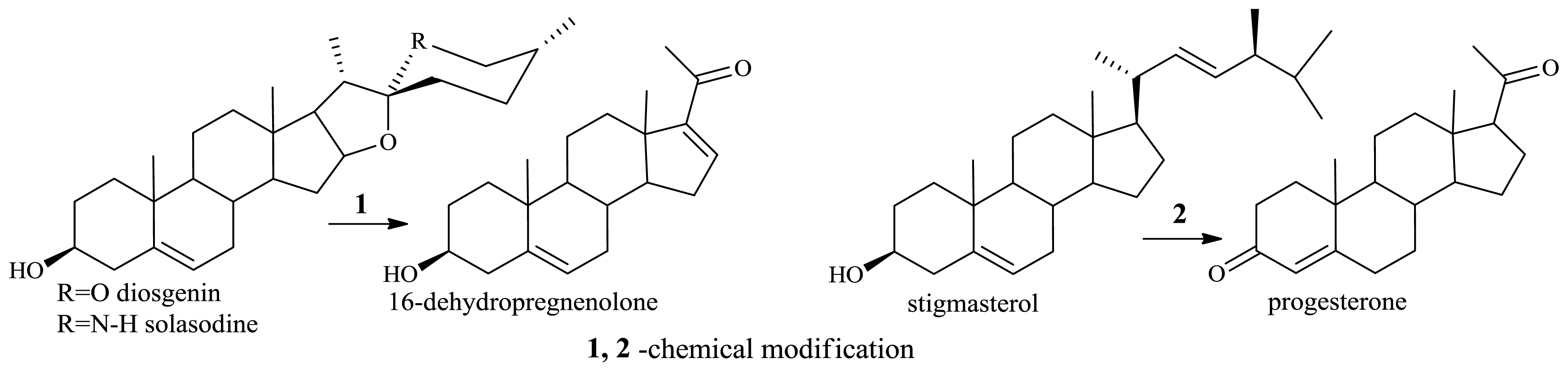
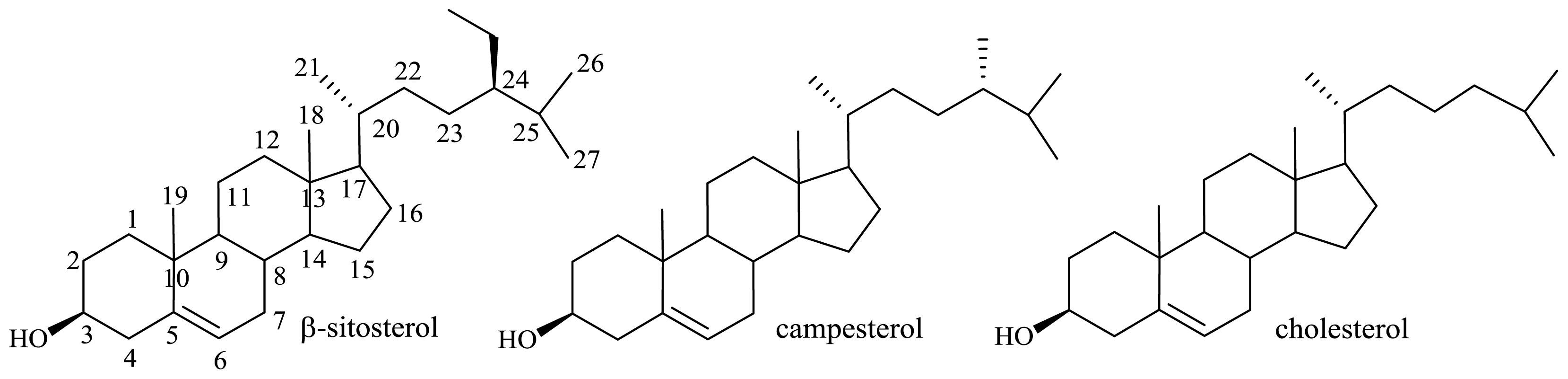
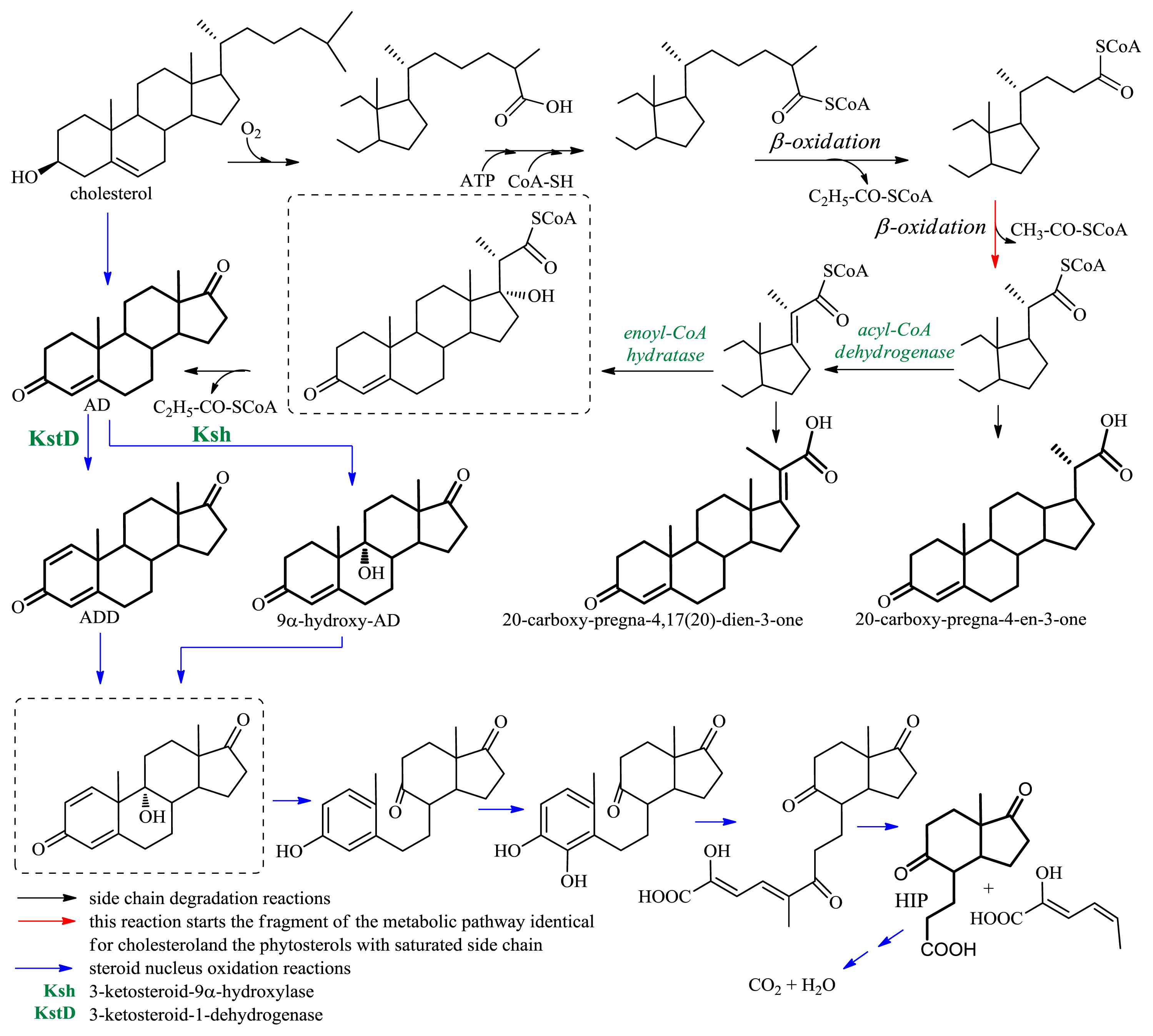
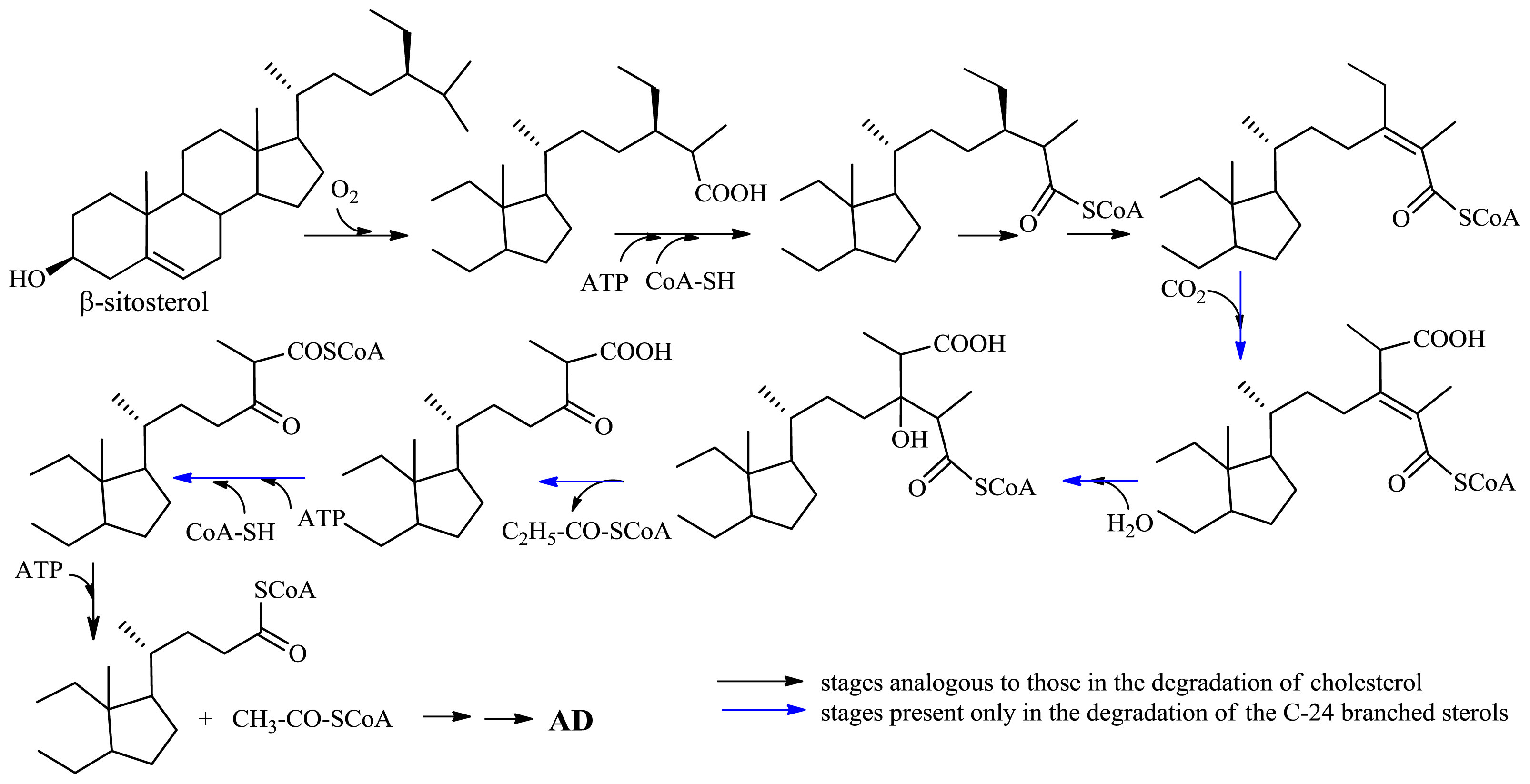
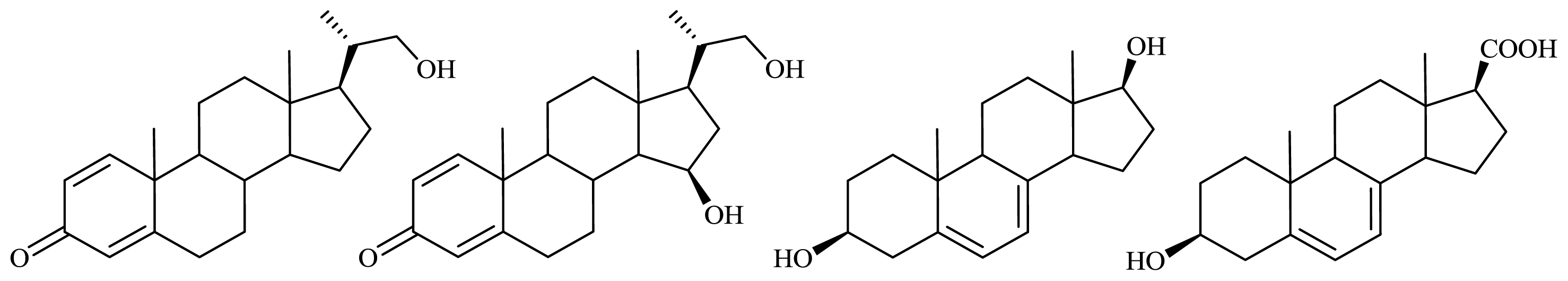
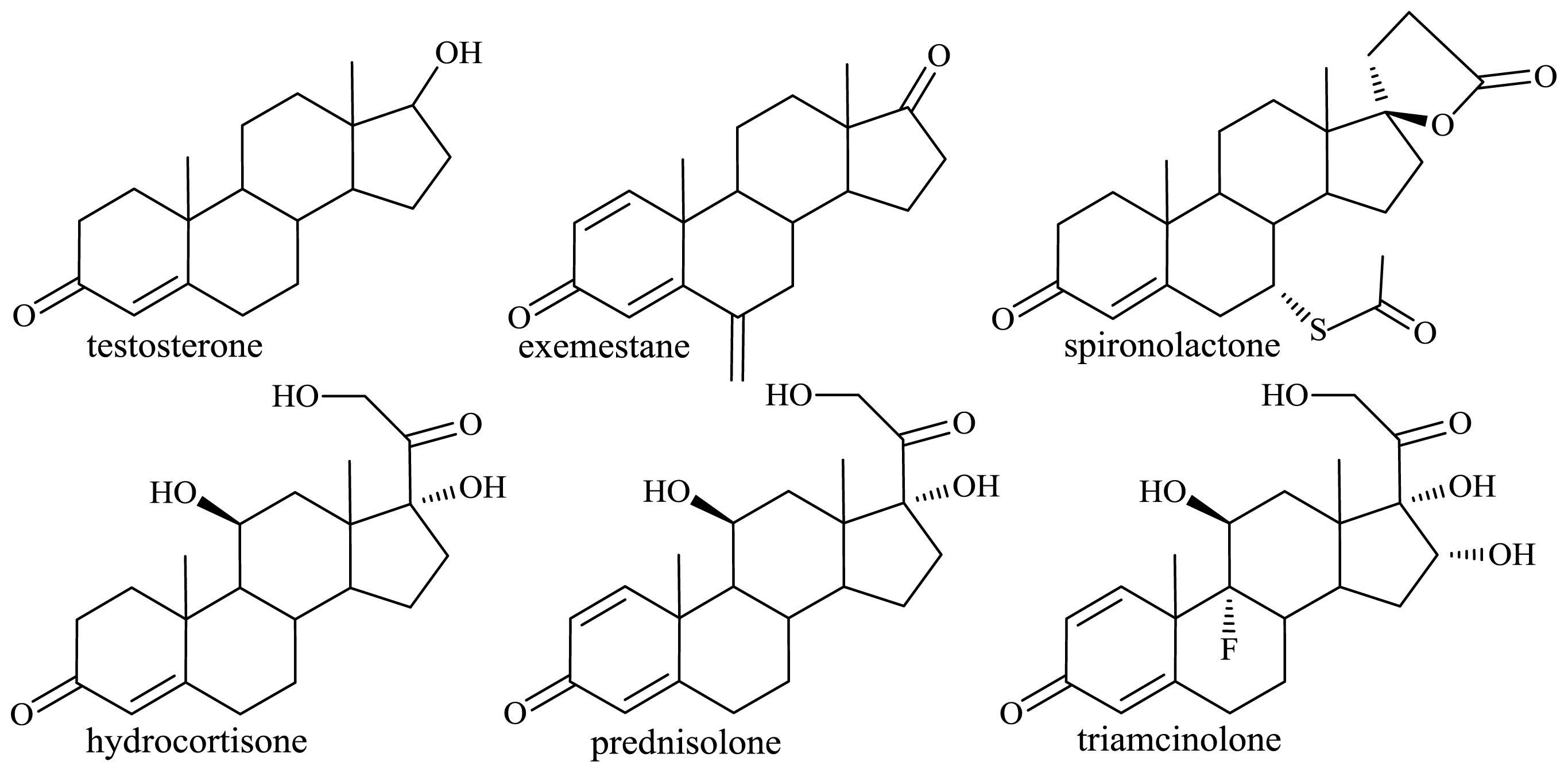
2.1.2. Microbiological Degradation of the Side Chain of Sterols
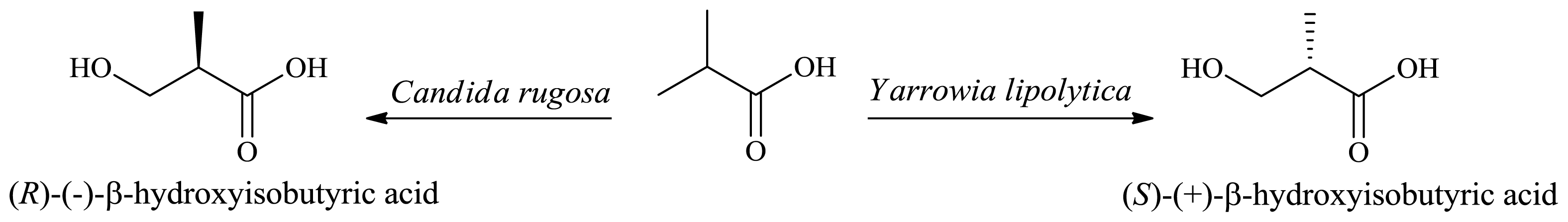
2.2. Production of Optically Active β-Hydroxyacids
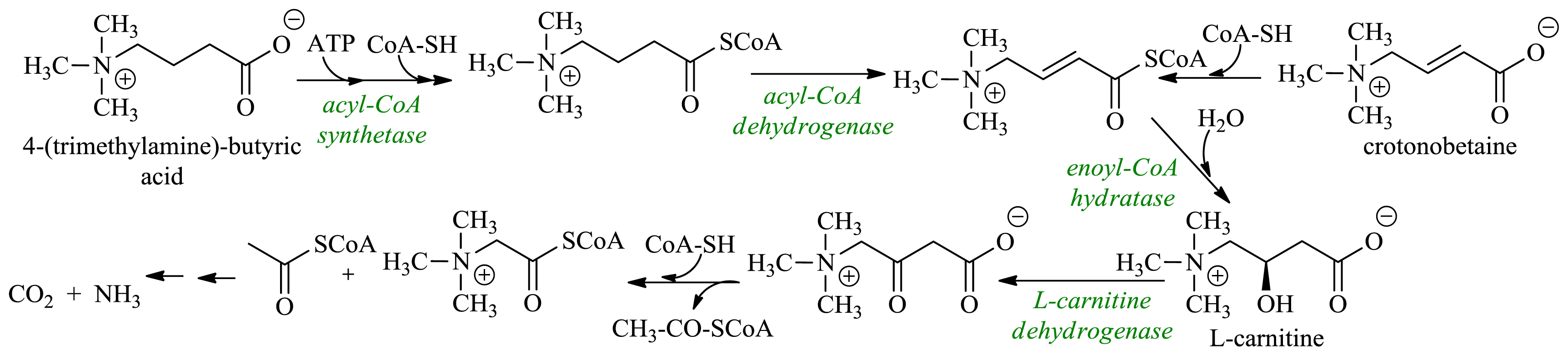
2.3. Production of l-Carnitine
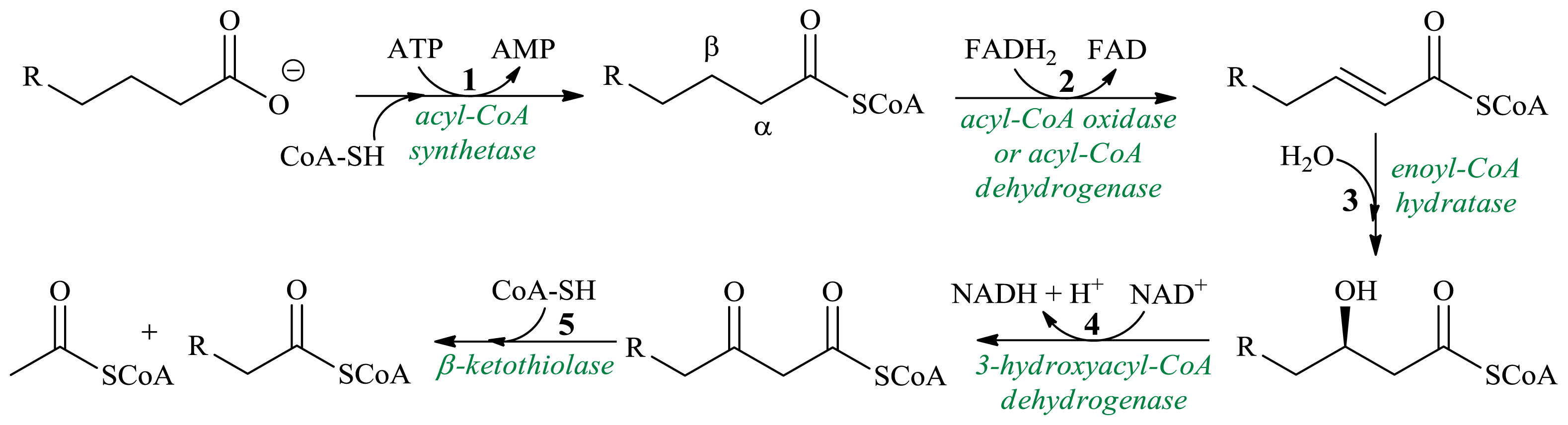
3. β-Oxidation in Synthesis of Achiral Products
3.1. Production of β-Hydroxy-β-Methylbutyric Acid
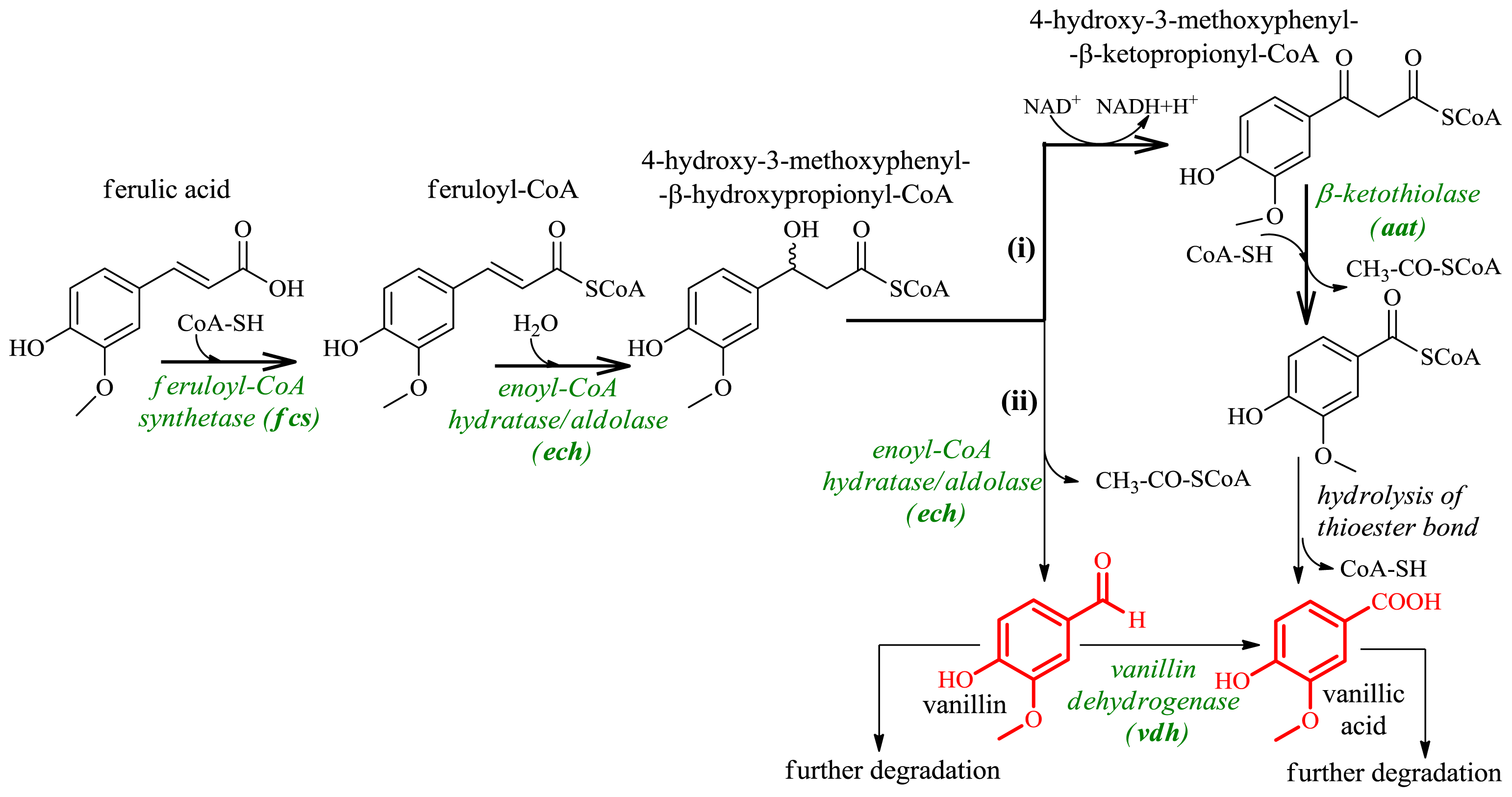
3.2. Production of Vanillic Acid and Vanillin
4. Conclusions
References
- Leffingwell, J.C. Chirality & bioactivity I.: Pharmacology. Leffingwell Rep 2003, 3, 1–27. [Google Scholar]
- Brenna, E.; Fuganti, C.; Serra, S. Enantioselective perception of chiral odorants. Tetrahedron: Asymmetry 2003, 14, 1–42. [Google Scholar]
- Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keβeler, M.; Stürmer, R.; Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Edit 2004, 43, 788–824. [Google Scholar]
- Schrader, J.; Etschmann, M.M.W.; Sell, D.; Hilmer, J.M.; Rabenhorst, H. Applied biocatalysis for the synthesis of natural flavor compound—current industrial processes and future prospects. Biotechnol. Lett 2004, 26, 463–472. [Google Scholar]
- Kieslich, K. Microbial side-chain degradation of sterols. J. Basic Microbiol 1985, 25, 461–474. [Google Scholar]
- Vyas, K.P.; Kari, P.H.; Pitzenberger, S.M.; Halpin, R.A.; Ramjit, H.G.; Arison, B.; Murphy, J.S.; Hoffman, W.F.; Schwartz, M.S.; Ulm, E.H. Biotransformation of lovastatin. I. Structure elucidation of in vitro and in vivo metabolites in the rat and mouse. Drug Metab. Dispos 1990, 18, 203–211. [Google Scholar]
- Prueksaritanont, T.; Ma, B.; Fang, X.; Subramanian, R.; Yu, J.; Lin, J.H. β-Oxidation of simvastatin in mouse liver preparations. Drug Metab. Dispos 2001, 29, 1251–1255. [Google Scholar]
- Walker, V.; Mills, G.A. Urine 4-heptanone: A β-oxidation product of 2-ethylhexanoic acid from plasticizers. Clin. Chim. Acta 2001, 306, 51–61. [Google Scholar]
- Magliano, P.; Flipphi, M.; Arpat, B.A.; Delessert, S.; Poirier, Y. Contributions of the peroxisome and β-oxidation cycle to biotin synthesis in fungi. J. Biol. Chem 2011, 286, 42133–42140. [Google Scholar]
- Rorije, E.; Peijnenburg, W.J.G.M.; Klopman, G. Structural requirements for anaerobic biodegradation of organic chemicals: A fragment model analysis. Environ. Toxicol. Chem 1998, 17, 1943–1950. [Google Scholar]
- Szentirmai, A. Microbial physiology of sidechain degradation of sterols. J. Ind. Microbiol 1990, 6, 101–116. [Google Scholar]
- Crueger, A.; Crueger, W. Carbohydrates. In Biotechnology, A Comprehensive Treatise in 8 Volumes; Rehm, H.J., Reed, G., Ebel, H.F., Eds.; Verlag-Chemie: Weinheim, Germany, 1984; Volume 6a, pp. 421–457. [Google Scholar]
- Schedel, M. Regioselective Oxidation of Aminosorbitol with Gluconobacter oxydans, Key Reaction in the Industrial 1-Deoxynojirimycin Synthesis. In Biotechnology, Biotransformations II; Rehm, H.J., Reed, G., Kelly,, D.R., Eds.; Verlag-Chemie: Weinheim, Germany, 2000; Volume 8b, pp. 295–311. [Google Scholar]
- Waché, Y.; Aguedo, M.; Nicaud, J.M.; Belin, J.M. Catabolism of hydroxyacids and biotechnological production of lactones by Yarrowia lipolytica. Appl. Microbiol. Biotechnol 2003, 61, 393–404. [Google Scholar]
