Differential Regulation of CD4+ T Cell Adhesion to Cerebral Microvascular Endothelium by the β-Chemokines CCL2 and CCL3

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
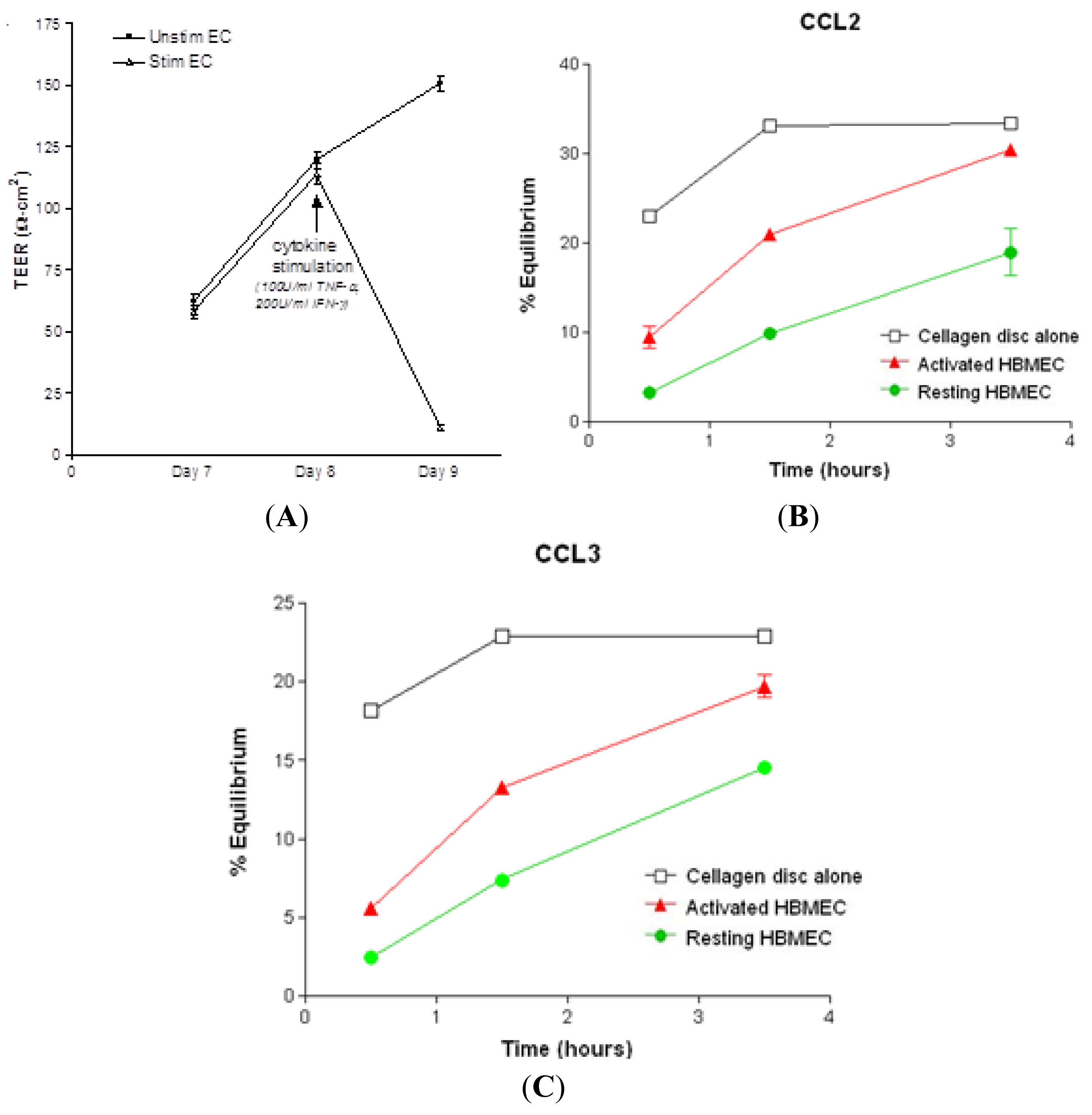
