Base Excision Repair in Physiology and Pathology of the Central Nervous System

Abstract
:1. Introduction
2. DNA Glycosylases
2.1. The Helix-Hairpin-Helix DNA Glycosylases
2.1.1. OGG1
2.1.1.1. Parkinson’s Disease
2.1.1.2. Amyotrophic Lateral Sclerosis
2.1.1.3. Triplet Repeat Expansion Diseases
2.1.1.4. Stroke/Ischemia/Hypoxia
2.1.1.5. Alzheimer’s Disease
2.1.1.6. Involvement of OGG1 in other Neurodegenerative Disorders
2.1.2. MUTYH
2.1.2.1. Parkinson’s Disease
2.1.2.2. Stroke/Ischemia/Hypoxia
2.1.2.3. Involvement of MUTYH in Other Neurodegenerative Disorders
2.1.3. MBD4
2.1.4. NTHL1
2.2. The Endonuclease VIII-Like Glycosylases
2.2.1. NEIL1
2.2.1.1. Involvement of NEIL1 in Neurodegenerative Disorders
2.2.1.2. Stroke/Ischemia/Hypoxia
2.2.2. NEIL2
2.2.3. NEIL3
2.2.3.1. Stroke/Ischemia/Hypoxia
2.3. The Alkyladenine DNA Glycosylase
2.3.1. Involvement of AAG in Neurodegenerative Disorders
2.4. The Uracil DNA Glycosylases
2.4.1. UNG
2.4.1.1. Involvement of UNG in Neurodegenerative Disorders
2.4.2. TDG
3. BER Proteins other than DNA Glycosylases
3.1. Apurinic/Apyrimidinic Endonuclease 1
3.1.1. Alzheimer’s Disease
3.1.2. Involvement of APE1 in other Neurodegenerative Disorders
3.1.3. Stroke/Ischemia/Hypoxia
3.2. Polynucleotide Kinase
3.2.1. Involvement of PNK in Neurodegenerative Disorders
3.3. DNA Polymerase β
3.3.1. Alzheimer’s and Parkinson Disease
3.3.2. Triplet Repeat Expansion Diseases
3.3.3. Stroke/Ischemia/Hypoxia
3.4. DNA Polymerases δ and ɛ
3.5. X-Ray Repair Cross Complementing 1 Protein
3.5.1. Alzheimer’s and Parkinson Disease
3.5.2. Stroke/Ischemia/Hypoxia
3.6. Flap Endonuclease 1
3.6.1. Triplet Repeat Expansion Diseases
3.7. DNA Ligase I/III
3.7.1. Stroke/Ischemia/Hypoxia
4. Conclusions and Future Perspectives
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar]
- Van Loon, B.; Markkanen, E.; Hubscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar]
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci 2009, 66, 1039–1056. [Google Scholar]
- Magistretti, P.J.; Pellerin, L. Cellular bases of brain energy metabolism and their relevance to functional brain imaging: Evidence for a prominent role of astrocytes. Cerebr. Cortex 1996, 6, 50–61. [Google Scholar]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging 2001, 18, 685–716. [Google Scholar]
- Jacobs, A.L.; Schar, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar]
- Matsumoto, Y.; Kim, K.; Bogenhagen, D.F. Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: An alternative pathway of base excision DNA repair. Mol. Cell. Biol 1994, 14, 6187–6197. [Google Scholar]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem 1996, 271, 9573–9578. [Google Scholar]
- Liu, Y.; Beard, W.A.; Shock, D.D.; Prasad, R.; Hou, E.W.; Wilson, S.H. DNA polymerase beta and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J. Biol. Chem 2005, 280, 3665–3674. [Google Scholar]
- Stucki, M.; Pascucci, B.; Parlanti, E.; Fortini, P.; Wilson, S.H.; Hubscher, U.; Dogliotti, E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene 1998, 17, 835–843. [Google Scholar]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell. Mol. Life Sci 2009, 66, 981–993. [Google Scholar]
- Nash, H.M.; Bruner, S.D.; Scharer, O.D.; Kawate, T.; Addona, T.A.; Spooner, E.; Lane, W.S.; Verdine, G.L. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol 1996, 6, 968–980. [Google Scholar]
- Van der Kemp, P.A.; Thomas, D.; Barbey, R.; de Oliveira, R.; Boiteux, S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-Nmethylformamidopyrimidine. Proc. Natl. Acad. Sci. USA 1996, 93, 5197–5202. [Google Scholar]
- Lu, R.; Nash, H.M.; Verdine, G.L. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol 1997, 7, 397–407. [Google Scholar]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar]
- Karahalil, B.; Girard, P.M.; Boiteux, S.; Dizdaroglu, M. Substrate specificity of the Ogg1 protein of Saccharomyces cerevisiae: Excision of guanine lesions produced in DNA by ionizing radiation- or hydrogen peroxide/metal ion-generated free radicals. Nucleic Acids Res 1998, 26, 1228–1233. [Google Scholar]
- Dherin, C.; Radicella, J.P.; Dizdaroglu, M.; Boiteux, S. Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res 1999, 27, 4001–4007. [Google Scholar]
- Audebert, M.; Radicella, J.P.; Dizdaroglu, M. Effect of single mutations in the OGG1 gene found in human tumors on the substrate specificity of the Ogg1 protein. Nucleic Acids Res 2000, 28, 2672–2678. [Google Scholar]
- Dherin, C.; Dizdaroglu, M.; Doerflinger, H.; Boiteux, S.; Radicella, J.P. Repair of oxidative DNA damage in Drosophila melanogaster: Identification and characterization of dOgg1, a second DNA glycosylase activity for 8-hydroxyguanine and formamidopyrimidines. Nucleic Acids Res 2000, 28, 4583–4592. [Google Scholar]
- Jensen, A.; Calvayrac, G.; Karahalil, B.; Bohr, V.A.; Stevnsner, T. Mammalian 8-oxoguanine DNA glycosylase 1 incises 8-oxoadenine opposite cytosine in nuclei and mitochondria, while a different glycosylase incises 8-oxoadenine opposite guanine in nuclei. J. Biol. Chem 2003, 278, 19541–19548. [Google Scholar]
- Morales-Ruiz, T.; Birincioglu, M.; Jaruga, P.; Rodriguez, H.; Roldan-Arjona, T.; Dizdaroglu, M. Arabidopsis thaliana Ogg1 protein excises 8-hydroxyguanine and 2,6-diamino-4-hydroxy-5-formamidopyrimidine from oxidatively damaged DNA containing multiple lesions. Biochemistry 2003, 42, 3089–3095. [Google Scholar]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar]
- Rolseth, V.; Runden-Pran, E.; Luna, L.; McMurray, C.; Bjoras, M.; Ottersen, O.P. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair 2008, 7, 1578–1588. [Google Scholar]
- Swain, U.; Subba Rao, K. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev 2011, 132, 374–381. [Google Scholar]
- Swain, U.; Rao, K.S. Age-dependent decline of DNA base excision repair activity in rat cortical neurons. Mech. Ageing Dev 2012, 133, 186–194. [Google Scholar]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol 2006, 26, 1654–1665. [Google Scholar]
- Koltai, E.; Zhao, Z.; Lacza, Z.; Cselenyak, A.; Vacz, G.; Nyakas, C.; Boldogh, I.; Ichinoseki-Sekine, N.; Radak, Z. Combined exercise and insulin-like growth factor-1 supplementation induces neurogenesis in old rats, but do not attenuate age-associated DNA damage. Rejuvenation Res 2011, 14, 585–596. [Google Scholar]
- Ogonovszky, H.; Berkes, I.; Kumagai, S.; Kaneko, T.; Tahara, S.; Goto, S.; Radak, Z. The effects of moderate-, strenuous- and over-training on oxidative stress markers, DNA repair, and memory, in rat brain. Neurochem. Int 2005, 46, 635–640. [Google Scholar]
- Chen, B.; Zhong, Y.; Peng, W.; Sun, Y.; Hu, Y.J.; Yang, Y.; Kong, W.J. Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of D-galactose-induced aging rats. Mol. Biol. Rep 2011, 38, 3635–3642. [Google Scholar]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar]
- La Maestra, S.; Kisby, G.E.; Micale, R.T.; Johnson, J.; Kow, Y.W.; Bao, G.; Sheppard, C.; Stanfield, S.; Tran, H.; Woltjer, R.L.; et al. Cigarette smoke induces DNA damage and alters base-excision repair and tau levels in the brain of neonatal mice. Toxicol. Sci 2011, 123, 471–479. [Google Scholar]
- Gu, A.; Shi, X.; Yuan, C.; Ji, G.; Zhou, Y.; Long, Y.; Song, L.; Wang, S.; Wang, X. Exposure to fenvalerate causes brain impairment during zebrafish development. Toxicol. Lett 2010, 197, 188–192. [Google Scholar]
- Stedeford, T.; Cardozo-Pelaez, F.; Nemeth, N.; Song, S.; Harbison, R.D.; Sanchez-Ramos, J. Comparison of base-excision repair capacity in proliferating and differentiated PC 12 cells following acute challenge with dieldrin. Free Radic. Biol. Med 2001, 31, 1272–1278. [Google Scholar]