- Meyer, J. γ-Decalactone microbial production from alkyl ricinoleate by hydrolysis, β-oxidation, and chemical cyclisation of 4-hydroxy decanoic acid produced for flavoring and perfume. German Patent DE 4 18 February 1993. [Google Scholar]
- Aguedo, M.; Ly, M.H.; Belo, I.; Teixeira, J.A.; Belin, J.M.; Waché, Y. The use of enzymes and microorganisms for the production of aroma compounds from lipids. Food Technol. Biotechnol 2004, 42, 327–336. [Google Scholar]
- Mitsuhashi, K.; Iimori, M. Method for producing lactone. U.S. Patent US7,129,067, 31 October 2006. [Google Scholar]
- Pagot, Y.; Endrizzi, A.; Nicaud, J.M.; Belin, J.M. Utilization of an auxotrophic strain of the yeast Yarrowia lipolytica to improve γ-decalactone production yields. Lett. Appl. Microbiol 1997, 25, 113–116. [Google Scholar]
- Rabenhorst, J.; Gatfield, I. Method of producing γ-decalactone using Yarrowia lipolytica strain HR 145 (DSM 12397). U.S. Patent US6,451,565, 17 September 2002. [Google Scholar]
- Escamilla-García, E.; Aguedo, M.; Gomes, N.; Choquet, A.; Belo, I.; Teixeira, J.A.; Belin, J.M.; Waché, Y. Production of 3-hydroxy-γ-decalactone, the precursor of two decanolides with flavouring properties, by the yeast Yarrowia lipolytica. J. Mol. Catal. B Enzym 2009, 57, 22–26. [Google Scholar]
- Romero-Guido, C.; Belo, I.; Ta, T.M.N.; Cao-Hoang, L.; Alchihab, M.; Gomes, N.; Thonart, P.; Teixeira, J.A.; Destain, J.; Waché, Y. Biochemistry of lactone formation in yeast and fungi and its utilisation for the production of flavour and fragrance compounds. Appl. Microbiol. Biotechnol 2011, 89, 535–547. [Google Scholar]
- Fickers, P.; Benetti, P.H.; Waché, Y.; Marty, A.; Mauersberger, S.; Smit, M.S.; Nicaud, J.M. Hydrophobic substrate utilisation by the yeast Yarrowia lipolytica, and its potential application. FEMS Yeast Res 2005, 5, 527–543. [Google Scholar]
- Waché, Y.; Laroche, C.; Bergmark, K.; Müller-Andersen, C.; Aguedo, M.; Le Dall, M.T.; Wang, H.; Nicaud, J.M.; Belin, J.M. Involvement of acyl coenzyme A oxidase isozymes in biotransformation of metyl ricinoleate into γ-decalactone by Yarrowia lipolytica. Appl. Environ. Microbiol 2000, 66, 1233–1236. [Google Scholar]
- Waché, Y.; Aguedo, M.; Choquet, A. Role of β-oxidation enzymes in γ-decalactone production by the yeast Yarrowia lipolytica. Appl. Environ. Microbiol 2001, 12, 5700–5704. [Google Scholar]
- Waché, Y.; Aguedo, M.; Le Dall, M.T.; Nicaud, J.M.; Belin, J.M. Optimization of Yarrowia lipolytica’s β-oxidation pathway for lactones production. J. Mol. Catal. B Enzym 2002, 153, 347–351. [Google Scholar]
- Groguenin, A.; Waché, Y.; Garcia, E.E.; Aguedo, M.; Husson, F.; LeDall, M.T.; Nicaud, J.M.; Belin, J.M. Genetic engineering of the β-oxidation pathway in the yeast Yarrowia lipolytica to increase the production of aroma compounds. J. Mol. Catal. B Enzym 2004, 28, 75–79. [Google Scholar]
- Guo, Y.; Song, H.; Wang, Z.; Ding, Y. Expression of POX2 gene and disruption of POX3 genes in the industrial Yarrowia lipolytica on the γ-decalactone production. Microbiol. Res 2012, 167, 246–252. [Google Scholar]
- Aguedo, M.; Beney, L.; Waché, Y.; Belin, J.M. Mechanism underlying the toxicity of lactone aroma compounds towards the producing yeast cells. J. Appl. Microbiol 2003, 94, 258–265. [Google Scholar]
- Feron, G.; Dufosse, L.; Pierard, E.; Bonnarme, P.; Quere, J.L.; Spinnler, H.E. Production, identification, and toxicity of γ-decalactone and 4-hydroxydecenoic acid from Sporidiobolus spp. Appl. Environ. Microbiol 1996, 62, 2826–2831. [Google Scholar]