2.1. Chemokine Diffusion Across HBMEC Monolayers
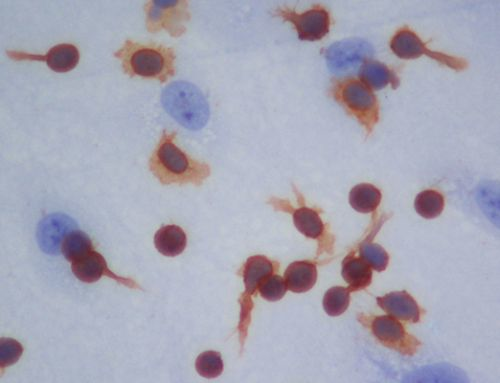
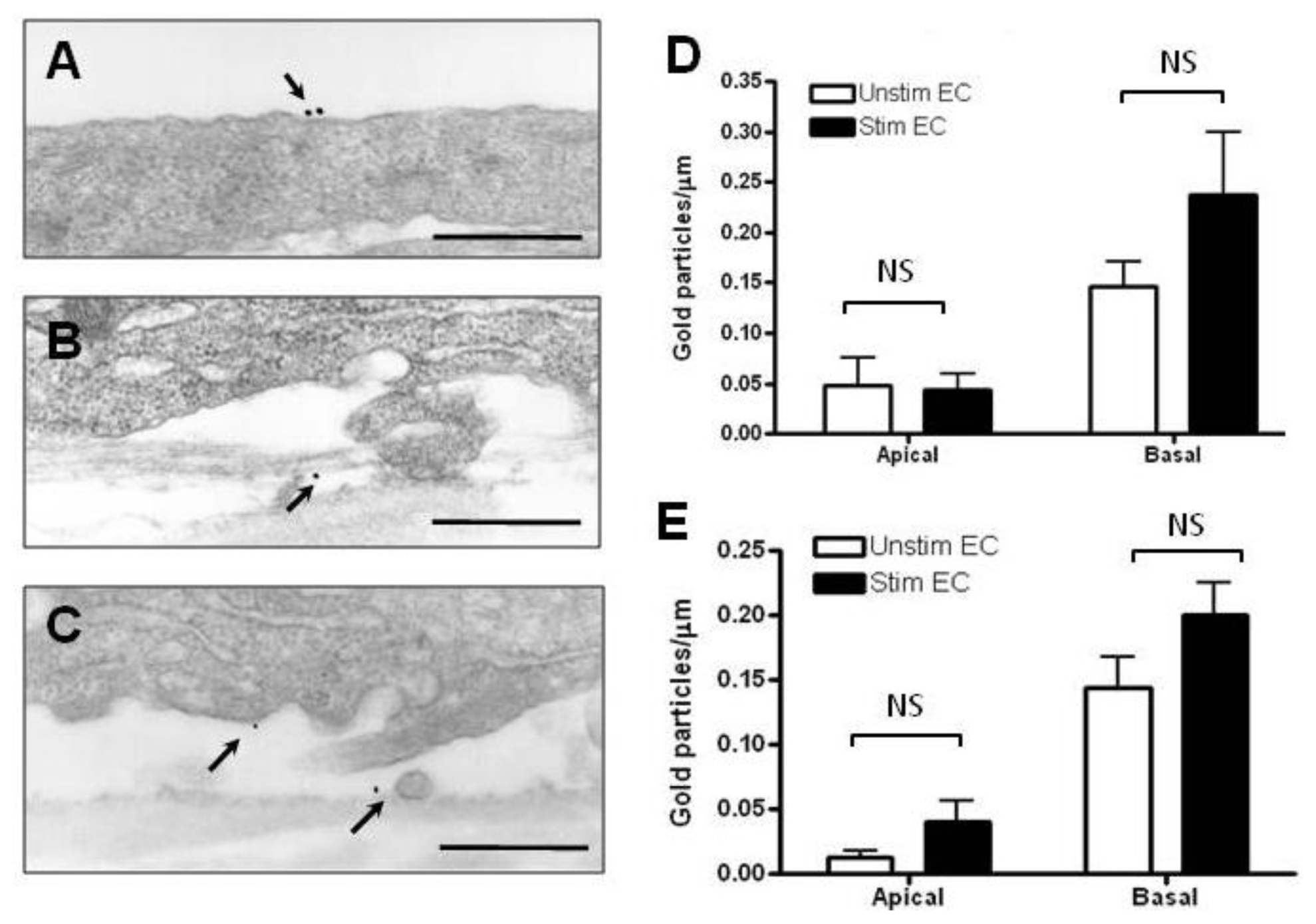
2.2. Localization and Binding of CCL2 and CCL3 to HBMEC