- Bolin, C.M.; Basha, R.; Cox, D.; Zawia, N.H.; Maloney, B.; Lahiri, D.K.; Cardozo-Pelaez, F. Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J 2006, 20, 788–790. [Google Scholar]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjoras, M.; Eide, L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci 2011, 31, 9746–9751. [Google Scholar]
- Reis, A.; Hermanson, O. The DNA glycosylases OGG1 and NEIL3 influence differentiation potential, proliferation, and senescence-associated signs in neural stem cells. Biochem. Biophys. Res. Commun 2012, 423, 621–626. [Google Scholar]
- Coelho, M.; Ferreira, J.J. Late-stage Parkinson disease. Nat. Rev. Neurol. 2012. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol 2011, 94, 166–200. [Google Scholar]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J 2012, 31, 3038–3062. [Google Scholar]
- Shimura-Miura, H.; Hattori, N.; Kang, D.; Miyako, K.; Nakabeppu, Y.; Mizuno, Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann. Neurol 1999, 46, 920–924. [Google Scholar]
- Fukae, J.; Mizuno, Y.; Hattori, N. Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 2007, 7, 58–62. [Google Scholar]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int 2012, 61, 721–730. [Google Scholar]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res 2007, 85, 919–934. [Google Scholar]
- Duan, W.X.; Hua, R.X.; Yi, W.; Shen, L.J.; Jin, Z.X.; Zhao, Y.H.; Yi, D.H.; Chen, W.S.; Yu, S.Q. The association between OGG1 Ser326Cys polymorphism and lung cancer susceptibility: A meta-analysis of 27 studies. PLoS One 2012, 7, e35970. [Google Scholar]
- Yuan, W.; Xu, L.; Feng, Y.; Yang, Y.; Chen, W.; Wang, J.; Pang, D.; Li, D. The hOGG1 Ser326Cys polymorphism and breast cancer risk: A meta-analysis. Breast Cancer Res. Treat 2010, 122, 835–842. [Google Scholar]
- Coppede, F.; Ceravolo, R.; Migheli, F.; Fanucchi, F.; Frosini, D.; Siciliano, G.; Bonuccelli, U.; Migliore, L. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson’s disease. Neurosci. Lett 2010, 473, 248–251. [Google Scholar]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar]
- Kikuchi, H.; Furuta, A.; Nishioka, K.; Suzuki, S.O.; Nakabeppu, Y.; Iwaki, T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol 2002, 103, 408–414. [Google Scholar]
- Coppede, F.; Mancuso, M.; Lo Gerfo, A.; Carlesi, C.; Piazza, S.; Rocchi, A.; Petrozzi, L.; Nesti, C.; Micheli, D.; Bacci, A.; et al. Association of the hOGG1 Ser326Cys polymorphism with sporadic amyotrophic lateral sclerosis. Neurosci. Lett 2007, 420, 163–168. [Google Scholar]
- Murakami, T.; Nagai, M.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Kurata, T.; Takehisa, Y.; Kamiya, T.; Abe, K. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res 2007, 1150, 182–189. [Google Scholar]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci 2007, 30, 575–621. [Google Scholar]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar]
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet 2010, 11, 786–799. [Google Scholar]
- Jarem, D.A.; Wilson, N.R.; Schermerhorn, K.M.; Delaney, S. Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair 2011, 10, 887–896. [Google Scholar]
- Lin, Y.; Wilson, J.H. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol. Cell. Biol 2007, 27, 6209–6217. [Google Scholar]
- Goula, A.V.; Berquist, B.R.; Wilson, D.M., 3rd; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet 2009, 5, e1000749. [Google Scholar]
- Coppede, F.; Migheli, F.; Ceravolo, R.; Bregant, E.; Rocchi, A.; Petrozzi, L.; Unti, E.; Lonigro, R.; Siciliano, G.; Migliore, L. The hOGG1 Ser326Cys polymorphism and Huntington’s disease. Toxicology 2010, 278, 199–203. [Google Scholar]
- Broussalis, E.; Killer, M.; McCoy, M.; Harrer, A.; Trinka, E.; Kraus, J. Current therapies in ischemic stroke. Part A. Recent developments in acute stroke treatment and in stroke prevention. Drug Discov. Today 2012, 17, 296–309. [Google Scholar]
- Bohr, V.A.; Ottersen, O.P.; Tonjum, T. Genome instability and DNA repair in brain, ageing and neurological disease. Neuroscience 2007, 145, 1183–1186. [Google Scholar]
- He, M.; Xing, S.; Yang, B.; Zhao, L.; Hua, H.; Liang, Z.; Zhou, W.; Zeng, J.; Pei, Z. Ebselen attenuates oxidative DNA damage and enhances its repair activity in the thalamus after focal cortical infarction in hypertensive rats. Brain Res 2007, 1181, 83–92. [Google Scholar]
- Rolseth, V.; Runden-Pran, E.; Neurauter, C.G.; Yndestad, A.; Luna, L.; Aukrust, P.; Ottersen, O.P.; Bjoras, M. Base excision repair activities in organotypic hippocampal slice cultures exposed to oxygen and glucose deprivation. DNA Repair 2008, 7, 869–878. [Google Scholar]
- Li, W.; Luo, Y.; Zhang, F.; Signore, A.P.; Gobbel, G.T.; Simon, R.P.; Chen, J. Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway. J. Cerebr. Blood Flow Metabol 2006, 26, 181–198. [Google Scholar]
- Liu, D.; Croteau, D.L.; Souza-Pinto, N.; Pitta, M.; Tian, J.; Wu, C.; Jiang, H.; Mustafa, K.; Keijzers, G.; Bohr, V.A.; et al. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J. Cerebr. Blood Flow Metabol 2011, 31, 680–692. [Google Scholar]
- Yang, Y.; Candelario-Jalil, E.; Thompson, J.F.; Cuadrado, E.; Estrada, E.Y.; Rosell, A.; Montaner, J.; Rosenberg, G.A. Increased intranuclear matrix metalloproteinase activity in neurons interferes with oxidative DNA repair in focal cerebral ischemia. J. Neurochem 2010, 112, 134–149. [Google Scholar]
- Hill, J.W.; Poddar, R.; Thompson, J.F.; Rosenberg, G.A.; Yang, Y. Intranuclear matrix metalloproteinases promote DNA damage and apoptosis induced by oxygen-glucose deprivation in neurons. Neuroscience 2012, 220, 277–290. [Google Scholar]
- Lin, R.; Roseborough, G.; Dong, Y.; Williams, G.M.; Wei, C. DNA damage and repair system in spinal cord ischemia. J. Vasc. Surg 2003, 37, 847–858. [Google Scholar]
- Dalen, M.L.; Alme, T.N.; Bjoras, M.; Munkeby, B.H.; Rootwelt, T.; Saugstad, O.D. Reduced expression of DNA glycosylases in post-hypoxic newborn pigs undergoing therapeutic hypothermia. Brain Res 2010, 1363, 198–205. [Google Scholar]
- Lovell, M.A.; Soman, S.; Bradley, M.A. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech. Ageing Dev 2011, 132, 443–448. [Google Scholar]
- Feng, J.F.; He, L.L.; Li, D.; Yuan, L.H.; Yu, H.L.; Ma, W.W.; Yang, Y.; Xi, Y.D.; Ding, J.; Xiao, Y.X.; et al. Antagonizing Effects of Soybean Isoflavones on beta-amyloid Peptides-induced Oxidative Damage in Neuron Mitochondria of Rats. Basic Clin. Pharmacol. Toxicol 2012, 111, 248–253. [Google Scholar]
- Wu, M.; Audet, A.; Cusic, J.; Seeger, D.; Cochran, R.; Ghribi, O. Broad DNA repair responses in neural injury are associated with activation of the IL-6 pathway in cholesterol-fed rabbits. J. Neurochem 2009, 111, 1011–1021. [Google Scholar]
- Forlenza, M.J.; Miller, G.E. Increased serum levels of 8-hydroxy-2’-deoxyguanosine in clinical depression. Psychosom. Med 2006, 68, 1–7. [Google Scholar]
- Teyssier, J.R.; Ragot, S.; Chauvet-Gelinier, J.C.; Trojak, B.; Bonin, B. Expression of oxidative stress-response genes is not activated in the prefrontal cortex of patients with depressive disorder. Psychiatr. Res 2011, 186, 244–247. [Google Scholar]
- Lin, Y.; Xu, J.; Cao, L.; Han, Y.; Gao, J.; Xie, N.; Zhao, X.; Jiang, H.; Chi, Z. Mitochondrial base excision repair pathway failed to respond to status epilepticus induced by pilocarpine. Neurosci. Lett 2010, 474, 22–25. [Google Scholar]
- Laposa, R.R.; Huang, E.J.; Cleaver, J.E. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc. Natl. Acad Sci. USA 2007, 104, 1389–1394. [Google Scholar]