- Alchihab, M.; Destain, J.; Aguedo, M.; Wathelet, J.P.; Thonart, P. The utilization of gum tragacanth to improve the growth of Rhodotorula aurantiaca and the production of γ-decalactone in large scale. Appl. Biochem. Biotechnol 2010, 162, 233–241. [Google Scholar]
- Boog, A.L.G.M.; Peters, A.L.J.; Roos, R. Process for producing δ-lactones from 11-hydroxy fatty acids. U.S. Patent US5,215,901, 1 June 1993. [Google Scholar]
- Cardillo, R.; Fuganti, C.; Barbieri, M.; Cabella, P.; Guarda, P.A.; Allegrone, G.A. Process for the microbiological production of γ- and δ-lactones. U.S. Patent US5,168,054, 1 December 1992. [Google Scholar]
- Gocho, S.; Tabogami, N.; Inagaki, M.; Kawabata, C.; Komai, T. Biotransformation of oleic acid to optically active γ-dodecalactone. Biosci. Biotechnol. Biochem 1995, 59, 1571–1572. [Google Scholar]
- Hosoi, K.; Okawa, T. Production of γ-dodecalactone. Japanese Patent JP3,479,337, 3 October 2003. [Google Scholar]
- Joo, Y.C.; Seo, E.S.; Kim, Y.S.; Kim, K.R.; Park, J.B.; Oh, D.K. Production of 10-hydroxystearic acid from oleic acid by whole cells of recombinant Escherichia coli containing oleate hydratase from Stenotrophomonas maltophilia. J. Biotechnol 2012, 158, 17–23. [Google Scholar]
- Saitoh, C.; Masuda, Y.; Yashiro, A.; Ishiguro, H. Process for producing hydroxylated fatty acid and δ-lactone. U.S. Patent US6,777,211, 17 August 2004. [Google Scholar]
- Serrano-Carreon, L.; Hathout, Y.; Bensoussan, M.; Belin, J.M. Metabolism of linoleic acid or mevalonate and 6-pentyl-alpha-pyrone biosynthesis by Trichoderma species. Appl. Environ. Microbiol 1993, 59, 2945–2950. [Google Scholar]
- Bonnarme, P.; Djian, A.; Latrasse, A.; Féron, G.; Giniès, C.; Durand, A.; Le Quéré, J.L. Production of 6-pentyl-α-pyrone by Trichoderma sp. from vegetable oils. J. Biotechnol 1997, 56, 143–150. [Google Scholar]
- Tong, W.Y.; Dong, X. Microbial biotransformation: Recent developments on steroid drugs. Recent Pat. Biotechnol 2009, 3, 141–153. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products, A Biosynthetic Approach; John Wiley & Sons: Chichester, UK, 2009; pp. 187–310. [Google Scholar]
- Fernandes, P.; Cabral, J.M.S. Phytosterols: Applications and recovery methods. Bioresour. Technol 2007, 98, 2335–2350. [Google Scholar]
- Smith, L.L. Steroids. In Biotechnology, A Comprehensive Treatise in 8 Volumes; Rehm, H.J., Reed, G., Ebel, H.F., Eds.; Verlag-Chemie: Weinheim, Germany, 1984; Volume 6a, pp. 31–78. [Google Scholar]
- Van der Waard, W.F. Process for the microbiological preparation of steroids. Netherlands Patent 6,513,718, 22 October 1965. [Google Scholar]
- Van der Geize, R.; Hessels, G.I.; Dijkhuizen, L. Method for the production of modified steroid degrading microorganisms and their use. U.S. Patent US0,189,390, 23 July 2009. [Google Scholar]
- Fujimoto, Y.; Chen, C.S.; Szeleczky, Z.; Ditullio, D.; Sih, C.J. Microbial degradation of the phytosterol side chain. Enzymic conversion of 3-oxo-24-ethylcholest-4-en-26-oic acid into 3-oxochol-4-en-24-oic acid and androst-4-ene-3,17-dione. J. Am. Chem. Soc 1982, 104, 4718–4720. [Google Scholar]
- Wang, F.Q.; Yao, K.; Wei, D.Z. From Soybean Phytosterols to Steroid Hormones. In Soybean and Health; El-Shemy, H.A., Ed.; InTech: Rijeka, Croatia, 2011; pp. 241–263. [Google Scholar]
- Van der Geize, R.; Hessels, G.I.; Dijkhuizen, L. Molecular and functional characterization of the kstD2 gene of Rhodococcus erythropolis SQ1 encoding a second 3-ketosteroid Δ1-dehydrogenase isoenzyme. Microbiology 2002, 148, 3285–3292. [Google Scholar]