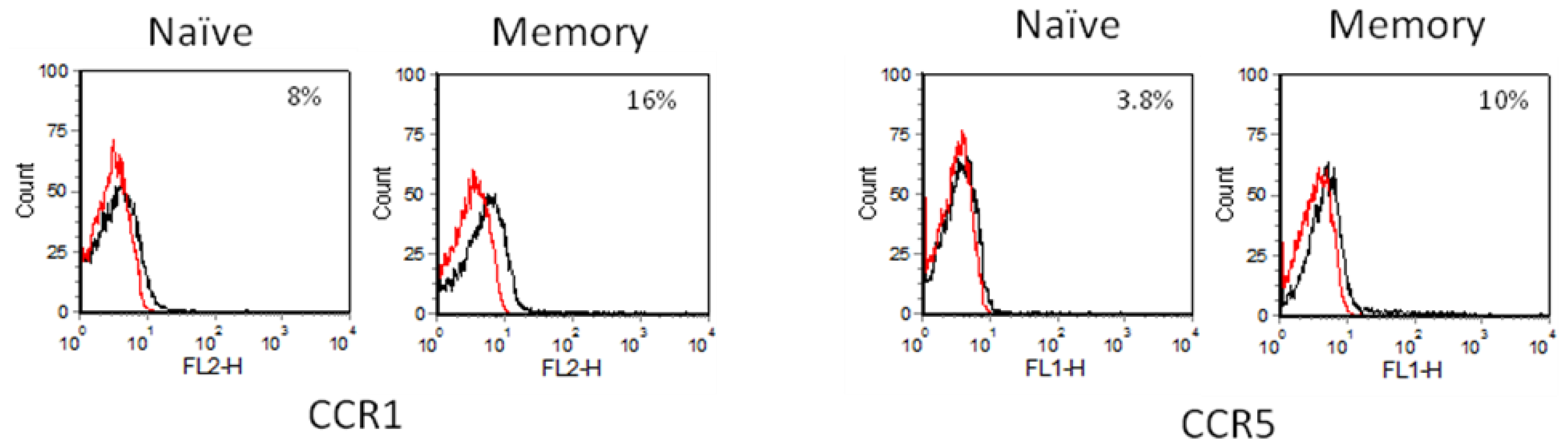
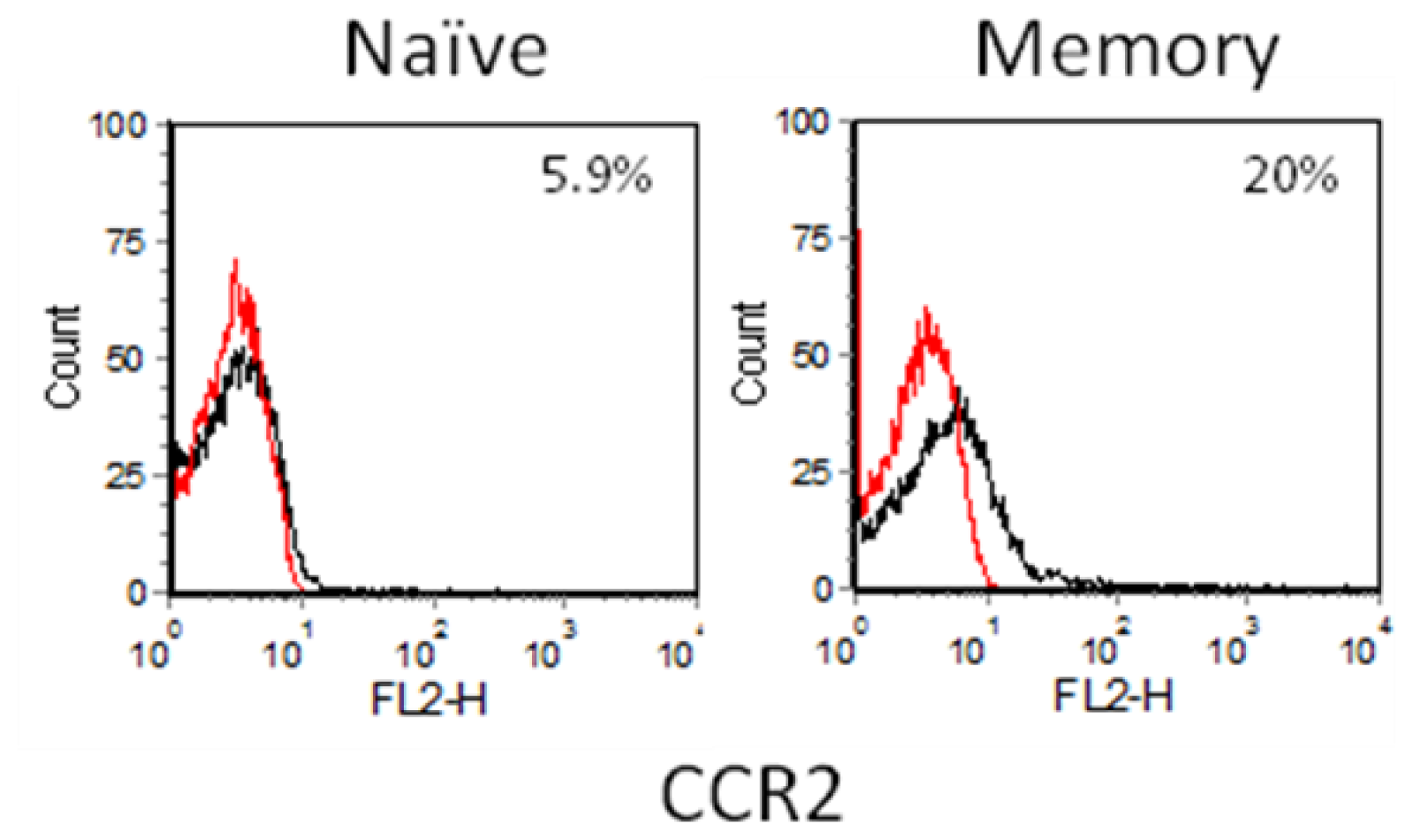
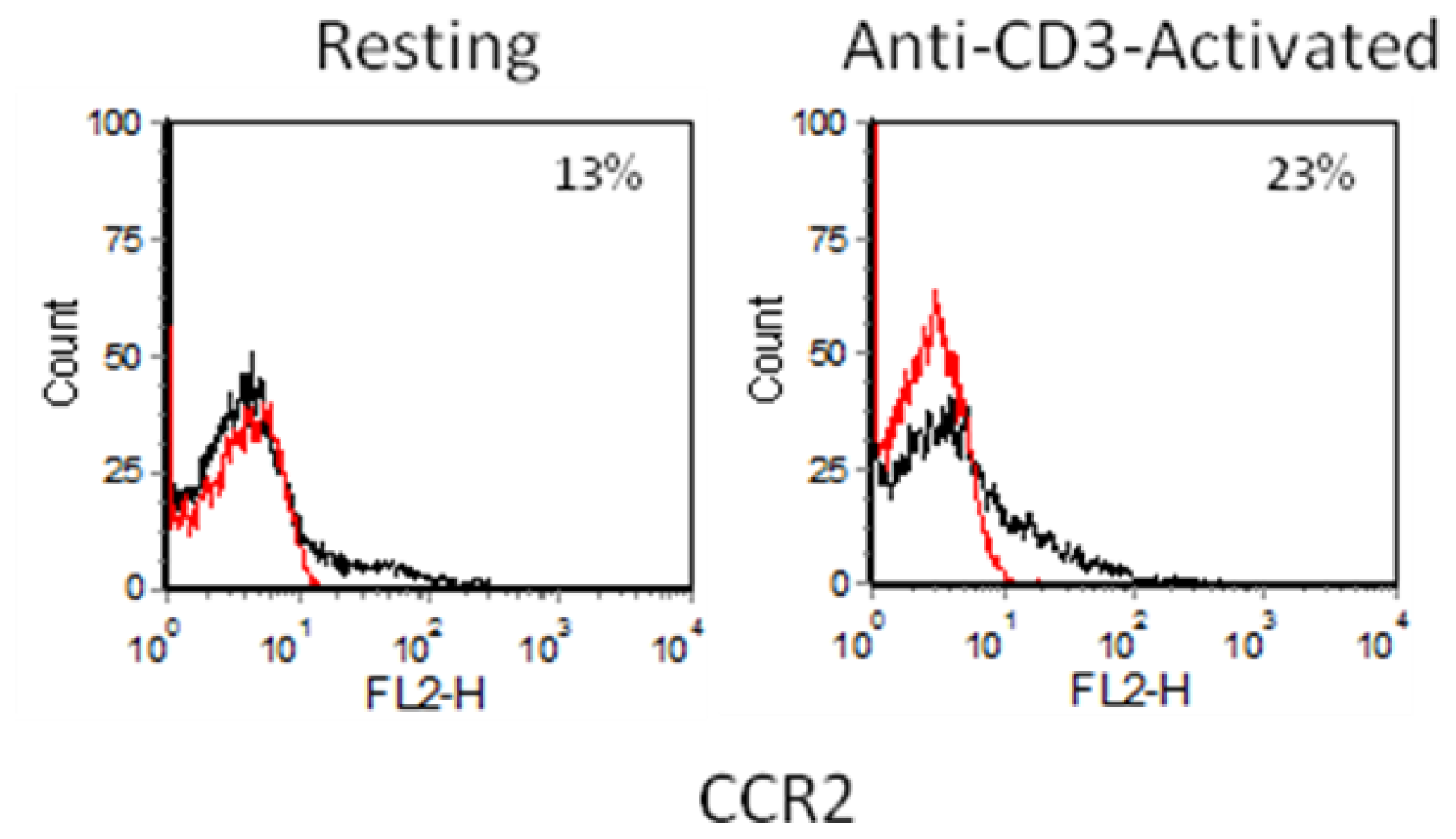
