Secondary Metabolites from the Roots of Beilschmiedia tsangii and Their Anti-Inflammatory Activities

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure Elucidation
2.2. Anti-inflammatory Activities
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Liao, J.C. Lauraceae. In Flora of Taiwan, 2nd ed; Editorial Committee of the Flora of Taiwan; Taipei, Taiwan1996; Volume 2, pp. 433–499.
- Chen, J.J.; Chou, E.T.; Duh, C.Y.; Yang, S.Z.; Chen, I.S. New cytotoxic tetrahydrofuran- and dihydrofuran-type lignans from the stem of Beilschmiedia tsangii. Planta Med 2006, 72, 351–357. [Google Scholar]
- Chen, J.J.; Chou, E.T.; Peng, C.F.; Chen, I.S.; Yang, S.Z.; Huang, H.Y. Novel epoxyfuranoid lignans and antitubercular constituents from the leaves of Beilschmiedia tsangii. Planta Med 2007, 73, 567–571. [Google Scholar]
- Huang, Y.T.; Chang, H.S.; Wang, G.J.; Chen, C.H.; Lin, C.H.; Yang, Y.Z.; Chen, I.S. Anti-inflammatory endiandric acid analogues from the roots of Beilschmiedia tsangii. J. Nat. Prod 2011, 74, 1875–1880. [Google Scholar]