- Van der Geize, R.; Hessels, G.I.; van Gerwen, R.; van der Meijden, P.; Dijkhuizen, L. Unmarked gene deletion mutagenesis of kstD, Encoding 3-Ketosteroid δ(1)-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counterselectable marker. FEMS Microbiol. Lett 2001, 205, 197–202. [Google Scholar]
- Van der Geize, R.; Hessels, G.I.; van Gerwen, R.; van der Meijden, R.; Dijkhuizen, L. Molecular and functional characterization of kshA and kshB, encoding two components of 3-ketosteroid-9- alpha-hydroxylase, a class IA monooxygenase, in Rhodococcus erythropolis strain SQ1. Mol. Microbiol 2002, 45, 1007–1018. [Google Scholar]
- Donova, M.V.; Egorova, O.V. Microbial steroid transformations: Current state and prospects. Appl. Microbiol. Biotechnol 2012, 94, 1423–1447. [Google Scholar]
- Van der Geize, R.; Yam, K.; Heuser, T.; Wilbrink, M.H.; Hara, H.; Anderton, M.C.; Sim, E.; Dijkhuizen, L.; Davies, J.E.; Mohn, W.W.; et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 1947–1952. [Google Scholar]
- Wilbrink, M.H.; Petrusma, M.; Dijkhuizen, L.; van der Geize, R. FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for degradation of C-24 branched sterol side chains. Appl. Environ. Microbiol 2011, 77, 4455–4464. [Google Scholar]
- Liu, Y.; Chen, G.; Ge, F.; Li, W.; Zeng, L.; Cao, W. Efficient biotransformation of cholesterol to androsta-1,4-diene-3,17-dione by a newly isolated actinomycete Gordonia neofelifaecis. World J. Microbiol. Biotechnol 2011, 27, 759–765. [Google Scholar]
- Wei, W.; Wang, F.Q.; Fan, S.Y.; Wei, D.Z. Inactivation and augmentation of the primary 3-ketosteroid-{γ}1-dehydrogenase in Mycobacterium neoaurum NwIB-01: Biotransformation of soybean phytosterols to 4-androstene-3,17-dione or 1,4-androstadiene-3,17-dione. Appl. Environ. Microbiol. 2010, 76, 4578–4582. [Google Scholar]
- Egorova, O.V.; Nikolayeva, V.M.; Sukhodolskaya, G.V.; Donova, M.V. Transformation of C19-steroids and testosterone production by sterol-transforming strains of Mycobacterium spp. J. Mol. Catal. B: Enzym 2009, 57, 198–203. [Google Scholar]
- Weber, A.; Kennecke, M. Process for the production of 4-androstene-3,17-dione and 1,4-androstadiene-3,17-dione from ergosterol with Mycobacterium. U.S. Patent US5,516,649, 14 May 1996. [Google Scholar]
- Dovbnya, D.V.; Egorova, O.V.; Donova, M.V. Microbial side-chain degradation of ergosterol and its 3-substituted derivatives: a new route for obtaining of deltanoids. Steroids 2010, 75, 653–658. [Google Scholar]
- Weber, A.; Kennekke, M.; Neef, G. Process for the production of 20-methyl-5,7-pregnadiene- 3β,21-diol derivatives using mycobacterium. U.S. Patent US5,429,934, 4 July 1995. [Google Scholar]
- Agoston, E.S. Anticancer agents. Med. Chem 2006, 6, 53–71. [Google Scholar]
- Steinmeyer, A.; Kirsch, G.; Neef, G.; Schwarz, K.; Thieroff-Ekerdt, R.; Wiesinger, H.; Haberey, M.; Fahnrich, M. Vitamin D derivatives with carbo- or heterocyclic substituents at C-25, a process for their production, intermediate products and their use for producing medicaments. U.S. Patent US6,600,058, 29 July 2003. [Google Scholar]
- Fernandes, P.; Cruz, A.; Angelova, B.; Pinheiro, H.M.; Cabral, J.M.S. Microbial conversion of steroid compounds: Recent developments. Enzyme Microb. Technol 2003, 32, 688–705. [Google Scholar]
- Malaviya, A.; Gomes, J. Androstenedione production by biotransformation of phytosterols. Bioresour. Technol 2008, 99, 6725–6737. [Google Scholar]
- Atrat, P.; Hösel, P.; Richter, W.; Meyer, H.; Hörhold, C. Interactions of Mycobacterium fortuitum with solid sterol substrate particles. J. Basic Microbiol 1991, 31, 413–422. [Google Scholar]