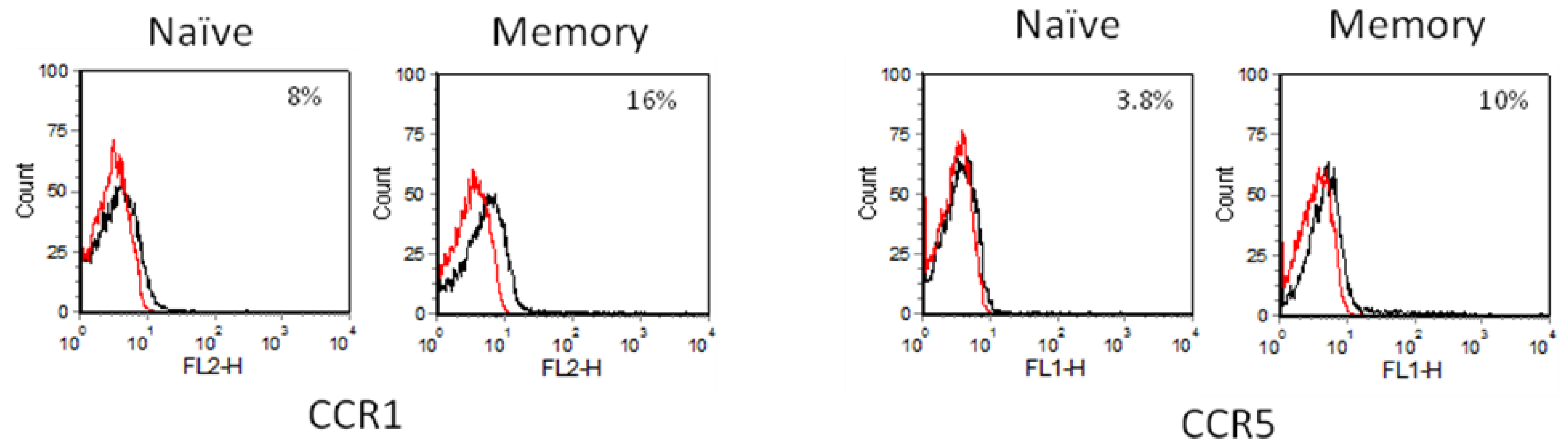
2.3. Chemokine Receptor Expression on Leukocyte Subsets