- McGoldrick, J.P.; Yeh, Y.C.; Solomon, M.; Essigmann, J.M.; Lu, A.L. Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell. Biol 1995, 15, 989–996. [Google Scholar]
- Slupska, M.M.; Baikalov, C.; Luther, W.M.; Chiang, J.H.; Wei, Y.F.; Miller, J.H. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol 1996, 178, 3885–3892. [Google Scholar]
- Van Loon, B.; Hubscher, U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc. Natl. Acad Sci. USA 2009, 106, 18201–18206. [Google Scholar]
- Markkanen, E.; van Loon, B.; Ferrari, E.; Hubscher, U. Ubiquitylation of DNA polymerase lambda. FEBS Lett. 2011. [Google Scholar] [CrossRef]
- Markkanen, E.; Hubscher, U.; van Loon, B. Regulation of oxidative DNA damage repair: The adenine:8-oxo-guanine problem. Cell. Cycle 2012, 11, 1070–1075. [Google Scholar]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res 2004, 32, 477–487. [Google Scholar]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet 2002, 30, 227–232. [Google Scholar]
- Win, A.K.; Cleary, S.P.; Dowty, J.G.; Baron, J.A.; Young, J.P.; Buchanan, D.D.; Southey, M.C.; Burnett, T.; Parfrey, P.S.; Green, R.C.; et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int. J. Cancer 2011, 129, 2256–2262. [Google Scholar]
- Russo, M.T.; de Luca, G.; Degan, P.; Parlanti, E.; Dogliotti, E.; Barnes, D.E.; Lindahl, T.; Yang, H.; Miller, J.H.; Bignami, M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res 2004, 64, 4411–4414. [Google Scholar]
- Lee, H.M.; Hu, Z.; Ma, H.; Greeley, G.H., Jr; Wang, C.; Englander, E.W. Developmental changes in expression and subcellular localization of the DNA repair glycosylase, MYH, in the rat brain. J. Neurochem. 2004, 88, 394–400. [Google Scholar]
- Arai, T.; Fukae, J.; Hatano, T.; Kubo, S.; Ohtsubo, T.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson’s disease. Acta Neuropathol 2006, 112, 139–145. [Google Scholar]
- He, M.X.; Zeng, J.S.; Hua, H.Y.; Xing, S.H.; Ba, Y.P. Changes in DNA repair enzymes in rat ventroposterior nucleus of the thalamus after cerebral cortex infarction. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 2010, 22, 587–590. [Google Scholar]
- Lee, H.M.; Wang, C.; Hu, Z.; Greeley, G.H.; Makalowski, W.; Hellmich, H.L.; Englander, E.W. Hypoxia induces mitochondrial DNA damage and stimulates expression of a DNA repair enzyme, the Escherichia coli MutY DNA glycosylase homolog (MYH), in vivo, in the rat brain. J. Neurochem 2002, 80, 928–937. [Google Scholar]
- Englander, E.W.; Hu, Z.; Sharma, A.; Lee, H.M.; Wu, Z.H.; Greeley, G.H. Rat MYH, a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J. Neurochem 2002, 83, 1471–1480. [Google Scholar]
- Wang, G.; Hazra, T.K.; Mitra, S.; Lee, H.M.; Englander, E.W. Mitochondrial DNA damage and a hypoxic response are induced by CoCl(2) in rat neuronal PC12 cells. Nucleic Acids Res 2000, 28, 2135–2140. [Google Scholar]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 2010, 31, 2002–2010. [Google Scholar]
- Brault, L.S.; Cooper, C.A.; Famula, T.R.; Murray, J.D.; Penedo, M.C. Mapping of equine cerebellar abiotrophy to ECA2 and identification of a potential causative mutation affecting expression of MUTYH. Genomics 2011, 97, 121–129. [Google Scholar]
- Hendrich, B.; Hardeland, U.; Ng, H.H.; Jiricny, J.; Bird, A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 1999, 401, 301–304. [Google Scholar]
- Petronzelli, F.; Riccio, A.; Markham, G.D.; Seeholzer, S.H.; Genuardi, M.; Karbowski, M.; Yeung, A.T.; Matsumoto, Y.; Bellacosa, A. Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): Fundamental role of the catalytic domain. J. Cell. Physiol 2000, 185, 473–480. [Google Scholar]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; Larue, L.; et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc. Natl. Acad Sci. USA 2003, 100, 15071–15076. [Google Scholar]
- Millar, C.B.; Guy, J.; Sansom, O.J.; Selfridge, J.; MacDougall, E.; Hendrich, B.; Keightley, P.D.; Bishop, S.M.; Clarke, A.R. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297, 403–405. [Google Scholar]
- Wong, E.; Yang, K.; Kuraguchi, M.; Werling, U.; Avdievich, E.; Fan, K.; Fazzari, M.; Jin, B.; Brown, A.M.; Lipkin, M.; et al. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc. Natl. Acad Sci. USA 2002, 99, 14937–14942. [Google Scholar]
- Benes, F.M.; Lim, B.; Subburaju, S. Site-specific regulation of cell cycle and DNA repair in post-mitotic GABA cells in schizophrenic versus bipolars. Proc. Natl. Acad Sci. USA 2009, 106, 11731–11736. [Google Scholar]
- Dizdaroglu, M.; Karahalil, B.; Senturker, S.; Buckley, T.J.; Roldan-Arjona, T. Excision of products of oxidative DNA base damage by human NTH1 protein. Biochemistry 1999, 38, 243–246. [Google Scholar]
- Luna, L.; Bjoras, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res 2000, 460, 95–104. [Google Scholar]
- Eide, L.; Luna, L.; Gustad, E.C.; Henderson, P.T.; Essigmann, J.M.; Demple, B.; Seeberg, E. Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry 2001, 40, 6653–6659. [Google Scholar]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad Sci. USA 2002, 99, 3523–3528. [Google Scholar]
- Jaruga, P.; Birincioglu, M.; Rosenquist, T.A.; Dizdaroglu, M. Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry 2004, 43, 15909–15914. [Google Scholar]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem 2005, 280, 40544–40551. [Google Scholar]
- Chan, M.K.; Ocampo-Hafalla, M.T.; Vartanian, V.; Jaruga, P.; Kirkali, G.; Koenig, K.L.; Brown, S.; Lloyd, R.S.; Dizdaroglu, M.; Teebor, G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 2009, 8, 786–794. [Google Scholar]
- Ikeda, S.; Biswas, T.; Roy, R.; Izumi, T.; Boldogh, I.; Kurosky, A.; Sarker, A.H.; Seki, S.; Mitra, S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem 1998, 273, 21585–21593. [Google Scholar]
- Ocampo, M.T.; Chaung, W.; Marenstein, D.R.; Chan, M.K.; Altamirano, A.; Basu, A.K.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Mol. Cell Biol 2002, 22, 6111–6121. [Google Scholar]
- Takao, M.; Kanno, S.; Kobayashi, K.; Zhang, Q.M.; Yonei, S.; van der Horst, G.T.; Yasui, A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J. Biol. Chem 2002, 277, 42205–42213. [Google Scholar]
- Briggs, F.B.; Goldstein, B.A.; McCauley, J.L.; Zuvich, R.L.; de Jager, P.L.; Rioux, J.D.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; Hauser, S.L.; et al. Variation within DNA repair pathway genes and risk of multiple sclerosis. Am. J. Epidemiol 2010, 172, 217–224. [Google Scholar]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem 2002, 277, 30417–30420. [Google Scholar]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res 2002, 30, 4926–4936. [Google Scholar]
- Rosenquist, T.A.; Zaika, E.; Fernandes, A.S.; Zharkov, D.O.; Miller, H.; Grollman, A.P. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 2003, 2, 581–591. [Google Scholar]
- Roy, L.M.; Jaruga, P.; Wood, T.G.; McCullough, A.K.; Dizdaroglu, M.; Lloyd, R.S. Human polymorphic variants of the NEIL1 DNA glycosylase. J. Biol. Chem 2007, 282, 15790–15798. [Google Scholar]
- Muftuoglu, M.; de Souza-Pinto, N.C.; Dogan, A.; Aamann, M.; Stevnsner, T.; Rybanska, I.; Kirkali, G.; Dizdaroglu, M.; Bohr, V.A. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem 2009, 284, 9270–9279. [Google Scholar]
- Jaruga, P.; Xiao, Y.; Vartanian, V.; Lloyd, R.S.; Dizdaroglu, M. Evidence for the involvement of DNA repair enzyme NEIL1 in nucleotide excision repair of (5′R)- and (5′S)-8,5′-cyclo-2′-deoxyadenosines. Biochemistry 2010, 49, 1053–1055. [Google Scholar]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar]