- Goetschel, R.; Bar, R. Formation of mixed crystals in microbial conversion of sterols and steroids. Enzyme Microb. Technol 1992, 14, 462–469. [Google Scholar]
- Perfumo, A.; Smyth, T.J.P.; Marchant, R.; Banat, I.M. Production and Roles of Biosurfactants and Bioemulsifiers in Accessing Hydrophobic Substrates. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin, Germany, 2010; pp. 1501–1512. [Google Scholar]
- Parales, R.E.; Ditty, J.L. Substrate Transport. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin, Germany, 2010; pp. 1545–1553. [Google Scholar]
- Heipieper, H.J.; Cornelissen, S.; Pepi, M. Surface Properties and Cellular Energetics of Bacteria in Response to the Presence of Hydrocarbons. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin, Germany, 2010; pp. 1615–1624. [Google Scholar]
- Korycka-Machala, M.; Rumijowska-Galewicz, A.; Dziadek, J. The effect of ethambutol on mycobacterial cell wall permeability to hydrophobic compounds. Pol. J. Microbiol 2005, 54, 5–11. [Google Scholar]
- Li, J.L.; Chen, B.H. Surfactant-mediated biodegradation of polycyclic aromatic hydrocarbons. Materials 2009, 2, 76–94. [Google Scholar]
- Mohn, W.W.; van der Geize, R.; Stewart, G.R.; Okamoto, S.; Liu, J.; Dijkhuizen, L.; Eltis, L.D. The actinobacterial mce4 locus encodes a steroid transporter. J. Biol. Chem 2008, 283, 35368–35374. [Google Scholar]
- Perez, C.; Falero, A.; Llanes, N.; Hung, B.R.; Herve, M.E.; Palmero, A; Martii, E. Resistance to androstanes as an approach for androstandienedione yield enhancement in industrial mycobacteria. J. Ind. Microbiol. Biotechnol. 2003, 30, 623–626. [Google Scholar]
- Huang, C.L.; Chen, Y.R.; Liu, W.H. Production of androstenones from phytosterol by mutants of Mycobacterium sp. Enzyme Microb. Technol 2006, 39, 296–300. [Google Scholar]
- Dumas, B.; Brocard-Masson, C.; Assemat-Lebrun, K.; Achstetter, T. Hydrocortisone made in yeast: Metabolic engineering turns a unicellular microorganism into a drug-synthesizing factory. Biotechnol. J 2006, 1, 299–307. [Google Scholar]
- Choudhary, M.I.; Erum, S.; Atif, M.; Malik, R.; Khan, N.T.; Atta-ur-Rahman. Biotransformation of (20S)-20-hydroxymethylpregna-1,4-dien-3-one by four filamentous fungi. Steroids 2011, 76, 1288–1296. [Google Scholar]
- Gates, S.; Loria, R.M. Compositions for regulation of immune responses. U.S. Patent US5,776,921, 7 July 1998. [Google Scholar]
- Kim, T.K.; Chen, J.; Li, W.; Zjawiony, J.; Miller, D.; Janjetovic, Z.; Tuckey, R.C.; Slominski, A. A new steroidal 5,7-diene derivative, 3β-hydroxyandrosta-5,7-diene-17β-carboxylic acid, shows potent anti-proliferative activity. Steroids 2010, 75, 230–239. [Google Scholar]
- Hasegawa, J.; Nagashima, N. Production of Chiral β-Hydroxy Acids and Its Application in Organic Synthesis. In Stereoselective Biocatalysis; Patel, R.N., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 343–363. [Google Scholar]
- Meyers, A.I.; Hudspeth, R.A. Enantioselective synthesis of C3–C10 fragment (northeastern zone) of maytansinoids with 4-chiral centers (4S,5S,6R,7S). Tetrahedron Lett 1981, 22, 3925–3928. [Google Scholar]
- Goodhoue, C.T.; Schafler, J.R. Preparation of l(+)-β-hydroxyisobutyric acid by bacterial oxidation of isobutyric acid. Biotechnol. Bioeng 1971, 13, 203–214. [Google Scholar]
- Hasegawa, J.; Hamaguchi, S.; Ogura, M.; Watanabe, K. Production of β-hydroxycarboxylic acids from aliphatic carboxylic acids by microorganisms. J. Ferment. Technol 1981, 59, 257–262. [Google Scholar]