2.3.1. Resting and Activated CD4+ T Cells
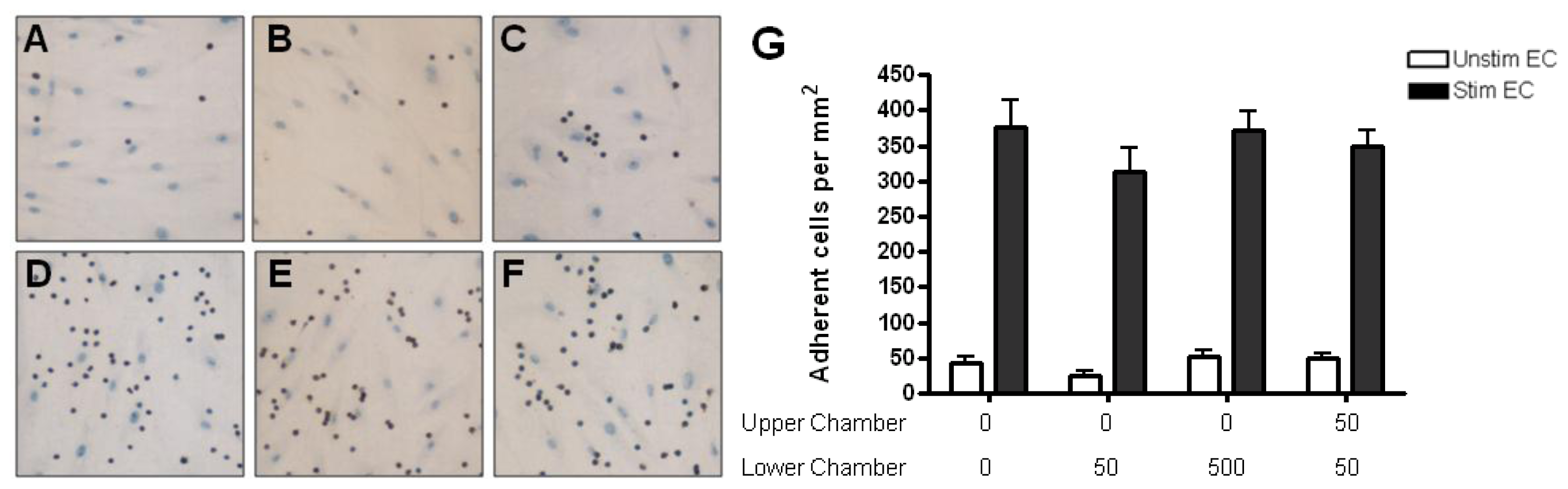