- Parsons, J.L.; Zharkov, D.O.; Dianov, G.L. NEIL1 excises 3′ end proximal oxidative DNA lesions resistant to cleavage by NTH1 and OGG1. Nucleic Acids Res 2005, 33, 4849–4856. [Google Scholar]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem 2003, 278, 49679–49684. [Google Scholar]
- Grin, I.R.; Zharkov, D.O. Eukaryotic endonuclease VIII-like proteins: new components of the base excision DNA repair system. Biochemistry 2011, 76, 80–93. [Google Scholar]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad Sci. USA 2006, 103, 1864–1869. [Google Scholar]
- Englander, E.W.; Ma, H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech. Ageing Dev 2006, 127, 64–69. [Google Scholar]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad Sci. USA 2012, 109, 14948–14953. [Google Scholar]
- Mollersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012. [Google Scholar] [CrossRef]
- Wallace, S.S.; Bandaru, V.; Kathe, S.D.; Bond, J.P. The enigma of endonuclease VIII. DNA Repair 2003, 2, 441–453. [Google Scholar]
- Takao, M.; Oohata, Y.; Kitadokoro, K.; Kobayashi, K.; Iwai, S.; Yasui, A.; Yonei, S.; Zhang, Q.M. Human Nei-like protein NEIL3 has AP lyase activity specific for single-stranded DNA and confers oxidative stress resistance in Escherichia coli mutant. Genes Cells 2009, 14, 261–270. [Google Scholar]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad Sci USA 2010, 107, 4925–4930. [Google Scholar]
- Torisu, K.; Tsuchimoto, D.; Ohnishi, Y.; Nakabeppu, Y. Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease VIII-like protein. J. Biochem 2005, 138, 763–772. [Google Scholar]
- Hildrestrand, G.A.; Neurauter, C.G.; Diep, D.B.; Castellanos, C.G.; Krauss, S.; Bjoras, M.; Luna, L. Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci 2009, 10, 45. [Google Scholar]
- Sejersted, Y.; Hildrestrand, G.A.; Kunke, D.; Rolseth, V.; Krokeide, S.Z.; Neurauter, C.G.; Suganthan, R.; Atneosen-Asegg, M.; Fleming, A.M.; Saugstad, O.D.; et al. Endonuclease VIII-like 3 (Neil3) DNA glycosylase promotes neurogenesis induced by hypoxia-ischemia. Proc. Natl. Acad Sci. USA 2011, 108, 18802–18807. [Google Scholar]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar]
- Lau, A.Y.; Wyatt, M.D.; Glassner, B.J.; Samson, L.D.; Ellenberger, T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl. Acad Sci. USA 2000, 97, 13573–13578. [Google Scholar]
- Lee, C.Y.; Delaney, J.C.; Kartalou, M.; Lingaraju, G.M.; Maor-Shoshani, A.; Essigmann, J.M.; Samson, L.D. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG). Biochemistry 2009, 48, 1850–1861. [Google Scholar]
- Miao, F.; Bouziane, M.; O’Connor, T.R. Interaction of the recombinant human methylpurine-DNA glycosylase (MPG protein) with oligodeoxyribonucleotides containing either hypoxanthine or abasic sites. Nucleic Acids Res 1998, 26, 4034–4041. [Google Scholar]
- Saparbaev, M.; Laval, J. Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc. Natl Acad Sci. USA 1994, 91, 5873–5877. [Google Scholar]
- O’Connor, T.R. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res 1993, 21, 5561–5569. [Google Scholar]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol 2010, 28, 1248–1250. [Google Scholar]
- Meira, L.B.; Moroski-Erkul, C.A.; Green, S.L.; Calvo, J.A.; Bronson, R.T.; Shah, D.; Samson, L.D. Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 888–893. [Google Scholar]
- Engelward, B.P.; Weeda, G.; Wyatt, M.D.; Broekhof, J.L.; de Wit, J.; Donker, I.; Allan, J.M.; Gold, B.; Hoeijmakers, J.H.; Samson, L.D. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 13087–13092. [Google Scholar] [Green Version]
- Hang, B.; Singer, B.; Margison, G.P.; Elder, R.H. Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine. Proc. Natl. Acad. Sci. USA 1997, 94, 12869–12874. [Google Scholar]
- Calvo, J.A.; Meira, L.B.; Lee, C.Y.; Moroski-Erkul, C.A.; Abolhassani, N.; Taghizadeh, K.; Eichinger, L.W.; Muthupalani, S.; Nordstrand, L.M.; Klungland, A.; et al. DNA repair is indispensable for survival after acute inflammation. J. Clin. Invest 2012, 122, 2680–2689. [Google Scholar]
- Kisby, G.E.; Olivas, A.; Park, T.; Churchwell, M.; Doerge, D.; Samson, L.D.; Gerson, S.L.; Turker, M.S. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair 2009, 8, 400–412. [Google Scholar] [Green Version]
- Roth, R.B.; Samson, L.D. 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance. Cancer Res 2002, 62, 656–660. [Google Scholar]
- Kisby, G.E.; Lesselroth, H.; Olivas, A.; Samson, L.; Gold, B.; Tanaka, K.; Turker, M.S. Role of nucleotide- and base-excision repair in genotoxin-induced neuronal cell death. DNA Repair 2004, 3, 617–627. [Google Scholar]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res 1997, 25, 750–755. [Google Scholar]
- Imam, S.Z.; Karahalil, B.; Hogue, B.A.; Souza-Pinto, N.C.; Bohr, V.A. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging 2006, 27, 1129–1136. [Google Scholar]
- Focher, F.; Mazzarello, P.; Verri, A.; Hubscher, U.; Spadari, S. Activity profiles of enzymes that control the uracil incorporation into DNA during neuronal development. Mutat. Res 1990, 237, 65–73. [Google Scholar]
- Lauritzen, K.H.; Dalhus, B.; Storm, J.F.; Bjoras, M.; Klungland, A. Modeling the impact of mitochondrial DNA damage in forebrain neurons and beyond. Mech. Ageing Dev 2011, 132, 424–428. [Google Scholar]
- Kronenberg, G.; Gertz, K.; Overall, R.W.; Harms, C.; Klein, J.; Page, M.M.; Stuart, J.A.; Endres, M. Folate deficiency increases mtDNA and D-1 mtDNA deletion in aged brain of mice lacking uracil-DNA glycosylase. Exp. Neurol 2011, 228, 253–258. [Google Scholar]
- Kronenberg, G.; Harms, C.; Sobol, R.W.; Cardozo-Pelaez, F.; Linhart, H.; Winter, B.; Balkaya, M.; Gertz, K.; Gay, S.B.; Cox, D.; et al. Folate deficiency induces neurodegeneration and brain dysfunction in mice lacking uracil DNA glycosylase. J. Neurosci 2008, 28, 7219–7230. [Google Scholar]
- Kruman, I.I.; Schwartz, E.; Kruman, Y.; Cutler, R.G.; Zhu, X.; Greig, N.H.; Mattson, M.P. Suppression of uracil-DNA glycosylase induces neuronal apoptosis. J. Biol. Chem 2004, 279, 43952–43960. [Google Scholar]
- Endres, M.; Biniszkiewicz, D.; Sobol, R.W.; Harms, C.; Ahmadi, M.; Lipski, A.; Katchanov, J.; Mergenthaler, P.; Dirnagl, U.; Wilson, S.H.; et al. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J. Clin. Invest 2004, 113, 1711–1721. [Google Scholar]
- Yang, G.; Wang, L.; Zhu, M.; Xu, D. Identification of non-Alzheimer’s disease tauopathies-related proteins by proteomic analysis. Neurol. Res 2008, 30, 613–622. [Google Scholar]
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 2007, 35, 5545–5555. [Google Scholar]
- Wiebauer, K.; Jiricny, J. In vitro correction of G.T mispairs to G.C pairs in nuclear extracts from human cells. Nature 1989, 339, 234–236. [Google Scholar]
- Neddermann, P.; Jiricny, J. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem 1993, 268, 21218–21224. [Google Scholar]
- Neddermann, P.; Jiricny, J. Efficient removal of uracil from G.U mispairs by the mismatch-specific thymine DNA glycosylase from HeLa cells. Proc. Natl. Acad. Sci. USA 1994, 91, 1642–1646. [Google Scholar]
- Neddermann, P.; Gallinari, P.; Lettieri, T.; Schmid, D.; Truong, O.; Hsuan, J.J.; Wiebauer, K.; Jiricny, J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem 1996, 271, 12767–12774. [Google Scholar]
- Hardeland, U.; Bentele, M.; Jiricny, J.; Schar, P. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem 2000, 275, 33449–33456. [Google Scholar]