- Chouna, J.R.; Nkeng-Efouet, P.A.; Lenta, B.N.; Wansi, J.D.; Kimbu, S.; Sewald, N. Endiandric acid derivatives from the stem bark of Beilschmiedia anacardioides. Phytochem. Lett 2010, 3, 13–16. [Google Scholar]
- Bandaranayake, W.M.; Banfield, J.E.; Black, D.S.C.; Fallon, G.D.; Gatehouse, B.M. Constituents of Endiandra species. I. endiandric acid, a novel carboxylic acid from Endiandra introrsa (Lauraceae), and a derived lactone. Aust. J. Chem 1981, 34, 1655–1667. [Google Scholar]
- Bandaranayake, W.M.; Banfield, J.E.; Black, D.S.C.; Fallon, G.D.; Gatehouse, B.M. Constituents of Endiandra species. III. 4-[(E,E)-5′-phenylpenta-2′,4′-dien-1′-yl]tetracyclo[5,4,0,02,5,03,9]undec-10-ene-8-carboxylic acid from Endiandra introrsa (Lauraceae). Aust. J. Chem 1982, 35, 567–579. [Google Scholar]
- Yang, P.S.; Cheng, M.J.; Chen, J.J.; Chen, I.S. Two new endiandric acid analogs, a new benzopyran, and a new benzenoid from the root of Beilschmiedia erythrophloia. Helv. Chim. Acta 2008, 91, 2130–2138. [Google Scholar]