- Hasegawa, J.; Ogura, M.; Kanema, H.; Noda, N.; Kawaharada, H.; Watanabe, K. Production of β-hydroxyisobutyric acid by Candida rugosa and its mutant. J. Ferment. Technol 1982, 60, 501–508. [Google Scholar]
- Lee, I.Y.; Hong, W.K.; Hwang, Y.B.; Kim, C.H.; Choi, E.S.; Rhee, S.K.; Park, Y.H. Production of d-β-hydroxybutyric acid from isobutyric acid by Candida rugosa. J. Ferment. Bioeng 1996, 81, 79–82. [Google Scholar]
- Kim, H.S.; Ju, J.Y.; Shin, C.S. Optimized fed-batch fermentation of l-β-hydroxyisobutyric acid by Yarrowia lipolytica. Bioproc. Biosyst. Eng 1999, 20, 189–193. [Google Scholar]
- Kim, C.H.; Hong, W.K.; Lee, I.Y.; Choi, E.S; Rhee, S.K. Enhanced production of d-β-hydroxyisobutyric acid through strain improvement. J. Biotechnol. 1999, 69, 75–79. [Google Scholar]
- Robinson, R.S.; Doremus, M.G. Method of preparing l-(+)-β-hydroxyisobutyric acid by fermentation. U.S. Patent US4,618,583, 21 October 1986. [Google Scholar]
- Kamal, A.; Krishnaji, T.; Khan, M.N.A. Lipase-catalysed resolution of N-(3-cyano-2-hydroxy propan-1-yl)phthalimide: Synthesis of (R)-GABOB and (R)-carnitine. J. Mol. Catal. B: Enzym 2007, 47, 1–5. [Google Scholar]
- Obón, J.M.; Maiquez, J.R.; Canovas, M.; Kleber, H.P.; Iborra, J.L. l(−)-Carnitine production with immobilized Escherichia coli cells in continuous reactors. Enzyme Microb. Technol 1997, 21, 531–536. [Google Scholar]
- Meier, P.J. d-Carnitine Harmlos? In Carnitine in der Medizin; Gitzelmann, R., Baerlocher, K., Steinmann, B., Eds.; Shattauer: Stuttgart, Germany, 1987; pp. 101–104. [Google Scholar]
- Kulla, H.G. Enzymatic hydroxylations in industrial application. Chimia 1991, 45, 81–85. [Google Scholar]
- Engemann, C.; Elssner, T.; Kleber, H.P. Biotransformation of crotonobetaine to l(−)-carnitine in Proteus sp. Arch. Microbiol 2001, 175, 353–359. [Google Scholar]
- Elssner, T.; Hennig, L.; Frauendorf, H.; Haferburg, D.; Kleber, H.P. Isolation, identification, and synthesis of γ-butyrobetainyl-CoA and crotonobetainyl-CoA, compounds involved in carnitine metabolism of E. coli. Biochemistry 2000, 39, 10761–10769. [Google Scholar]
- Guebel, D.V.; Torres, N.V.; Cánovas, M. Modeling analysis of the l(−)-carnitine production process by Escherichia coli. Process Biochem 2006, 41, 281–288. [Google Scholar]
- Cánovas, M.; Torroglosa, T.; Iborra, J.L. Permeabilization of Escherichia coli cells in the biotransformation of trimethylammonium compounds into l-carnitine. Enzyme Microb. Technol 2005, 37, 300–308. [Google Scholar]
- Bernal, V.; Masdemont, B.; Arense, P.; Cánovas, M.; Iborra, J.L. Redirecting metabolic fluxes through cofactor engineering: Role of CoA-esters pool during l(−)-carnitine production by Escherichia coli. J. Biotechnol 2007, 132, 110–117. [Google Scholar]
- Nissen, S.L.; Fuller, J.J.; Sell, J.; Ferket, P.R.; Rives, D.Y. The effect of β-hydroxy-β-methylbutyrate on growth, mortality and carcass qualities of broiler chickens. Poult. Sci 1994, 73, 137–155. [Google Scholar]
- Nissen, S.L.; Faidley, T.D.; Zimmerman, D.R.; Izard, R.; Fisher, C.T. Colostral milk fat percentage and pig performance are enhanced by feeding the leucine metabolite β-hydroxy-β-methyl butyrate to sows. J. Anim. Sci 1994, 72, 2332–2337. [Google Scholar]
- Wilson, G.J.; Wilson, J.M.; Manninen, A.H. Effects of β-hydroxy-β-methylbutyrate (HMB) on exercise performance and body composition across varying levels of age, sex, and training experience. Nutr. Metab 2008, 5, 1–17. [Google Scholar]
- Van Kovering, M.; Nissen, S.L. Oxidation of leucine and alphaketoisocaproate to β-hydroxy-β-methylbutyrate in vivo. Am. J. Physiol. Endocrinol. Metab 1992, 26, 27–31. [Google Scholar]