- Borys-Brzywczy, E.; Arczewska, K.D.; Saparbaev, M.; Hardeland, U.; Schar, P.; Kusmierek, J.T. Mismatch dependent uracil/thymine-DNA glycosylases excise exocyclic hydroxyethano and hydroxypropano cytosine adducts. Acta Biochim. Pol 2005, 52, 149–165. [Google Scholar]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem 2011, 286, 35334–35338. [Google Scholar]
- Hashimoto, H.; Hong, S.; Bhagwat, A.S.; Zhang, X.; Cheng, X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: Its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef]
- Cortazar, D.; Kunz, C.; Selfridge, J.; Lettieri, T.; Saito, Y.; MacDougall, E.; Wirz, A.; Schuermann, D.; Jacobs, A.L.; Siegrist, F.; et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470, 419–423. [Google Scholar]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar]
- Marietta, C.; Palombo, F.; Gallinari, P.; Jiricny, J.; Brooks, P.J. Expression of long-patch and short-patch DNA mismatch repair proteins in the embryonic and adult mammalian brain. Brain Res. Mol 1998, 53, 317–320. [Google Scholar]
- Niederreither, K.; Harbers, M.; Chambon, P.; Dolle, P. Expression of T:G mismatch-specific thymidine-DNA glycosylase and DNA methyl transferase genes during development and tumorigenesis. Oncogene 1998, 17, 1577–1585. [Google Scholar]
- Chen, D.S.; Herman, T.; Demple, B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res 1991, 19, 5907–5914. [Google Scholar]
- Demple, B.; Herman, T.; Chen, D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: Definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 11450–11454. [Google Scholar]
- Robson, C.N.; Hickson, I.D. Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli. xth (exonuclease III) mutants. Nucleic Acids Res 1991, 19, 5519–5523. [Google Scholar]
- Xanthoudakis, S.; Curran, T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J 1992, 11, 653–665. [Google Scholar]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 1992, 11, 3323–3335. [Google Scholar]
- Jayaraman, L.; Murthy, K.G.; Zhu, C.; Curran, T.; Xanthoudakis, S.; Prives, C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev 1997, 11, 558–570. [Google Scholar]
- Chou, K.M.; Cheng, Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar]
- Wong, D.; DeMott, M.S.; Demple, B. Modulation of the 3′→5′-exonuclease activity of human apurinic endonuclease (Ape1) by its 5′-incised Abasic DNA product. J. Biol. Chem 2003, 278, 36242–36249. [Google Scholar]
- Parsons, J.L.; Dianov, G.L. Monitoring base excision repair proteins on damaged DNA using human cell extracts. Biochem. Soc. Trans 2004, 32, 962–963. [Google Scholar]
- Evans, A.R.; Limp-Foster, M.; Kelley, M.R. Going APE over ref-1. Mutat. Res 2000, 461, 83–108. [Google Scholar]
- Vasko, M.R.; Guo, C.; Kelley, M.R. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair 2005, 4, 367–379. [Google Scholar]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res 2008, 68, 6425–6434. [Google Scholar]
- Jiang, Y.; Guo, C.; Fishel, M.L.; Wang, Z.Y.; Vasko, M.R.; Kelley, M.R. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: Use of APE1 small molecule inhibitors to delineate APE1 functions. DNA Repair 2009, 8, 1273–1282. [Google Scholar]
- Edwards, M.; Rassin, D.K.; Izumi, T.; Mitra, S.; Perez-Polo, J.R. APE/Ref-1 responses to oxidative stress in aged rats. J. Neurosci. Res 1998, 54, 635–638. [Google Scholar]
- Tan, Z.; Sun, N.; Schreiber, S.S. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer’s hippocampus. Neuroreport 1998, 9, 2749–2752. [Google Scholar]
- Davydov, V.; Hansen, L.A.; Shackelford, D.A. Is DNA repair compromised in Alzheimer’s disease? Neurobiol. Aging 2003, 24, 953–968. [Google Scholar]
- Marcon, G.; Tell, G.; Perrone, L.; Garbelli, R.; Quadrifoglio, F.; Tagliavini, F.; Giaccone, G. APE1/Ref-1 in Alzheimer’s disease: An immunohistochemical study. Neurosci. Lett 2009, 466, 124–127. [Google Scholar]
- Schubert, D.; Behl, C.; Lesley, R.; Brack, A.; Dargusch, R.; Sagara, Y.; Kimura, H. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 1989–1993. [Google Scholar]
- Tan, Z.; Shi, L.; Schreiber, S.S. Differential Expression of Redox Factor-1 Associated with Beta-Amyloid-Mediated Neurotoxicity. Open Neurosci. J 2009, 3, 26–34. [Google Scholar]
- Huang, E.; Qu, D.; Zhang, Y.; Venderova, K.; Haque, M.E.; Rousseaux, M.W.; Slack, R.S.; Woulfe, J.M.; Park, D.S. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat. Cell. Biol 2010, 12, 563–571. [Google Scholar]
- Kim, M.H.; Kim, H.B.; Acharya, S.; Sohn, H.M.; Jun, J.Y.; Chang, I.Y.; You, H.J. Ape1/Ref-1 induces glial cell-derived neurotropic factor (GDNF) responsiveness by upregulating GDNF receptor alpha1 expression. Mol. Cell. Biol 2009, 29, 2264–2277. [Google Scholar]
- Mantha, A.K.; Dhiman, M.; Taglialatela, G.; Perez-Polo, R.J.; Mitra, S. Proteomic study of amyloid beta (25–35) peptide exposure to neuronal cells: Impact on APE1/Ref-1’s protein-protein interaction. J. Neurosci. Res 2012, 90, 1230–1239. [Google Scholar]
- Parildar-Karpuzoglu, H.; Dogru-Abbasoglu, S.; Hanagasi, H.A.; Karadag, B.; Gurvit, H.; Emre, M.; Uysal, M. Single nucleotide polymorphisms in base-excision repair genes hOGG1, APE1 and XRCC1 do not alter risk of Alzheimer’s disease. Neurosci. Lett 2008, 442, 287–291. [Google Scholar]
- Harris, J.L.; Jakob, B.; Taucher-Scholz, G.; Dianov, G.L.; Becherel, O.J.; Lavin, M.F. Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum. Mol. Genet 2009, 18, 4102–4117. [Google Scholar]
- Date, H.; Onodera, O.; Tanaka, H.; Iwabuchi, K.; Uekawa, K.; Igarashi, S.; Koike, R.; Hiroi, T.; Yuasa, T.; Awaya, Y.; et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet 2001, 29, 184–188. [Google Scholar]
- Moreira, M.C.; Barbot, C.; Tachi, N.; Kozuka, N.; Uchida, E.; Gibson, T.; Mendonca, P.; Costa, M.; Barros, J.; Yanagisawa, T.; et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet 2001, 29, 189–193. [Google Scholar]
- Hirano, M.; Asai, H.; Kiriyama, T.; Furiya, Y.; Iwamoto, T.; Nishiwaki, T.; Yamamoto, A.; Mori, T.; Ueno, S. Short half-lives of ataxia-associated aprataxin proteins in neuronal cells. Neurosci. Lett 2007, 419, 184–187. [Google Scholar]
- Kisby, G.E.; Milne, J.; Sweatt, C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport 1997, 8, 1337–1340. [Google Scholar]
- Olkowski, Z.L. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. Neuroreport 1998, 9, 239–242. [Google Scholar]
- Pioro, E.P.; Antel, J.P.; Cashman, N.R.; Arnold, D.L. Detection of cortical neuron loss in motor neuron disease by proton magnetic resonance spectroscopic imaging in vivo. Neurology 1994, 44, 1933–1938. [Google Scholar]
- Rowland, L.P. Amyotrophic lateral sclerosis: human challenge for neuroscience. Proc. Natl. Acad. Sci. USA 1995, 92, 1251–1253. [Google Scholar]
- Hayward, C.; Colville, S.; Swingler, R.J.; Brock, D.J. Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology 1999, 52, 1899–1901. [Google Scholar]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular Med 2002, 2, 47–60. [Google Scholar]
- Quach, N.; Chan, T.; Lu, T.A.; Schreiber, S.S.; Tan, Z. Induction of DNA repair proteins, Ref-1 and XRCC1, in adult rat brain following kainic acid-induced seizures. Brain Res 2005, 1042, 236–240. [Google Scholar]
- Morita-Fujimura, Y.; Fujimura, M.; Kawase, M.; Chan, P.H. Early decrease in apurinic/apyrimidinic endonuclease is followed by DNA fragmentation after cold injury-induced brain trauma in mice. Neuroscience 1999, 93, 1465–1473. [Google Scholar]
- Lewen, A.; Sugawara, T.; Gasche, Y.; Fujimura, M.; Chan, P.H. Oxidative cellular damage and the reduction of APE/Ref-1 expression after experimental traumatic brain injury. Neurobiol. Dis 2001, 8, 380–390. [Google Scholar]