- Yang, P.S.; Cheng, M.J.; Peng, C.F.; Chen, J.J.; Chen, I.S. Endiandric acid analogues from the roots of Beilschmiedia erythrophloia. J. Nat. Prod 2009, 72, 53–58. [Google Scholar]
- Adio, A.M.; Konig, W.A. Sesquiterpenoids and norsesquiterpenoids from three liverworts. Tetrahedron 2007, 18, 1693–1700. [Google Scholar]
- Collado, I.G.; Hanson, J.R.; Hitchcock, P.B.; Macias-Sanchez, A.J. Stereochemistry of epoxidation of some caryophyllenols. J. Org. Chem 1997, 62, 1965–1969. [Google Scholar]
- Pouchert, C.J.; Behnke, J. The Aldrich Library of13C and1H FT NMR Spectra, 1st ed; Aldrich Chemical Co., Inc.: Wisconsin, WI, USA, 1993; Volume 2, p. 964. [Google Scholar]
- Iijima, T.; Yaoita, Y.; Kikuchi, M. Five new sesquiterpenoids and a new diterpenoid from Erigeron annuus (L.) Pers., Erigeron philadelphicus L. and Erigeron sumatrensis Retz. Chem. Pharm. Bull 2003, 51, 545–549. [Google Scholar]