- Lee, I.Y.; Nissen, S.L.; Rosazza, J.P.N. Conversion of β-methylbutyric acid to β-hydroxy-β-methylbutyric acid by Galactomyces reessii. Appl. Environ. Microbiol 1997, 63, 4191–4195. [Google Scholar]
- Lee, I.Y.; Rosazza, J.P.N. Enzyme analyses demonstrate that β-methylbutyric acid is converted to β-hydroxy-β-methylbutyric acid via the leucine catabolic pathway by Galactomyces reessii. Arch. Microbiol 1998, 169, 257–262. [Google Scholar]
- Dhar, A.; Dhar, K.; Rosazza, J.P.N. Purification and properties of an Galactomyces reessii hydratase that converts 3-methylcrotonic acid to 3-hydroxy-3-methylbutyric acid. J. Ind. Microbiol. Biotechnol 2002, 28, 81–87. [Google Scholar]
- Ashengroph, M.; Nahvi, I.; Zarkesh-Esfahani, H.; Momenbeik, F. Candida galli strain PGO6: A novel isolated yeast strain capable of transformation of isoeugenol into vanillin and vanillic acid. Curr. Microbiol 2011, 62, 990–998. [Google Scholar]
- Kim, M.C.; Kim, S.J.; Kim, D.S.; Jeon, Y.D.; Park, S.J.; Lee, H.S.; Um, J.Y.; Hong, S.H. Vanillic acid inhibits inflammatory mediators by suppressing NF-κB in lipopolysaccharide-stimulated mouse peritoneal macrophages. Immunopharmacol. Immunotoxicol 2011, 33, 525–532. [Google Scholar]
- Priefert, H.; Rabenhorst, J.; Steinbüchel, A. Biotechnological production of vanillin. Appl. Microbiol. Biotechnol 2001, 56, 296–314. [Google Scholar]
- Yoon, S.H.; Li, C.; Lee, Y.M.; Lee, S.H.; Kim, S.H.; Choi, M.S.; Seo, W.T.; Yang, J.-K.; Kim, J-Y.; Kim, S-W. Production of vanillin from ferulic acid using recombinant strains of Escherichia coli. Biotechnol. Bioprocess Eng. 2005, 10, 378–384. [Google Scholar]
- Di Gioia, D.; Luziatelli, F.; Negroni, A.; Ficca, A.G.; Fava, F.; Ruzzi, M. Metabolic engineering of Pseudomonas fluorescens for the production of vanillin from ferulic acid. J. Biotechnol 2011, 156, 309–316. [Google Scholar]
- Converti, A.; Aliakbarian, B.; Domínguez, J.M.; Bustos Vázquez, G.; Perego, P. Microbial production of biovanillin. Braz. J. Microbiol 2010, 41, 519–530. [Google Scholar]
- Ashengroph, M.; Nahvi, I.; Zarkesh-Esfahani, H.; Momenbeik, F. Novel strain of Bacillus licheniformis SHL1 with potential converting ferulic acid into vanillic acid. Ann. Microbiol 2012, 62, 553–558. [Google Scholar]
- Muheim, A.; Münch, T.; Wetli, M. Microbiological process for producing vanillin. U.S. Patent US6,235,507, 22 May 2001. [Google Scholar]
- Rabenhorst, J.; Hoop, R. Process for the preparation of vanillin and microorganisms suitable therefor. U.S. Patent US6,133,003, 17 October 2000. [Google Scholar]
- Hua, D.; Ma, C.; Song, L.; Lin, S.; Zhang, Z.; Deng, Z.; Xu, P. Enhanced vanillin production from ferulic acid using adsorbent resin. Appl. Microbiol. Biotechnol 2007, 74, 783–790. [Google Scholar]
- Overhage, J.; Priefert, H.; Steinbüchel, A. Biochemical and genetic analyses of ferulic acid catabolism in Pseudomonas sp. strain HR199. Appl. Environ. Microbiol 1999, 65, 4837–4847. [Google Scholar]
- Huang, Z.; Dostal, L.; Rosazza, J.P.N. Mechanism of ferulic acid conversions to vanillic acid and guaiacol by Rhodotorula rubra. J. Biol. Chem 1993, 268, 23954–23958. [Google Scholar]
- Plaggenborg, R.; Overhage, J.; Loos, A.; Archer, J.A.C.; Lessard, P.; Sinskey, A.J.; Steinbüchel, A.; Priefert, H. Potential of Rhodococcus strains for biotechnological vanillin production from ferulic acid and eugenol. Appl. Microbiol. Biotechnol 2006, 72, 745–755. [Google Scholar]