- Edwards, M.; Kent, T.A.; Rea, H.C.; Wei, J.; Quast, M.; Izumi, T.; Mitra, S.; Perez-Polo, J.R. APE/Ref-1 responses to ischemia in rat brain. Neuroreport 1998, 9, 4015–4018. [Google Scholar]
- Walton, M.; Lawlor, P.; Sirimanne, E.; Williams, C.; Gluckman, P.; Dragunow, M. Loss of Ref-1 protein expression precedes DNA fragmentation in apoptotic neurons. Mol. Brain Res 1997, 44, 167–170. [Google Scholar]
- Kawase, M.; Fujimura, M.; Morita-Fujimura, Y.; Chan, P.H. Reduction of apurinic/apyrimidinic endonuclease expression after transient global cerebral ischemia in rats: Implication of the failure of DNA repair in neuronal apoptosis. Stroke 1999, 30, 441–448. [Google Scholar]
- Stetler, R.A.; Gao, Y.; Zukin, R.S.; Vosler, P.S.; Zhang, L.; Zhang, F.; Cao, G.; Bennett, M.V.; Chen, J. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3204–3209. [Google Scholar]
- Sakurai, M.; Nagata, T.; Abe, K.; Horinouchi, T.; Itoyama, Y.; Tabayashi, K. Oxidative damage and reduction of redox factor-1 expression after transient spinal cord ischemia in rabbits. J. Vasc. Surg 2003, 37, 446–452. [Google Scholar]
- Vasko, M.R.; Guo, C.; Thompson, E.L.; Kelley, M.R. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair 2011, 10, 942–952. [Google Scholar]
- Yang, J.L.; Tadokoro, T.; Keijzers, G.; Mattson, M.P.; Bohr, V.A. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J. Biol. Chem 2010, 285, 28191–28199. [Google Scholar]
- Jilani, A.; Ramotar, D.; Slack, C.; Ong, C.; Yang, X.M.; Scherer, S.W.; Lasko, D.D. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem 1999, 274, 24176–24186. [Google Scholar]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar]
- Karimi-Busheri, F.; Daly, G.; Robins, P.; Canas, B.; Pappin, D.J.; Sgouros, J.; Miller, G.G.; Fakhrai, H.; Davis, E.M.; Le Beau, M.M.; et al. Molecular characterization of a human DNA kinase. J. Bio. Chem 1999, 274, 24187–24194. [Google Scholar]
- Loizou, J.I.; El-Khamisy, S.F.; Zlatanou, A.; Moore, D.J.; Chan, D.W.; Qin, J.; Sarno, S.; Meggio, F.; Pinna, L.A.; Caldecott, K.W. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 2004, 117, 17–28. [Google Scholar]
- Breslin, C.; Caldecott, K.W. DNA 3′-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol. Cell. Biol 2009, 29, 4653–4662. [Google Scholar]
- Shen, J.; Gilmore, E.C.; Marshall, C.A.; Haddadin, M.; Reynolds, J.J.; Eyaid, W.; Bodell, A.; Barry, B.; Gleason, D.; Allen, K.; et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet 2010, 42, 245–249. [Google Scholar]
- Reynolds, J.J.; Walker, A.K.; Gilmore, E.C.; Walsh, C.A.; Caldecott, K.W. Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic Acids Res 2012, 40, 6608–6619. [Google Scholar]
- Lindahl, T. New class of enzymes acting on damaged DNA. Nature 1976, 259, 64–66. [Google Scholar]
- Singhal, R.K.; Wilson, S.H. Short gap-filling synthesis by DNA polymerase beta is processive. J. Biol. Chem 1993, 268, 15906–15911. [Google Scholar]
- Prasad, R.; Beard, W.A.; Wilson, S.H. Studies of gapped DNA substrate binding by mammalian DNA polymerase beta. Dependence on 5′-phosphate group. J. Biol. Chem 1994, 269, 18096–18101. [Google Scholar]
- Matsumoto, Y.; Kim, K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 1995, 269, 699–702. [Google Scholar]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schar, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J 1996, 15, 6662–6670. [Google Scholar]
- Piersen, C.E.; Prasad, R.; Wilson, S.H.; Lloyd, R.S. Evidence for an imino intermediate in the DNA polymerase beta deoxyribose phosphate excision reaction. J. Biol. Chem 1996, 271, 17811–17815. [Google Scholar]
- Sobol, R.W.; Horton, J.K.; Kuhn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 1996, 379, 183–186. [Google Scholar]
- Prasad, R.; Beard, W.A.; Strauss, P.R.; Wilson, S.H. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem 1998, 273, 15263–15270. [Google Scholar]
- Dianov, G.L. Monitoring base excision repair by in vitro assays. Toxicology 2003, 193, 35–41. [Google Scholar]
- Dianova, I.I.; Sleeth, K.M.; Allinson, S.L.; Parsons, J.L.; Breslin, C.; Caldecott, K.W.; Dianov, G.L. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res 2004, 32, 2550–2555. [Google Scholar]
- Baril, E.F.; Brown, O.E.; Jenkins, M.D.; Laszlo, J. Deoxyribonucleic acid polymerase with rat liver ribosomes and smooth membranes. Purification and properties of the enzymes. Biochemistry 1971, 10, 1981–1992. [Google Scholar]
- Berger, H., Jr; Huang, R.C.; Irvin, J.L. Purification and characterization of a deoxyribonucleic acid polymerase from rat liver. J. Biol. Chem. 1971, 246, 7275–7283. [Google Scholar]
- Chang, L.M.; Bollum, F.J. Low molecular weight deoxyribonucleic acid polymerase in mammalian cells. J. Biol. Chem 1971, 246, 5835–5837. [Google Scholar]
- Haines, M.E.; Holmes, A.M.; Johnston, I.R. Distinct cytoplasmic and nuclear DNA polymerases from rat liver. FEBS Lett 1971, 17, 63–67. [Google Scholar]
- Weissbach, A.; Schlabach, A.; Fridlender, B.; Bolden, A. DNA polymerases from human cells. Nat. New Biol 1971, 231, 167–170. [Google Scholar]
- Hubscher, U.; Kuenzle, C.C.; Spadari, S. Functional roles of DNA polymerases beta and gamma. Proc. Natl. Acad. Sci. USA 1979, 76, 2316–2320. [Google Scholar]
- Waser, J.; Hubscher, U.; Kuenzle, C.C.; Spadari, S. DNA polymerase beta from brain neurons is a repair enzyme. Eur. J. Biochem 1979, 97, 361–368. [Google Scholar]
- Hardt, N.; Pedrali-Noy, G.; Focher, F.; Spadari, S. Aphidicolin does not inhibit DNA repair synthesis in ultraviolet-irradiated HeLa cells. A radioautographic study. Biochem. J 1981, 199, 453–455. [Google Scholar]
- Maga, G.; Hubscher, U. Repair and translesion DNA polymerases as anticancer drug targets. Anticancer Agents Med. Chem 2008, 8, 431–447. [Google Scholar]
- Hirose, F.; Hotta, Y.; Yamaguchi, M.; Matsukage, A. Difference in the expression level of DNA polymerase beta among mouse tissues: High expression in the pachytene spermatocyte. Exp. Cell. Res 1989, 181, 169–180. [Google Scholar]
- Sugo, N.; Aratani, Y.; Nagashima, Y.; Kubota, Y.; Koyama, H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J 2000, 19, 1397–1404. [Google Scholar]
- Sugo, N.; Niimi, N.; Aratani, Y.; Takiguchi-Hayashi, K.; Koyama, H. p53 Deficiency rescues neuronal apoptosis but not differentiation in DNA polymerase beta-deficient mice. Mol. Cell. Biol 2004, 24, 9470–9477. [Google Scholar]
- Niimi, N.; Sugo, N.; Aratani, Y.; Koyama, H. Genetic interaction between DNA polymerase beta and DNA-PKcs in embryogenesis and neurogenesis. Cell. Death. Differ 2005, 12, 184–191. [Google Scholar]
- Krishna, T.H.; Mahipal, S.; Sudhakar, A.; Sugimoto, H.; Kalluri, R.; Rao, K.S. Reduced DNA gap repair in aging rat neuronal extracts and its restoration by DNA polymerase beta and DNA-ligase. J. Neurochem 2005, 92, 818–823. [Google Scholar]
- Cabelof, D.C.; Raffoul, J.J.; Yanamadala, S.; Ganir, C.; Guo, Z.; Heydari, A.R. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res 2002, 500, 135–145. [Google Scholar]
- Cabelof, D.C.; Yanamadala, S.; Raffoul, J.J.; Guo, Z.; Soofi, A.; Heydari, A.R. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA repair 2003, 2, 295–307. [Google Scholar]
- Copani, A.; Condorelli, F.; Caruso, A.; Vancheri, C.; Sala, A.; Giuffrida Stella, A.M.; Canonico, P.L.; Nicoletti, F.; Sortino, M.A. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J 1999, 13, 2225–2234. [Google Scholar]
- Copani, A.; Sortino, M.A.; Caricasole, A.; Chiechio, S.; Chisari, M.; Battaglia, G.; Giuffrida-Stella, A.M.; Vancheri, C.; Nicoletti, F. Erratic expression of DNA polymerases by beta-amyloid causes neuronal death. FASEB J 2002, 16, 2006–2008. [Google Scholar]