- Zhang, H.J.; Tan, G.T.; Santarsiero, B.D.; Mesecar, A.D.; Van Hung, N.; Cuong, N.M.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H.S. New sesquiterpenes from Litsea verticillata. J. Nat. Prod 2003, 66, 609–615. [Google Scholar]
- Kamel, A. 7-epi-Eudesmanes from Teucrium polium. J. Nat. Prod 1995, 58, 428–431. [Google Scholar]
- Bilia, A.R.; Mendez, J.; Morelli, I. Phytochemical investigations of Licania genus. Flavonoids and triterpenoids from Licania carii. Pharm. Acta Helv 1996, 71, 191–197. [Google Scholar]
- Min, B.S.; Kim, Y.H.; Lee, S.M.; Jung, H.J.; Lee, J.S.; Na, M.K.; Lee, C.O.; Lee, J.P.; Bae, K.H. Cytotoxic triterpenes from Crataegus pinnatifida. Arch. Pharm. Res 2000, 23, 155–158. [Google Scholar]
- Banfield, J.E.; Black, D.S.C.; Collins, D.J.; Hyland, B.P.M.; Lee, J.J.; Pranowo, S.R. Constituents of some species of Beilschmiedia and Endiandra (Lauraceae): New endiandric acid and benzopyran derivatives isolated from B.oligandra. Aust. J. Chem 1994, 47, 587–607. [Google Scholar]