- Krings, U.; Pilawa, S.; Theobald, C.; Berger, R.G. Phenyl propenoic side chain degradation of ferulic acid by Pycnoporus cinnabarinus—elucidation of metabolic pathways using [5-2H]-ferulic acid. J. Biotechnol 2001, 85, 305–314. [Google Scholar]
- Muheim, A.; Lerch, K. Towards a high-yield bioconversion of ferulic acid to vanillin. Appl. Microbiol. Biotechnol 1999, 51, 456–461. [Google Scholar]
- Zheng, L.; Zheng, P.; Sun, Z.; Bai, Y.; Wang, J.; Guo, X. Production of vanillin from waste residua of rice brain oil by Aspergillus niger and Pycnoporus cinnabarinus. Bioresour. Technol. 2007, 98, 1115–1119. [Google Scholar]
- Calisti, C.; Ficca, A.G.; Barghini, P.; Ruzzi, M. Regulation of ferulic catabolic genes in Pseudomonas fluorescens BF13: Involvement of a MarR family regulator. Appl. Microbiol. Biotechnol 2008, 80, 475–483. [Google Scholar]
- Ghosh, S.; Sachan, A.; Sen, S.K.; Mitra, A. Microbial transformation of ferulic acid to vanillic acid by Streptomyces sannanensis MTCC 6637. J. Ind. Microbiol. Biotechnol 2007, 34, 131–138. [Google Scholar]
- Achterholt, S.; Priefert, H.; Steinbüchel, A. Identification of Amycolatopsis sp. strain HR167 genes, involved in the bioconversion of ferulic acid to vanillin. Appl. Microbiol. Biotechnol 2000, 54, 799–807. [Google Scholar]
- Yoon, S.-H.; Li, C.; Kim, J.-E.; Lee, S.-H.; Yoon, J.-Y.; Choi, M.-S.; Seo, W.-T.; Yang, J.-K.; Kim, J.-Y.; Kim, S.-W. Production of vanillin by metabolically engineered Escherichia coli. Biotechnol. Lett 2005, 27, 1829–1832. [Google Scholar]
- Barghini, P.; Di Gioia, D.; Fava, F.; Ruzzi, M. Vanillin production using metabolically engineered Escherichia coli under non-growing conditions. Microb. Cell Fact. 2007, 6. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty acid (substrate) | Lactone (product) | Odor description |
|---|---|---|
| ricinoleic | γ-decalactone | fatty, creamy, coconut, buttery, vanilla sweet |
| 3-hydroxy-γ-decalactone | odorless | |
| dec-3-en-4-olide | fruity, oily, fatty | |
| dec-2-en-4-olide | mushroom | |
| jalapinolic or coriolic or linoleic | δ-decalactone | sweet, creamy, milky, peach, nut; peach, buttery on dilution |
| ipurolic | δ-octalactone | sweet, creamy, fatty with tropical and dairy nuances |
| linoleic | 6-pentyl-2-pyrone | coconut, creamy, sweet, brown with a fatty waxy nuance |
| α-linolenic | δ-jasminlactone | powerful, creamy, jasmine-peachy-coconut |
| oleic | γ-dodecalactone | fatty, fruity, peach |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Świzdor, A.; Panek, A.; Milecka-Tronina, N.; Kołek, T. Biotransformations Utilizing β-Oxidation Cycle Reactions in the Synthesis of Natural Compounds and Medicines. Int. J. Mol. Sci. 2012, 13, 16514-16543. https://doi.org/10.3390/ijms131216514
Świzdor A, Panek A, Milecka-Tronina N, Kołek T. Biotransformations Utilizing β-Oxidation Cycle Reactions in the Synthesis of Natural Compounds and Medicines. International Journal of Molecular Sciences. 2012; 13(12):16514-16543. https://doi.org/10.3390/ijms131216514
Chicago/Turabian StyleŚwizdor, Alina, Anna Panek, Natalia Milecka-Tronina, and Teresa Kołek. 2012. "Biotransformations Utilizing β-Oxidation Cycle Reactions in the Synthesis of Natural Compounds and Medicines" International Journal of Molecular Sciences 13, no. 12: 16514-16543. https://doi.org/10.3390/ijms131216514
APA StyleŚwizdor, A., Panek, A., Milecka-Tronina, N., & Kołek, T. (2012). Biotransformations Utilizing β-Oxidation Cycle Reactions in the Synthesis of Natural Compounds and Medicines. International Journal of Molecular Sciences, 13(12), 16514-16543. https://doi.org/10.3390/ijms131216514