- Copani, A.; Hoozemans, J.J.; Caraci, F.; Calafiore, M.; van Haastert, E.S.; Veerhuis, R.; Rozemuller, A.J.; Aronica, E.; Sortino, M.A.; Nicoletti, F. DNA polymerase-beta is expressed early in neurons of Alzheimer’s disease brain and is loaded into DNA replication forks in neurons challenged with beta-amyloid. J. Neurosci 2006, 26, 10949–10957. [Google Scholar]
- Calafiore, M.; Copani, A.; Deng, W. DNA polymerase-beta mediates the neurogenic effect of beta-amyloid protein in cultured subventricular zone neurospheres. J. Neurosci. Res 2012, 90, 559–567. [Google Scholar]
- Zhang, Z.; Cao, X.; Xiong, N.; Wang, H.; Huang, J.; Sun, S.; Liang, Z.; Wang, T. DNA polymerase-beta is required for 1-methyl-4-phenylpyridinium-induced apoptotic death in neurons. Apoptosis 2010, 15, 105–115. [Google Scholar]
- Liu, Y.; Prasad, R.; Beard, W.A.; Hou, E.W.; Horton, J.K.; McMurray, C.T.; Wilson, S.H. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J. Biol. Chem 2009, 284, 28352–28366. [Google Scholar]
- Goula, A.V.; Pearson, C.E.; Della Maria, J.; Trottier, Y.; Tomkinson, A.E.; Wilson, D.M., 3rd; Merienne, K. The nucleotide sequence, DNA damage location, and protein stoichiometry influence the base excision repair outcome at CAG/CTG repeats. Biochemistry 2012, 51, 3919–3932. [Google Scholar]
- Mishra, O.P.; Akhter, W.; Ashraf, Q.M.; Delivoria-Papadopoulos, M. Hypoxia-induced modification of poly (ADP-ribose) polymerase and dna polymerase beta activity in cerebral cortical nuclei of newborn piglets: Role of nitric oxide. Neuroscience 2003, 119, 1023–1032. [Google Scholar]
- Lan, J.; Li, W.; Zhang, F.; Sun, F.Y.; Nagayama, T.; O’Horo, C.; Chen, J. Inducible repair of oxidative DNA lesions in the rat brain after transient focal ischemia and reperfusion. J. Cereb. Blood Flow Metab 2003, 23, 1324–1339. [Google Scholar]
- Li, N.; Wu, H.; Yang, S.; Chen, D. Ischemic preconditioning induces XRCC1, DNA polymerase-beta, and DNA ligase III and correlates with enhanced base excision repair. DNA Repair 2007, 6, 1297–1306. [Google Scholar]
- Hubscher, U.; Maga, G.; Spadari, S. Eukaryotic DNA polymerases. Annu. Rev. Biochem 2002, 71, 133–63. [Google Scholar]
- McCulloch, S.D.; Kunkel, T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell. Res 2008, 18, 148–161. [Google Scholar]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar]
- Schmitt, M.W.; Matsumoto, Y.; Loeb, L.A. High fidelity and lesion bypass capability of human DNA polymerase delta. Biochimie 2009, 91, 1163–1172. [Google Scholar]
- Hubscher, U. DNA replication fork proteins. Meth. Mol. Biol 2009, 521, 19–33. [Google Scholar]
- Podlutsky, A.J.; Dianova, II; Podust, V.N.; Bohr, V.A.; Dianov, G.L. Human DNA polymerase beta initiates DNA synthesis during long-patch repair of reduced AP sites in DNA. EMBO J. 2001, 20, 1477–1482. [Google Scholar]
- Raji, N.S.; Krishna, T.H.; Rao, K.S. DNA-polymerase alpha, beta, delta and epsilon activities in isolated neuronal and astroglial cell fractions from developing and aging rat cerebral cortex. Int J. Dev. Neurosci 2002, 20, 491–496. [Google Scholar]
- Bhattacharyya, S.; Lahue, R.S. Saccharomyces cerevisiae Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol. Cell. Biol 2004, 24, 7324–7330. [Google Scholar]
- Chan, N.L.; Hou, C.; Zhang, T.; Yuan, F.; Machwe, A.; Huang, J.; Orren, D.K.; Gu, L.; Li, G.M. The Werner Syndrome Protein Promotes CAG/CTG Repeat Stability by Resolving Large (CAG)n/(CTG)n Hairpins. J. Biol. Chem 2012, 287, 30151–30156. [Google Scholar]
- Caldecott, K.W. Mammalian DNA single-strand break repair: An X-ra(y)ted affair. Bioessays 2001, 23, 447–455. [Google Scholar]
- Caldecott, K.W.; Aoufouchi, S.; Johnson, P.; Shall, S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res 1996, 24, 4387–4394. [Google Scholar]
- Caldecott, K.W.; McKeown, C.K.; Tucker, J.D.; Ljungquist, S.; Thompson, L.H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol 1994, 14, 68–76. [Google Scholar]
- Cappelli, E.; Taylor, R.; Cevasco, M.; Abbondandolo, A.; Caldecott, K.; Frosina, G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem 1997, 272, 23970–23975. [Google Scholar]
- Lindahl, T.; Wood, R.D. Quality control by DNA repair. Science 1999, 286, 1897–1905. [Google Scholar]
- Fang-Kircher, S.G.; Labudova, O.; Kitzmueller, E.; Rink, H.; Cairns, N.; Lubec, G. Increased steady state mRNA levels of DNA-repair genes XRCC1, ERCC2 and ERCC3 in brain of patients with Down syndrome. Life Sci 1999, 64, 1689–1699. [Google Scholar]
- Busciglio, J.; Yankner, B.A. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 1995, 378, 776–779. [Google Scholar]
- Kulkarni, A.; McNeill, D.R.; Gleichmann, M.; Mattson, M.P.; Wilson, D.M., 3rd. XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008, 36, 5111–5121. [Google Scholar]
- Lee, Y.; Katyal, S.; Li, Y.; El-Khamisy, S.F.; Russell, H.R.; Caldecott, K.W.; McKinnon, P.J. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat. Neurosci 2009, 12, 973–980. [Google Scholar]
- Aronica, E.; Gorter, J.A. Gene expression profile in temporal lobe epilepsy. Neuroscientist 2007, 13, 100–108. [Google Scholar]
- Gluck, M.R.; Jayatilleke, E.; Shaw, S.; Rowan, A.J.; Haroutunian, V. CNS oxidative stress associated with the kainic acid rodent model of experimental epilepsy. Epilepsy Res 2000, 39, 63–71. [Google Scholar]
- Shen, M.R.; Jones, I.M.; Mohrenweiser, H. Nonconservative amino acid substitution variants exist at polymorphic frequency in DNA repair genes in healthy humans. Cancer Res 1998, 58, 604–608. [Google Scholar]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar]
- Dogru-Abbasoglu, S.; Aykac-Toker, G.; Hanagasi, H.A.; Gurvit, H.; Emre, M.; Uysal, M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer’s disease. Neurol. Sci 2007, 28, 31–34. [Google Scholar]
- Qian, Y.; Chen, W.; Wu, J.; Tao, T.; Bi, L.; Xu, W.; Qi, H.; Wang, Y.; Guo, L. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer’s disease and age of onset in elderly Han Chinese. J. Neurol. Sci 2010, 295, 62–65. [Google Scholar]
- Gencer, M.; Dasdemir, S.; Cakmakoglu, B.; Cetinkaya, Y.; Varlibas, F.; Tireli, H.; Kucukali, C.I.; Ozkok, E.; Aydin, M. DNA repair genes in Parkinson’s disease. Genet. Test. Mol. Biomark 2012, 16, 504–507. [Google Scholar]
- Fujimura, M.; Morita-Fujimura, Y.; Sugawara, T.; Chan, P.H. Early decrease of XRCC1, a DNA base excision repair protein, may contribute to DNA fragmentation after transient focal cerebral ischemia in mice. Stroke 1999, 30, 2456–2463. [Google Scholar]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal 2011, 14, 1505–1517. [Google Scholar]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar]
- Fujimura, M.; Morita-Fujimura, Y.; Noshita, N.; Yoshimoto, T.; Chan, P.H. Reduction of the DNA base excision repair protein, XRCC1, may contribute to DNA fragmentation after cold injury-induced brain trauma in mice. Brain Res 2000, 869, 105–111. [Google Scholar]
- Harrington, J.J.; Lieber, M.R. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J 1994, 13, 1235–1246. [Google Scholar]
- Murante, R.S.; Huang, L.; Turchi, J.J.; Bambara, R.A. The calf 5′- to 3′-exonuclease is also an endonuclease with both activities dependent on primers annealed upstream of the point of cleavage. J. Biol. Chem 1994, 269, 1191–1196. [Google Scholar]
- Li, X.; Li, J.; Harrington, J.; Lieber, M.R.; Burgers, P.M. Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem 1995, 270, 22109–22112. [Google Scholar]
- Bornarth, C.J.; Ranalli, T.A.; Henricksen, L.A.; Wahl, A.F.; Bambara, R.A. Effect of flap modifications on human FEN1 cleavage. Biochemistry 1999, 38, 13347–13354. [Google Scholar]