- Chouna, J.R.; Nkeng-Efouet, P.A.; Lenta, B.N.; Devkota, K.P.; Neumann, B.; Stammler, H.G.; Kimbu, S.; Sewald, N. Antibacterial endiandric acid derivatives from Beilschmiedia anacardioides. Phytochemistry 2009, 70, 684–688. [Google Scholar]
- Harborne, J.B.; Mendez, J. Flavonoids of Beilschmiedia miersii. Phytochemistry 1969, 8, 763–764. [Google Scholar]
- Lenta, B.N.; Tantangmo, F.; Devkota, K.P.; Wansi, J.D.; Chouna, J.R.; Soh, R.C.F.; Neumann, B.; Stammler, H.G.; Tsamo, E.; Sewald, N. Bioactive constituents of the stem bark of Beilschmiedia zenkeri. J. Nat. Prod 2009, 72, 2130–2134. [Google Scholar]
- Kitagawa, I.; Minagawa, K.; Zhang, R.S.; Hori, K.; Doi, M.; Inoue, M.; Ishida, T.; Kimura, M.; Uji, T.; Shibuya, H. Dehatrine, an antimalarial bisbenzylisoquinoline alkaloid from the Indonesian medicinal plant Beilschmiedia madang, isolated as a mixture of two rotational isomers. Chem. Pharm. Bull 1993, 41, 997–999. [Google Scholar]