- Liu, Y.; Kao, H.I.; Bambara, R.A. Flap endonuclease 1: A central component of DNA metabolism. Annu. Rev. Biochem 2004, 73, 589–615. [Google Scholar]
- Tomlinson, C.G.; Atack, J.M.; Chapados, B.; Tainer, J.A.; Grasby, J.A. Substrate recognition and catalysis by flap endonucleases and related enzymes. Biochem. Soc. Trans 2010, 38, 433–437. [Google Scholar]
- Nazarkina, Z.K.; Lavrik, O.I.; Khodyreva, S.N. Flap endonuclease-1 and its role in the processes of DNA metabolism in eucaryotic cells. Mol. Biol 2008, 42, 405–421. [Google Scholar]
- Liu, Y.; Wilson, S.H. DNA base excision repair: A mechanism of trinucleotide repeat expansion. Trends Biochem. Sci 2012, 37, 162–172. [Google Scholar]
- Levin, D.S.; Bai, W.; Yao, N.; O’Donnell, M.; Tomkinson, A.E. An interaction between DNA ligase I and proliferating cell nuclear antigen: Implications for Okazaki fragment synthesis and joining. Proc. Natl. Acad. Sci. USA 1997, 94, 12863–12868. [Google Scholar]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem 2008, 77, 313–338. [Google Scholar]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet 2002, 32, 267–272. [Google Scholar]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar]
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar]
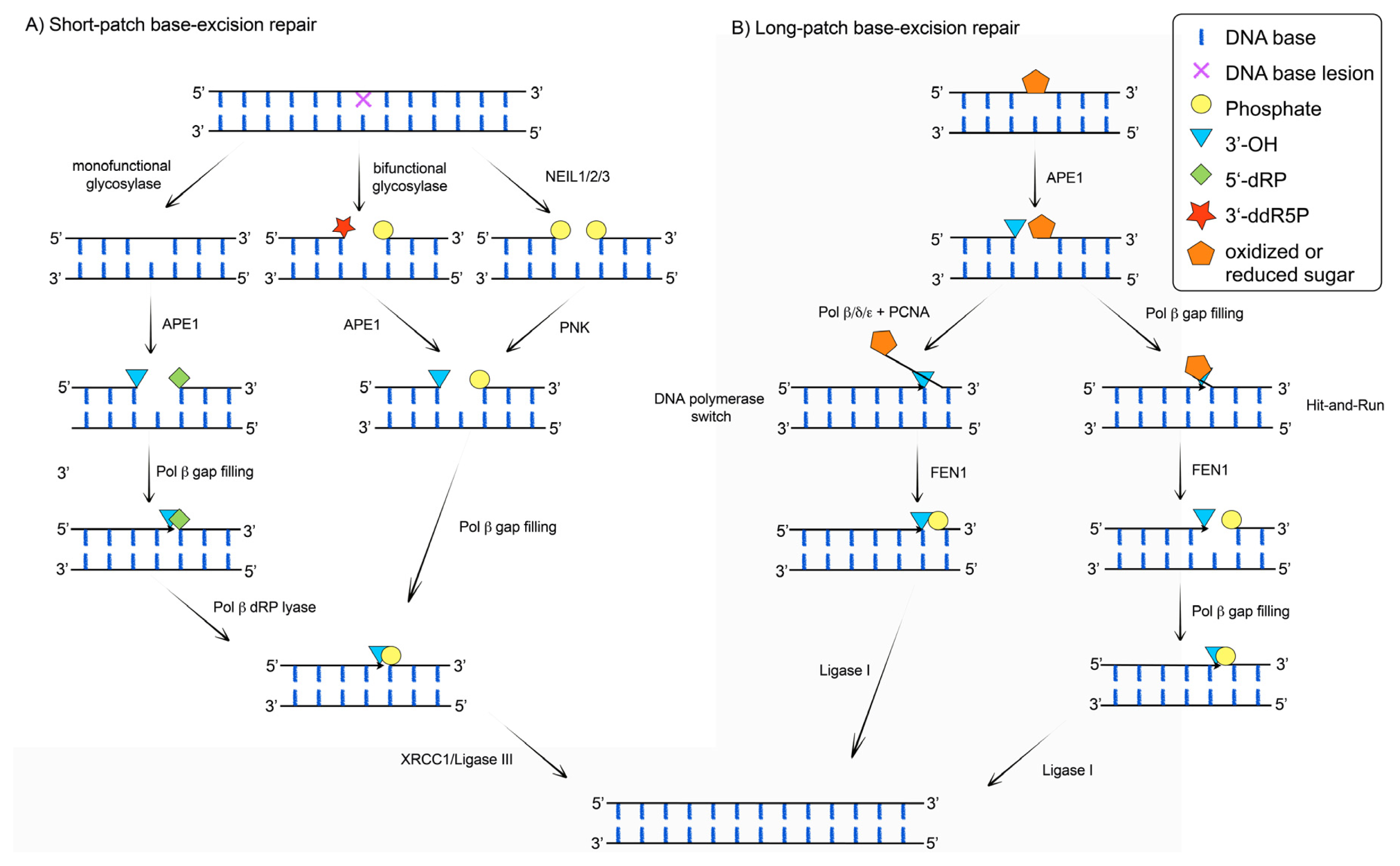

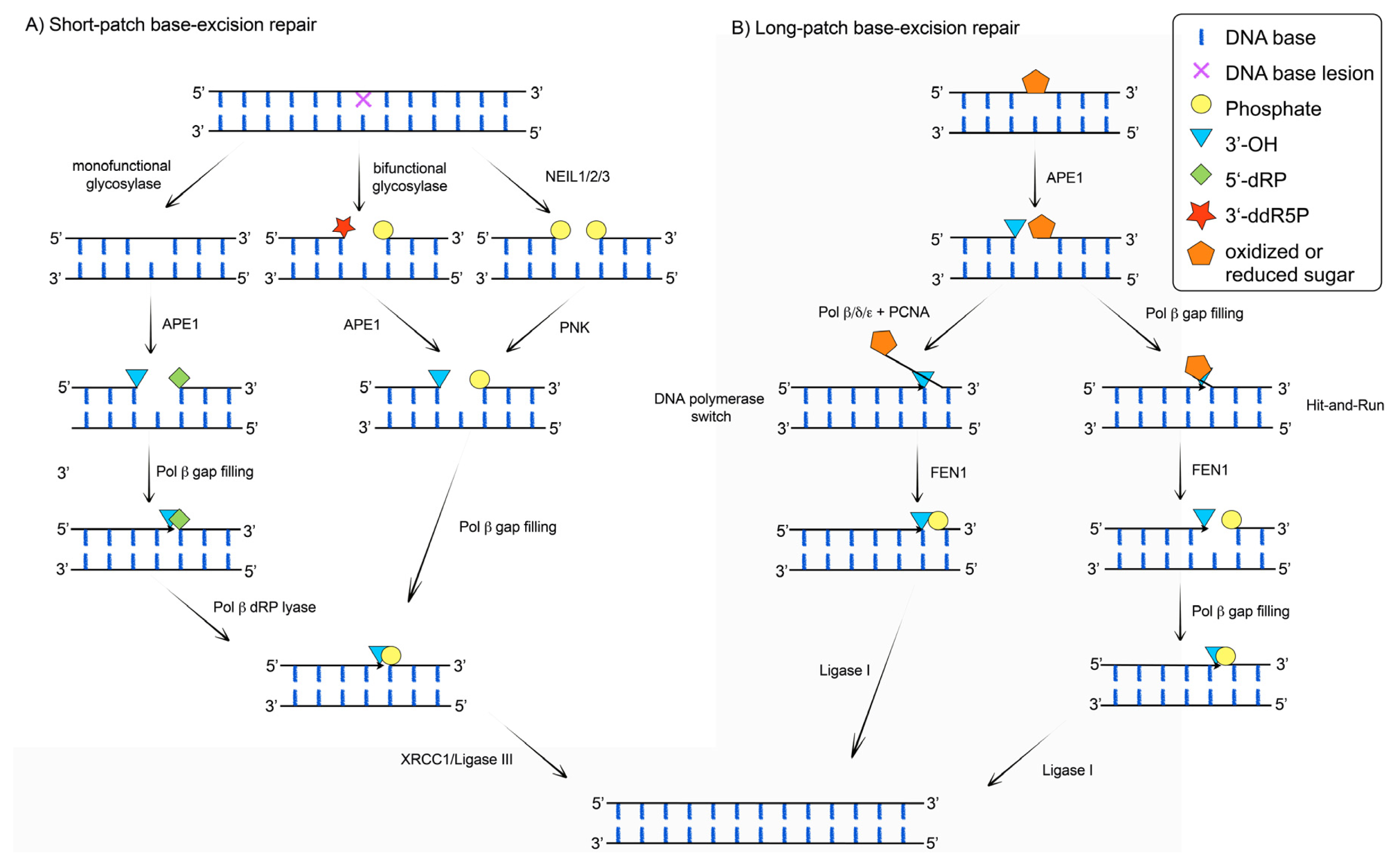{kind=link}
| Protein | Physiological expression in brain | Expression changes induced by | Changes associated with neurodgenerative disorders | Brain specific effect of knockout/knockdown | ||||
|---|---|---|---|---|---|---|---|---|
| Protein family | ||||||||
| DNA glycosylases | Helix-hairpin-helix family | OGG1 | - ↓ postnatal - ↑ from 8 weeks - ↓ age-dependently | ↑ | - cigarette smoke - dieldrin-proliferating cells - SIF in murine brains | PD | - ↑ | - differentiation shiftneural to astrocytic lineage - mild PD phenotype with age - ↑ sensitivity to dopaminergic substances and ischemia-induced DNA damage - combination with CSB kd—no effect on CS phenotype |
| ALS | - S326C increased risk - ↑ in presymptomatic SOD1 mice | |||||||
| ↓ | - dieldrin-differentiated cells - fenvalerate | HD | - OGG1 increases TNR instability, especially the S326C - ↓ in striatum of HD mice | |||||
| No change | - lead (Pb) | Stroke/Ischemia | - various effects depending on the model used | |||||
| AD | - ↑ but also ↓ observed | |||||||
| MUTYH | - ↑ in neonate and adult brain - ↓ with age | None reported | PD | - ↑ | None reported | |||
| Stroke/Ischemia | - mainly ↑ | |||||||
| Other disorders | - possibly ↓ in equine cerebellar abiotrophy | |||||||
| MBD4 | None reported | None reported | Diverse disorders | - ↑ in schizophrenia and bipolar disorder patients | None reported | |||
| NTHL1 | None reported | None reported | Diverse disorders | - no association with MS risk | None reported | |||
| Endonuclease VIII-like | NEIL1 | - ↑ mid-age, during differentiation - ↓ with age - minor changes in hippocampal mitochondria over lifespan | None reported | Stroke/Ischemia | - no changes by OGD in hippocampal slice cultures, ↓ by hypothermia | - impaired memory and increased brain damage after ischemia/reperfusion in ko mice | ||
| NEIL2 | - ↑ during differentiation | None reported | Stroke/Ischemia | - no changes after OGD | None reported | |||
| NEIL3 | - stem cell rich regions, also in early embryos - ↓ with age | None reported | Stroke/Ischemia | - ↓ in hypoxia | - ko with ↓ neuronal progenitors and NSC differentiation ability | |||
| AAG | - highly expressed in several brain regions | None reported | None reported | - ko results in suppression, while Tg in increase of toxicity induced by alkylating agents | ||||
| UDG | UNG | - varying expression depending on brain region and age | None reported | AD/TNR disorders | - changed in tauopathies and ↓ in AD patients | - ko and Tg with neurodegeneration - ko ↑ ischemic infarct size | ||
| TDG | None reported | None reported | None reported | - ko embryonic lethal | ||||
| Endonucleases | APE1 | - ↓ with age | ↑ | - 100% O2 in brains of young rats, but not in old ones | AD | - ↑ levels in patients, varying expression upon Aβ treatment - ↑ levels of p-APE1 (less active) - no significant correlation with D148E | None reported | |
| PD | - ↑ levels of p-APE1 (less active) | |||||||
| HD | - 2-fold increase in cerebellum HD mice | |||||||
| Stroke/ischemia | - ↓ in several models of hypoxia, hypothermia, stroke and trauma | |||||||
| Other diseases | - ↓ in AOA patients - both ↑ and ↓ in ALS patients detected - association of missense mutations, D148E - ↑ in epilepsia model | |||||||
| FEN1 | None reported | None reported | HD | - implicated in TNR expansion, increased in cerebellum of HD mice | None reported | |||
| PNK | - low expression | None reported | MCSZ | - multiple mutations associated | None reported | |||
| DNA polymerases | Pol β | - constitutive expression - ↓ activity with age | None reported | AD | - Aβ induced Pol β-mediated cell cycle reentrance, neuronal loss and differentiation of neural progenitors to neuronal lineage - MPP + induces Pol ββ-mediated cell cycle reentrance and cell death | - neonatal lethal, altered neurogenesis in ko mice, which is p53 dependent and more pronounced in a DNA-PKcs ko background | ||
| HD | - Pol β accumulation along CAG repeats in striatum of HD mice | |||||||
| Stroke/ischemia | - ↑ in several models | |||||||
| Pol δ + Pol ε | None reported | None reported | HD | - Pol δ blocks TNR expansion together with Srs2 and resolves srs1 and resolves TNR-based hairpin structures together with WRN | None reported | |||
| Scaffolding | XRCC1 | None reported | None reported | AD | - R194W and R399 ↑ risk, no effect by R280H/R399Q | - XRCC1nes−cre ko mice age-dependent accumulation of DNA damage, loss of certain neurons in the cerebellum and altered hippocampal homeostasis | ||
| HD | - 2-fold increase in cerebellum HD mice | |||||||
| Stroke/ischemia | - ↓ in several models of ischemia, hypothermia | |||||||
| Other diseases | - ↑ levels in some parts of the brains of Down’s syndrome patients, and ↓ in others - ↑ levels in a rat epilepsia model | |||||||
| DNA ligases | DNA ligase I | - moderate in cerebellum, lateral ventricle and cerebral cortex - ↓ in hippocampus and striatum | HD | - 2-fold ↑ in cerebellum HD mice | - essential for embryonic development | |||
| DNA ligase III | - ↑ in cerebellum and cerebral cortex - moderate in hippocampus and lateral ventricle | SCAN | - association due to interaction with TDP1? | - essential for embryonic development | ||||
| AOA1 | - association? | |||||||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bosshard, M.; Markkanen, E.; Van Loon, B. Base Excision Repair in Physiology and Pathology of the Central Nervous System. Int. J. Mol. Sci. 2012, 13, 16172-16222. https://doi.org/10.3390/ijms131216172
Bosshard M, Markkanen E, Van Loon B. Base Excision Repair in Physiology and Pathology of the Central Nervous System. International Journal of Molecular Sciences. 2012; 13(12):16172-16222. https://doi.org/10.3390/ijms131216172
Chicago/Turabian StyleBosshard, Matthias, Enni Markkanen, and Barbara Van Loon. 2012. "Base Excision Repair in Physiology and Pathology of the Central Nervous System" International Journal of Molecular Sciences 13, no. 12: 16172-16222. https://doi.org/10.3390/ijms131216172
APA StyleBosshard, M., Markkanen, E., & Van Loon, B. (2012). Base Excision Repair in Physiology and Pathology of the Central Nervous System. International Journal of Molecular Sciences, 13(12), 16172-16222. https://doi.org/10.3390/ijms131216172