- Tillequin, F.; Koch, M.; Pusset, J.; Chauviere, G. Two new morphinane alkaloids from Beilschmiedia oreophila Schlechter (Lauraceae). Heterocycles 1985, 23, 1357–1361. [Google Scholar]
- Johns, S.R.; Lamberton, J.A.; Sioumis, A.A.; Tweeddale, H.J. New aporphine alkaloids from Beilschmiedia podagrica. Aust. J. Chem 1969, 22, 1277–1281. [Google Scholar]
- Kumamoto, J.; Scora, R.W. Structure of sarisan, an isomer of myristicin, isolated from the leaf oil of Beilschmiedia miersii. J. Agric. Food Chem 1970, 18, 544–545. [Google Scholar]
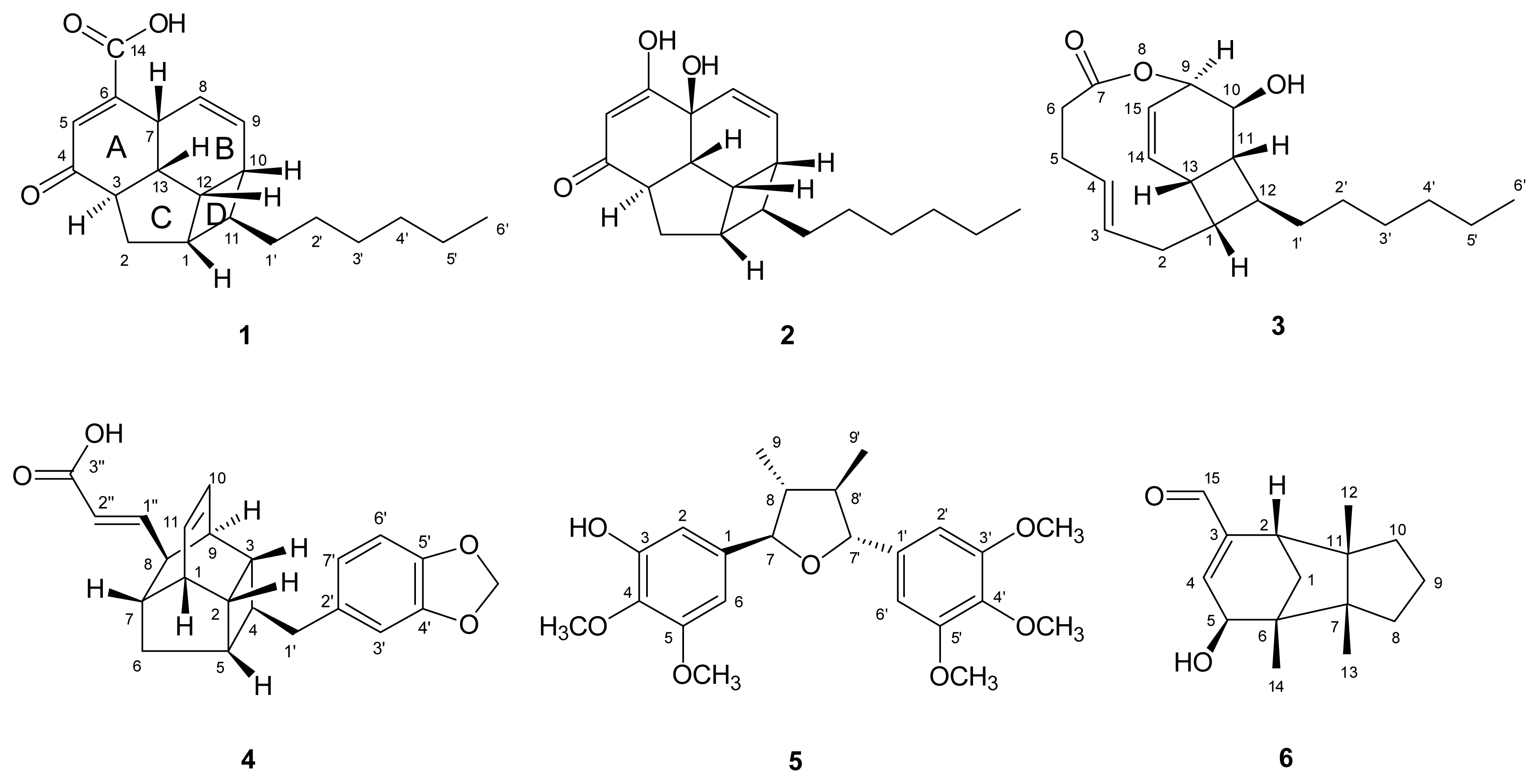
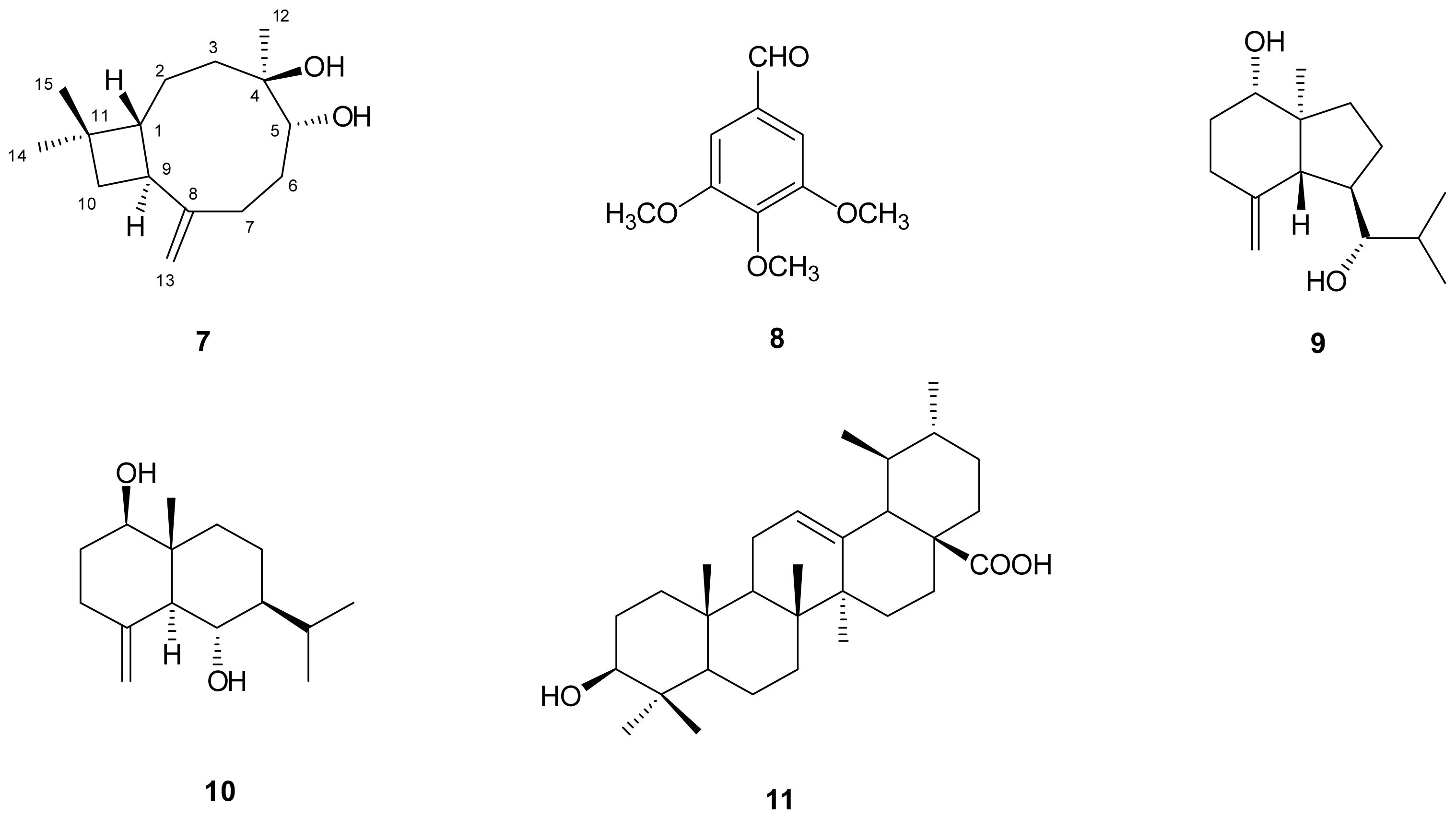
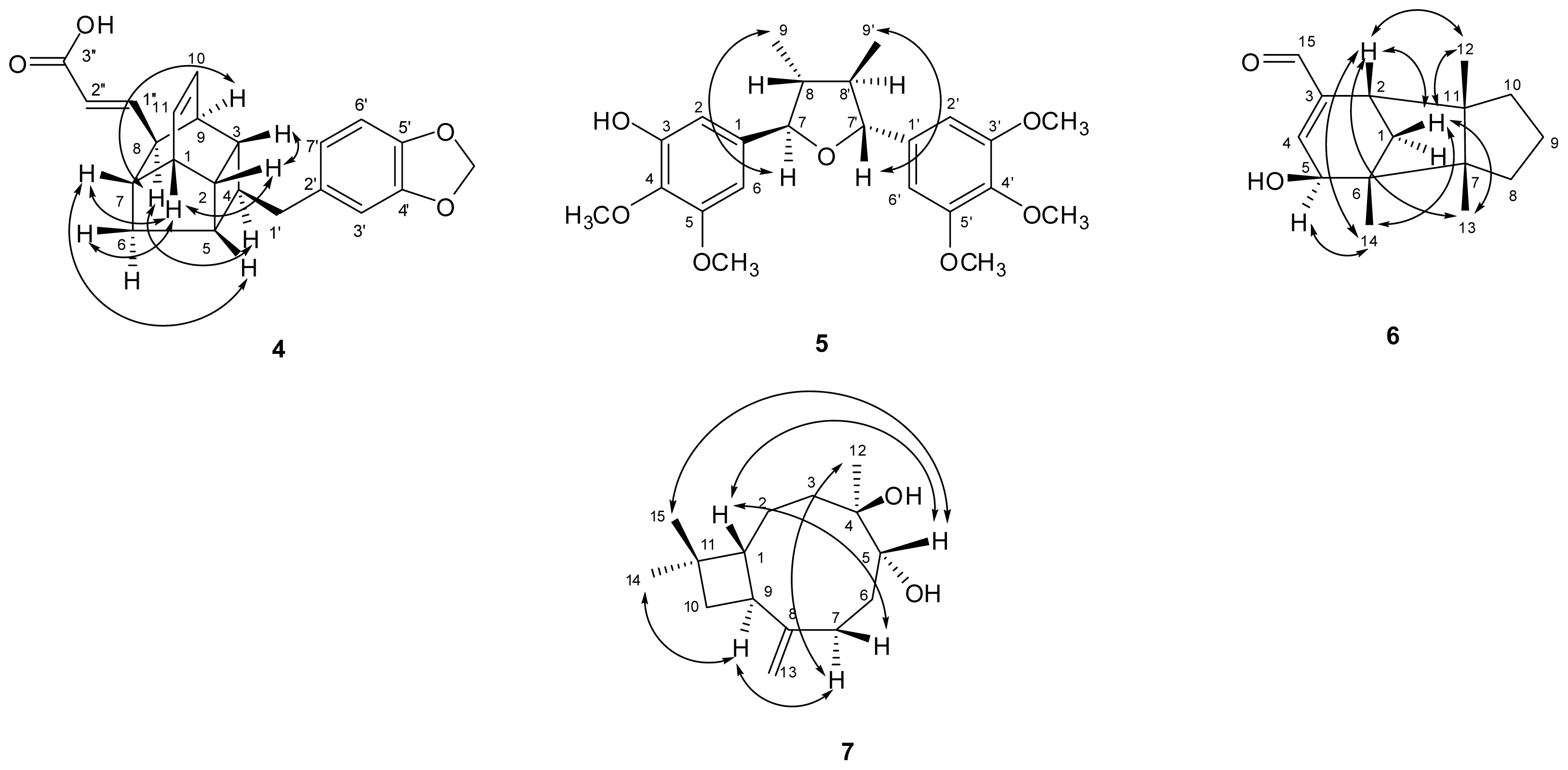
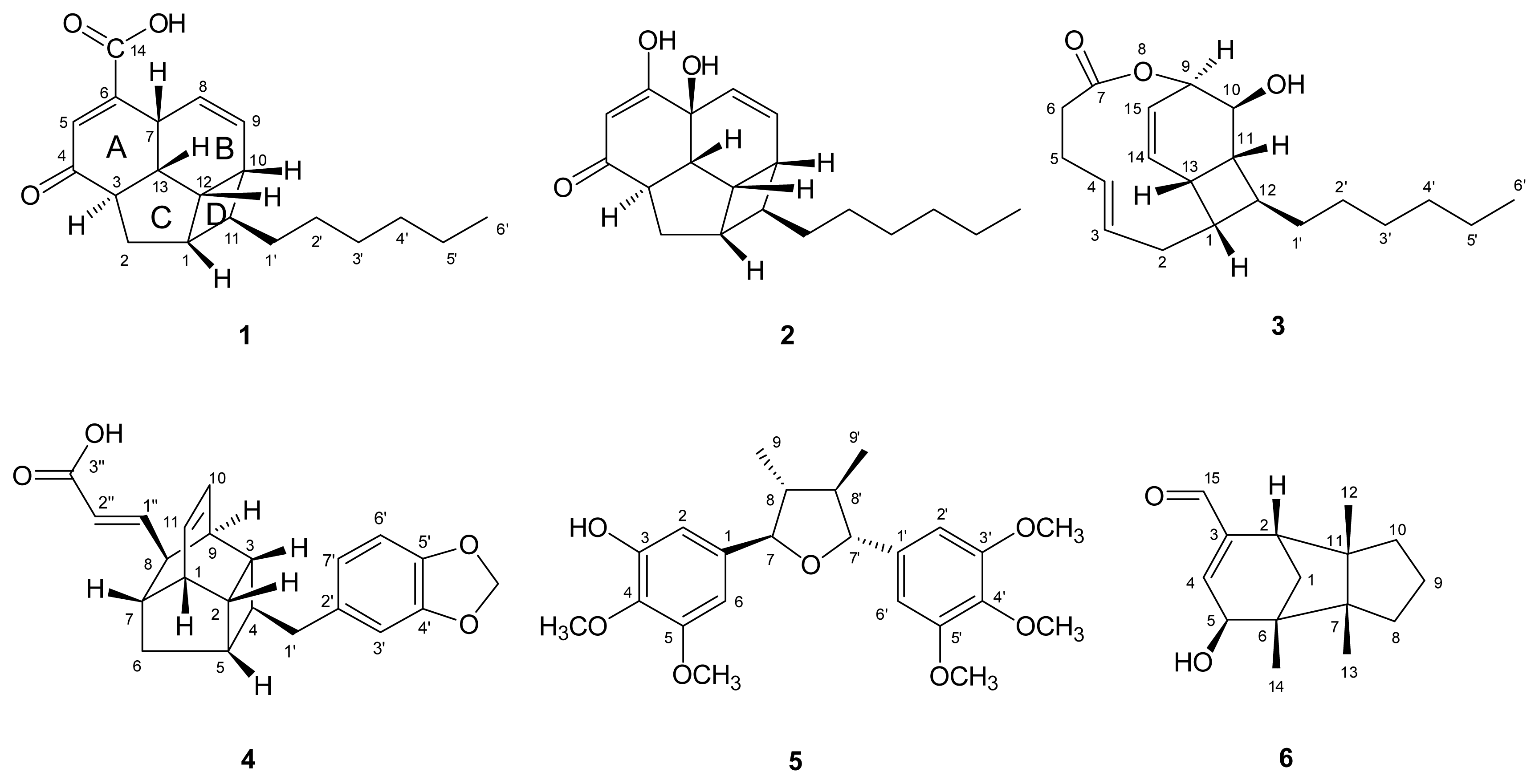
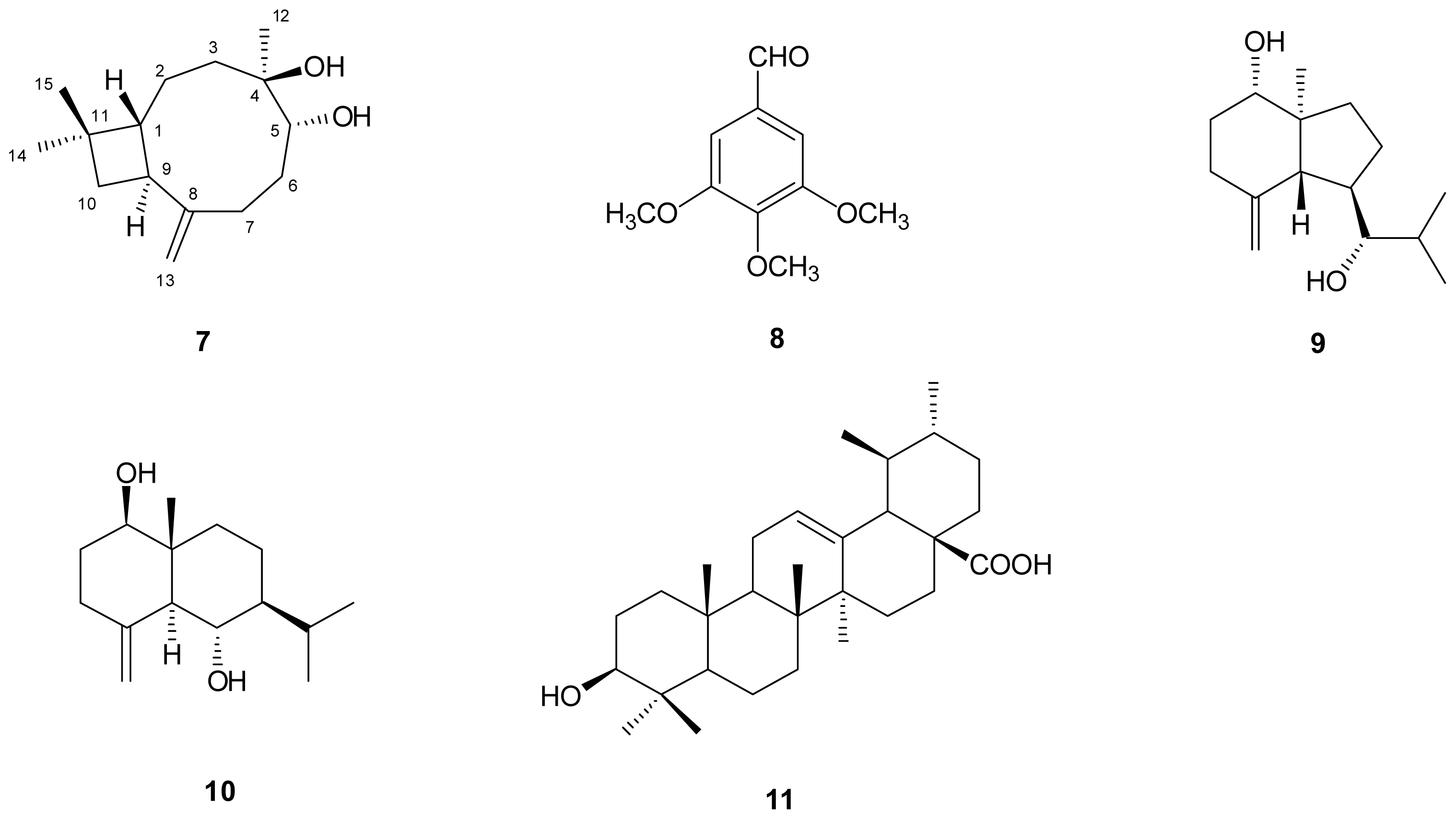
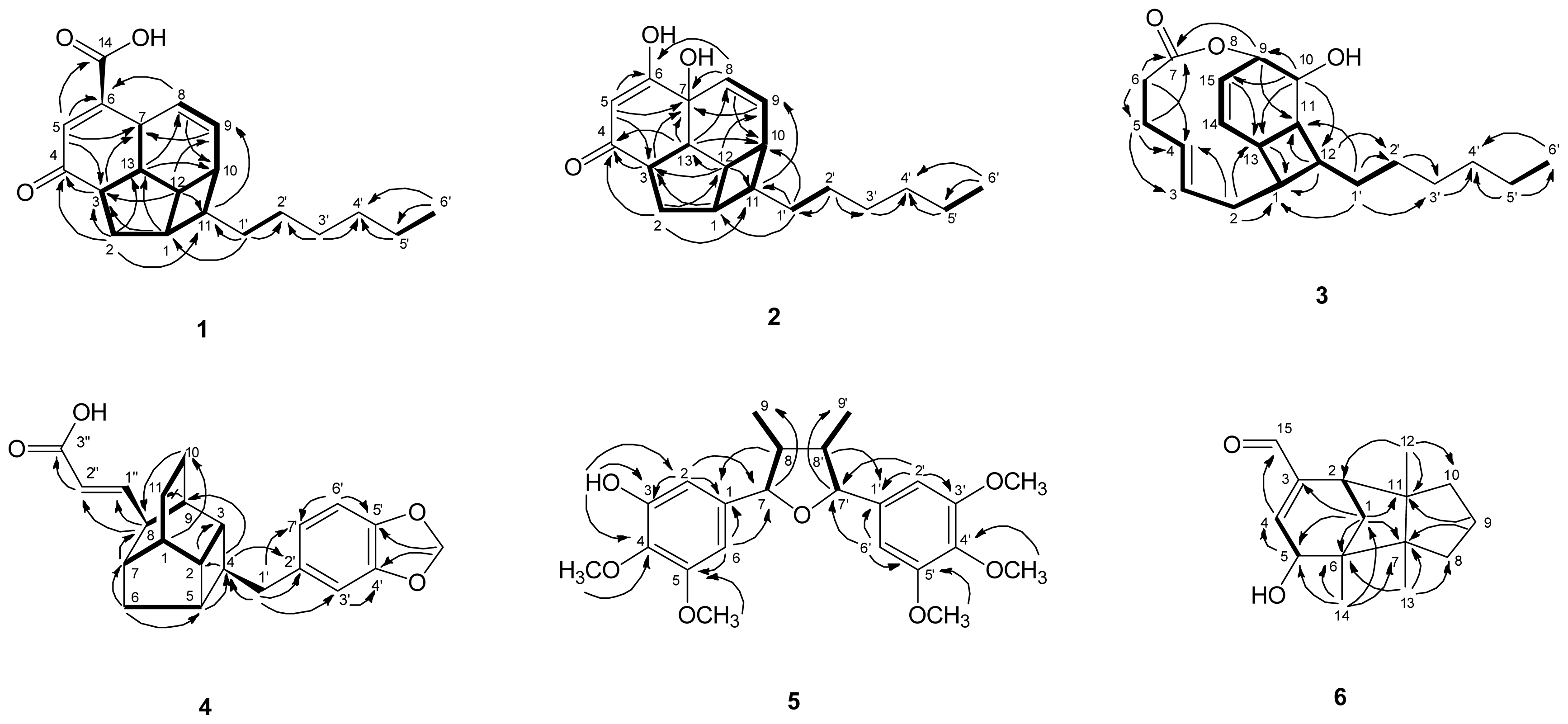
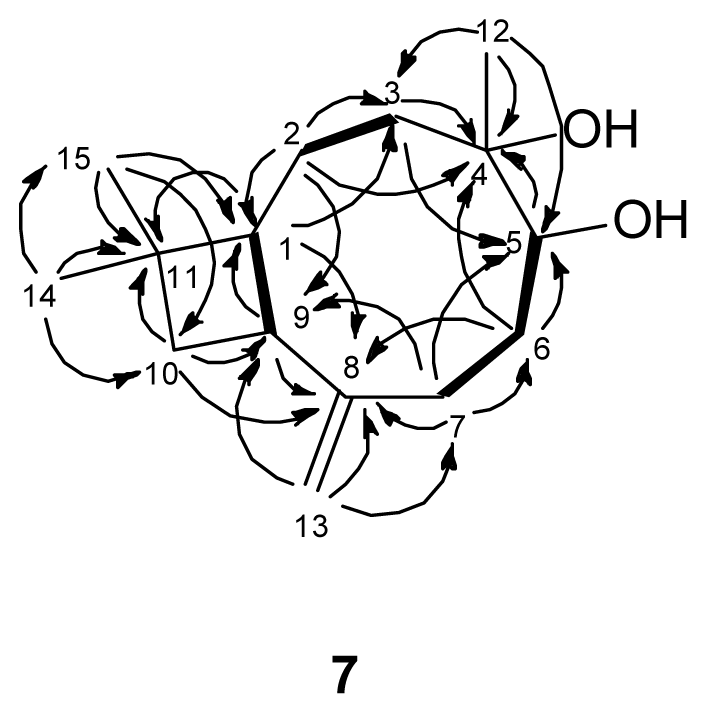
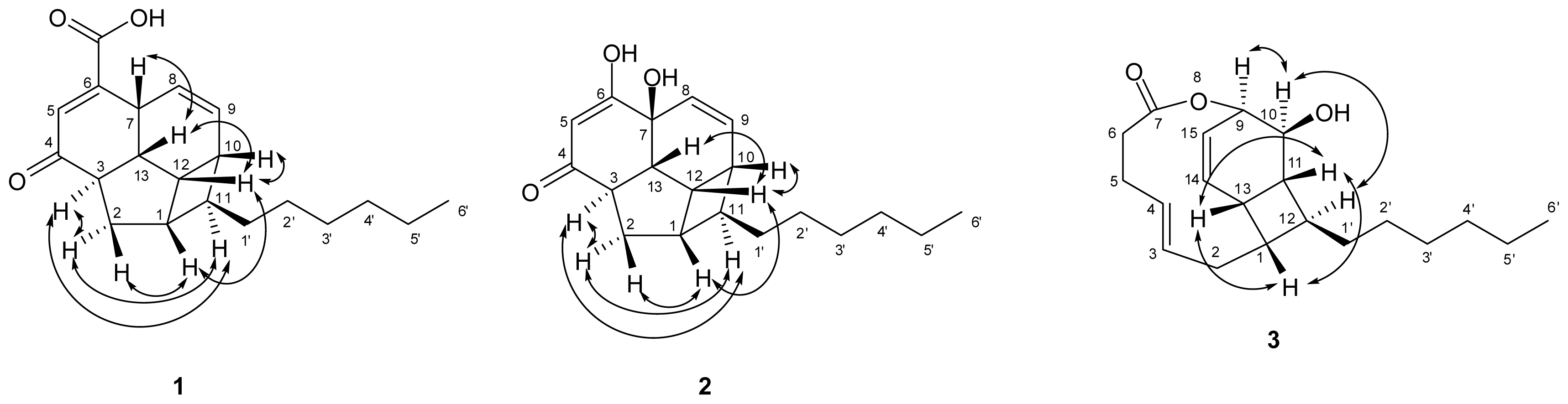
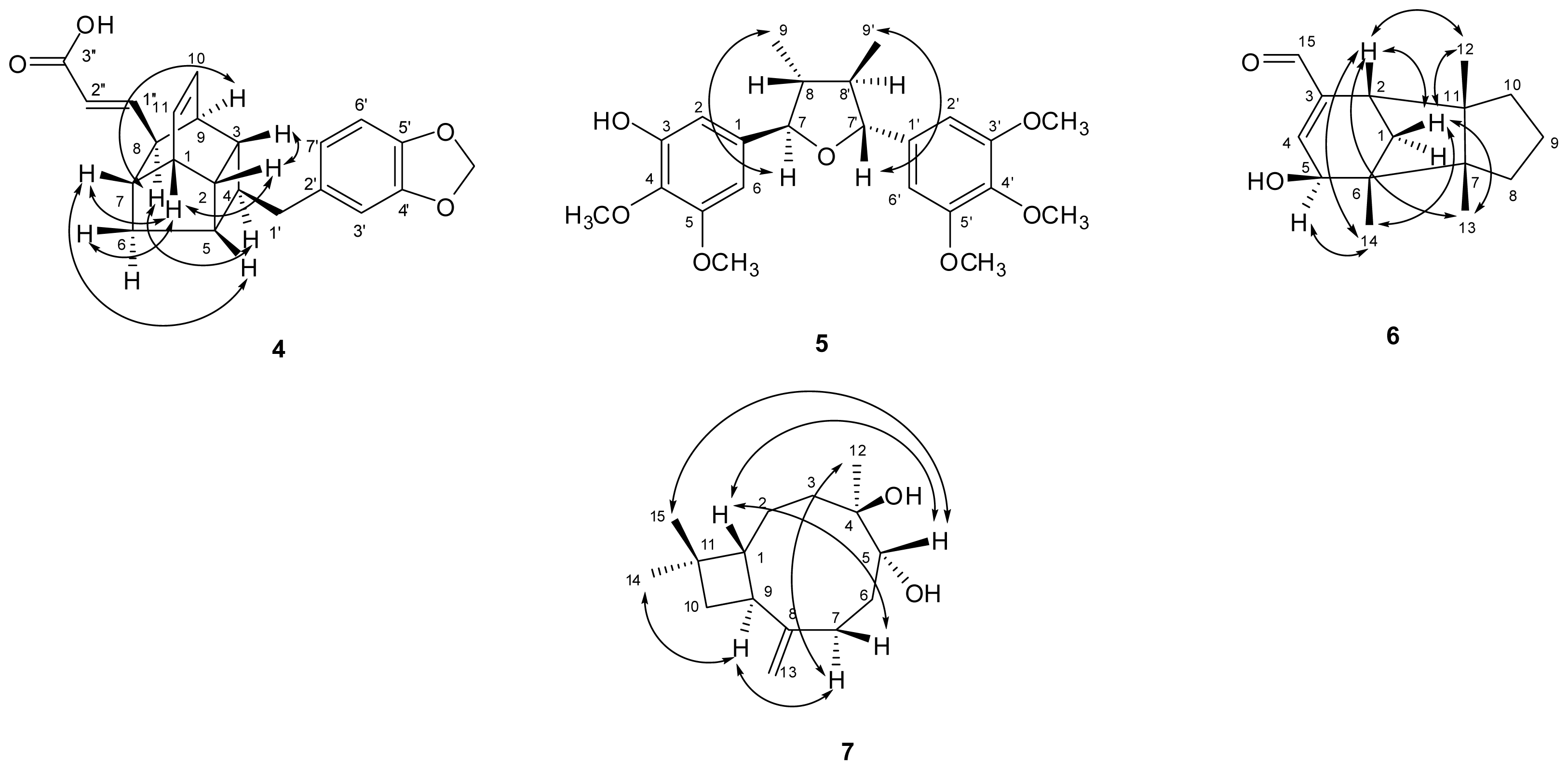
 ) connectivities for compounds 1–7.
) connectivities for compounds 1–7.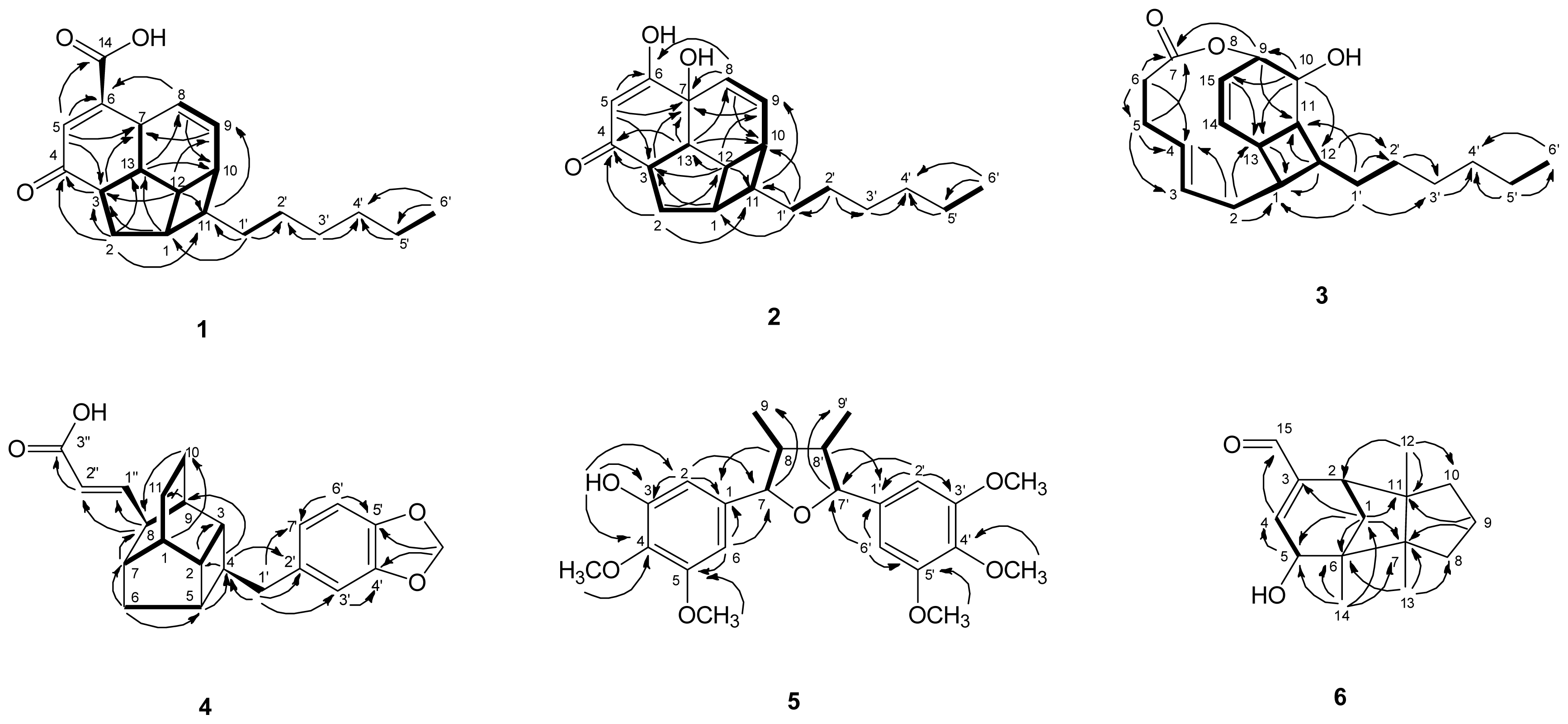
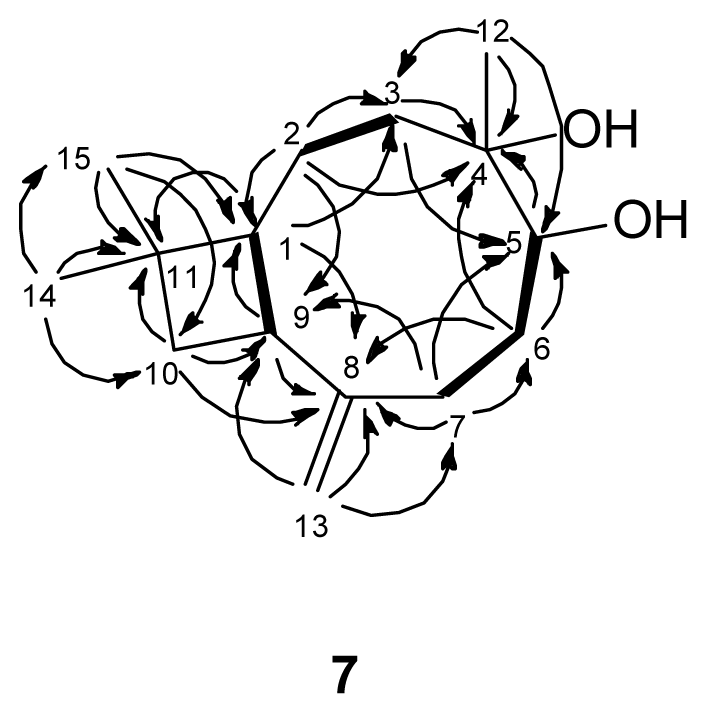
 ) connectivities for compounds 1–7.
) connectivities for compounds 1–7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| position | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 41.6 (CH) | 2.36, m | 42.0 (CH) | 2.41, br dd (12.0, 6.0) |
| 2 | 31.5 (CH2) | 1.56, td (12.0, 6.0) 1.65, br dd (12.0, 5.4) | 31.0 (CH2) | 1.57, td (12.0, 6.0) 1.62, br dd (12.0, 5.4) |
| 3 | 50.5 (CH) | 3.05, ddd (13.5, 12.0, 5.4) | 54.3 (CH) | 2.94, ddd (13.2, 12.0, 5.2) |
| 4 | 201.6 (C) | - | 203.7 (C) | - |
| 5 | 135.8 (CH) | 6.70, d (1.2) | 132.3 (CH) | 6.54, s |
| 6 | 151.5 (C) | - | 158.5 (C) | - |
| 7 | 36.2 (CH) | 3.51, br s | 71.2 (C) | - |
| 8 | 123.5 (CH) | 5.56, ddd (10.2, 3.0, 1.8) | 125.9 (CH) | 6.01, br d (10.2) |
| 9 | 131.5 (CH) | 5.85, ddd (10.2, 4.2, 3.0) | 132.6 (CH) | 5.87, dd (10.2, 4.2) |
| 10 | 35.8 (CH) | 2.40, m | 35.9 (CH) | 2.47, m |
| 11 | 48.0 (CH) | 1.47, m | 47.9 (CH) | 1.45, m |
| 12 | 35.2 (CH) | 2.87, m | 32.9 (CH) | 2.97, br dd (8.4, 6.0) |
| 13 | 44.8 (CH) | 2.35, m | 51.9 (CH) | 2.24, ddd (13.2, 8.4, 1.2) |
| 14 | 168.3 (C) | - | - | - |
| 1′ | 38.4 (CH2) | 1.54, m | 38.2 (CH2) | 1.54, m |
| 2′ | 28.3 (CH2) | 1.28, m | 28.0 (CH2) | 1.25, m |
| 3′ | 31.0 (CH2) | 1.28, m | 30.4 (CH2) | 1.28, m |
| 4′ | 33.2 (CH2) | 1.28, m | 33.0 (CH2) | 1.28, m |
| 5′ | 23.9 (CH2) | 1.28, m | 23.7 (CH2) | 1.30, m |
| 6′ | 15.0 (CH3) | 0.88, t (7.2) | 14.4 (CH3) | 0.90, t (6.9) |
| position | 3 | 4 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 41.9 (CH) | 2.37, m | 43.7 (CH) | 2.72, br dd (12.0, 5.4) |
| 2 | 31.8 (CH2) | 1.28, m 1.99, ddd (14.4, 9.6, 4.8) | 41.3 (CH) | 2.45, dt (9.0, 5.4) |
| 3 | 132.3 (CH) | 5.50, ddd (15.6, 9.6, 6.0) | 41.1 (CH) | 1.72, m |
| 4 | 130.1 (CH) | 5.24, ddd (15.6, 9.6, 5.4) | 41.2 (CH) | 2.25, br t (7.8) |
| 5 | 31.2 (CH2) | 2.38, m | 41.0 (CH) | 2.36, m |
| 6 | 34.3 (CH2) | 2.29, ddd (11.4, 5.4, 2.4) 2.22, m | 39.9 (CH2) | 1.63, br d (12.3) 1.87, br dd (12.3, 5.4) |
| 7 | 173.5 (C) | - | 43.4 (CH) | 1.89, br dd (9.0, 5.4) |
| 8 | - | - | 48.2 (CH) | 2.91, br dd (8.2, 3.6) |
| 9 | 70.1 (CH) | 4.87, dt (5.6, 2.4) | 38.8 (CH) | 2.53, m |
| 10 | 67.2 (CH) | 3.96, br d (7.2) | 134.2 (CH) | 6.19, br t (7.2) |
| 11 | 40.8 (CH) | 2.21, m | 131.9 (CH) | 6.25, br t (7.2) |
| 12 | 33.3 (CH) | 3.18, br dt (15.6, 7.5) | - | - |
| 13 | 30.9 (CH) | 3.02, m | - | - |
| 14 | 136.0 (CH) | 6.16, dd (10.4, 6.0) | - | - |
| 15 | 123.9 (CH) | 6.25, dd (10.4, 5.6) | - | - |
| 1′ | 36.3 (CH2) | 1.47, q (7.5) | 43.1 (CH2) | 2.78, dd (15.6, 7.8) 2.81, br d (7.8) |
| 2′ | 27.7 (CH2) | 1.27–1.33, m | 136.5 (C) | - |
| 3′ | 29.5 (CH2) | 1.27–1.33, m | 110.5 (CH) | 6.75, d (1.8) |
| 4′ | 31.9 (CH2) | 2.24, m | 149.2 (C) | - |
| 5′ | 22.7 (CH2) | 1.27–1.33, m | 147.3 (C) | - |
| 6′ | 14.1 (CH3) | 0.90, t (6.9) | 109.3 (CH) | 6.74, d (7.8) |
| 7′ | - | - | 123.1 (CH) | 6.68, dd (7.8, 1.8) |
| 1″ | - | - | 155.3 (CH) | 6.72, dd (15.6, 8.2) |
| 2″ | - | - | 121.3 (CH) | 5.71, dd (15.6, 0.9) |
| 3″ | - | - | 168.4 (C) | - |
| OCH2O | - | - | 102.3 (CH2) | 5.94, s |
| position | 6 | 7 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 36.9 (CH2) | 1.37, br d (12.0) 1.69, dd (12.0, 4.2) | 56.9 (CH) | 1.65, ddd (10.2, 3.6, 1.8) |
| 2 | 42.8 (CH) | 2.64, d (4.2) | 23.2 (CH2) | 1.33, m 1.66, m |
| 3 | 147.7 (C) | - | 40.8 (CH2) | 1.56, ddd (15.0, 8.4, 1.8) 1.93, ddd (15.0, 11.1, 2.1) |
| 4 | 147.5 (CH) | 6.60, dd (3.6, 0.6) | 75.1 (C) | - |
| 5 | 71.2 (CH) | 4.09, d (3.6) | 73.3 (CH) | 3.60, br dd (6.0, 2.4) |
| 6 | 48.7 (C) | - | 32.6 (CH2) | 1.57, m 1.75, m |
| 7 | 56.9 (C) | - | 34.7 (CH2) | 2.05, m 2.43, ddd (13.2, 9.0, 4.2) |
| 8 | 36.2 (CH2) | 1.10, m 1.58, m | 151.8 (C) | - |
| 9 | 38.6 (CH2) | 1.25, m | 42.3 (CH) | 2.37, q (10.2) |
| 10 | 27.1 (CH2) | 1.32, br dd (6.9, 0.9) 1.54, m | 36.0 (CH2) | 1.58, m 1.74, t (10.2) |
| 11 | 54.9 (C) | - | 34.1 (C) | - |
| 12 | 26.9 (CH3) | 1.089, s | 21.5 (CH3) | 1.14, s |
| 13 | 24.2 (CH3) | 0.96, s | 110.5 (CH2) | 4.92, d (1.2) 4.94, d (1.2) |
| 14 | 18.7 (CH3) | 1.087, s | 22.1 (CH3) | 0.98, s |
| 15 | 193.3 (CH) | 9.54, s | 30.1 (CH3) | 1.00, s |
| OH | - | - | - | 2.07, br s |
| OH | - | - | - | 2.29, br d (3.0) |
| Compounds | Emax (%) a | IC50 (μM) b |
|---|---|---|
| endiandric acid M (4) | 97.03 ± 1.30 | 31.70 ± 0.25 |
| beilschminol B (5) | 55.62 ± 1.96 | 95.37 ± 0.52 |
| ursolic acid (11) | 36.55 ± 4.66 | >100 |
| 6β-hydroxystigmast-4-en-3-one c | 11.69 ± 2.91 | >100 |
| rel-(7S,8S,7′R,8′R)-3,4,5,3′,4′,5′-hexmethoxylignanc | 52.67 ± 5.05 | 98.26 ± 0.13 |
| aminoguanidine d (a selective iNOS inhibitor) | 80.35 ± 0.26 | 26.55 ± 0.48 |
| Nω-nitro-L-arginine d (a nonselective iNOS inhibitor) | 43.72 ± 0.76 | 152.46 ± 10.53 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, Y.-T.; Chang, H.-S.; Wang, G.-J.; Lin, C.-H.; Chen, I.-S. Secondary Metabolites from the Roots of Beilschmiedia tsangii and Their Anti-Inflammatory Activities. Int. J. Mol. Sci. 2012, 13, 16430-16443. https://doi.org/10.3390/ijms131216430
Huang Y-T, Chang H-S, Wang G-J, Lin C-H, Chen I-S. Secondary Metabolites from the Roots of Beilschmiedia tsangii and Their Anti-Inflammatory Activities. International Journal of Molecular Sciences. 2012; 13(12):16430-16443. https://doi.org/10.3390/ijms131216430
Chicago/Turabian StyleHuang, Yun-Ting, Hsun-Shuo Chang, Guei-Jane Wang, Chu-Hung Lin, and Ih-Sheng Chen. 2012. "Secondary Metabolites from the Roots of Beilschmiedia tsangii and Their Anti-Inflammatory Activities" International Journal of Molecular Sciences 13, no. 12: 16430-16443. https://doi.org/10.3390/ijms131216430