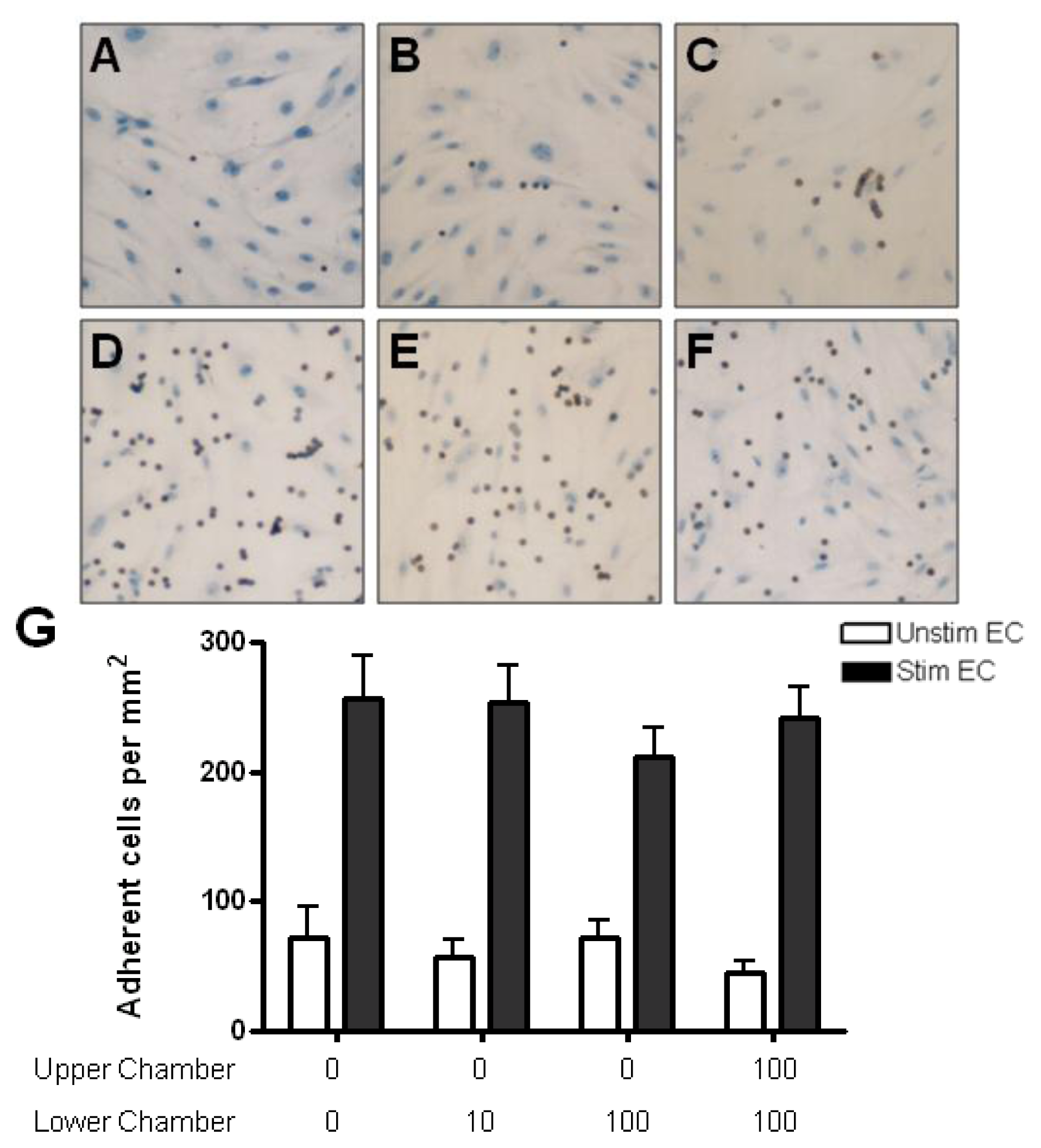
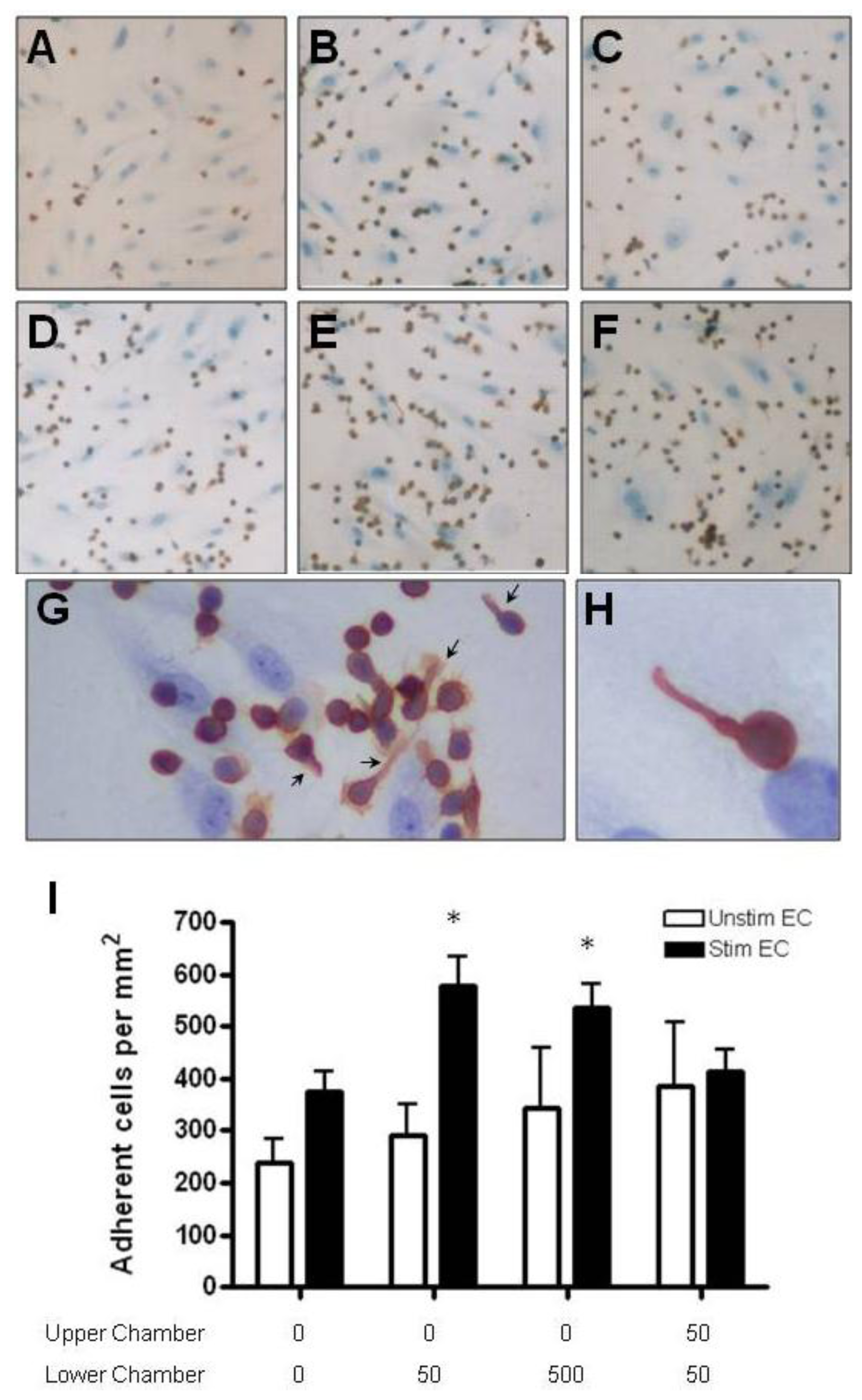
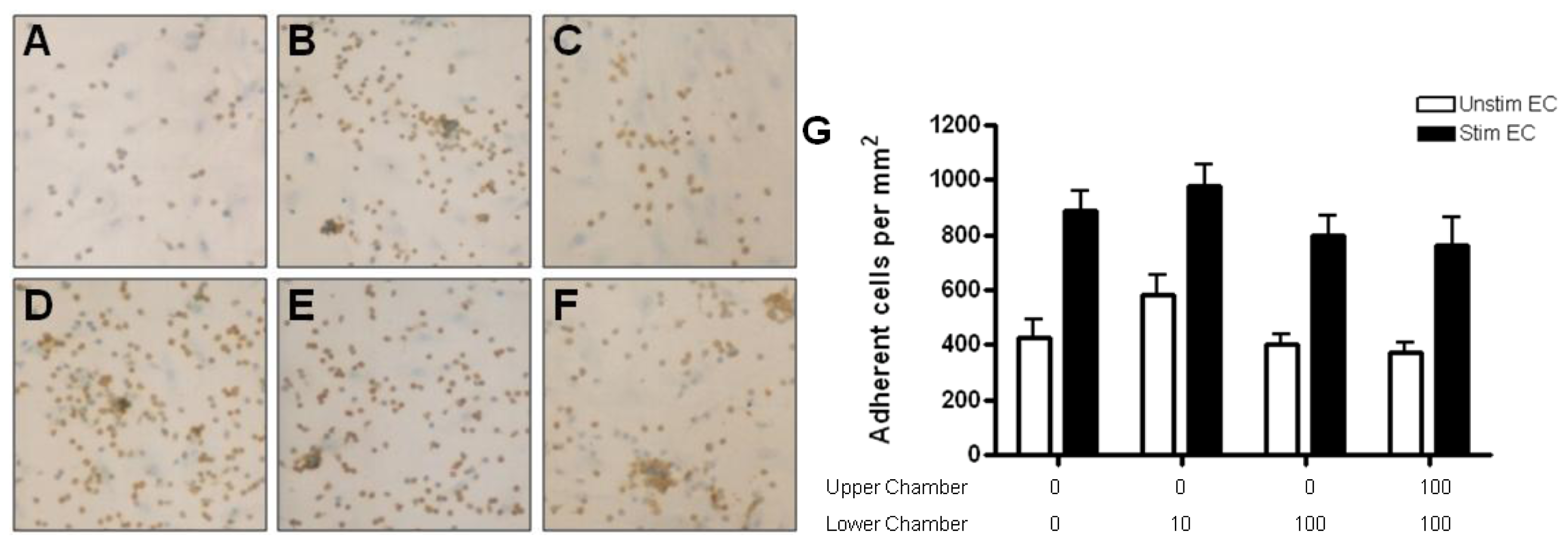
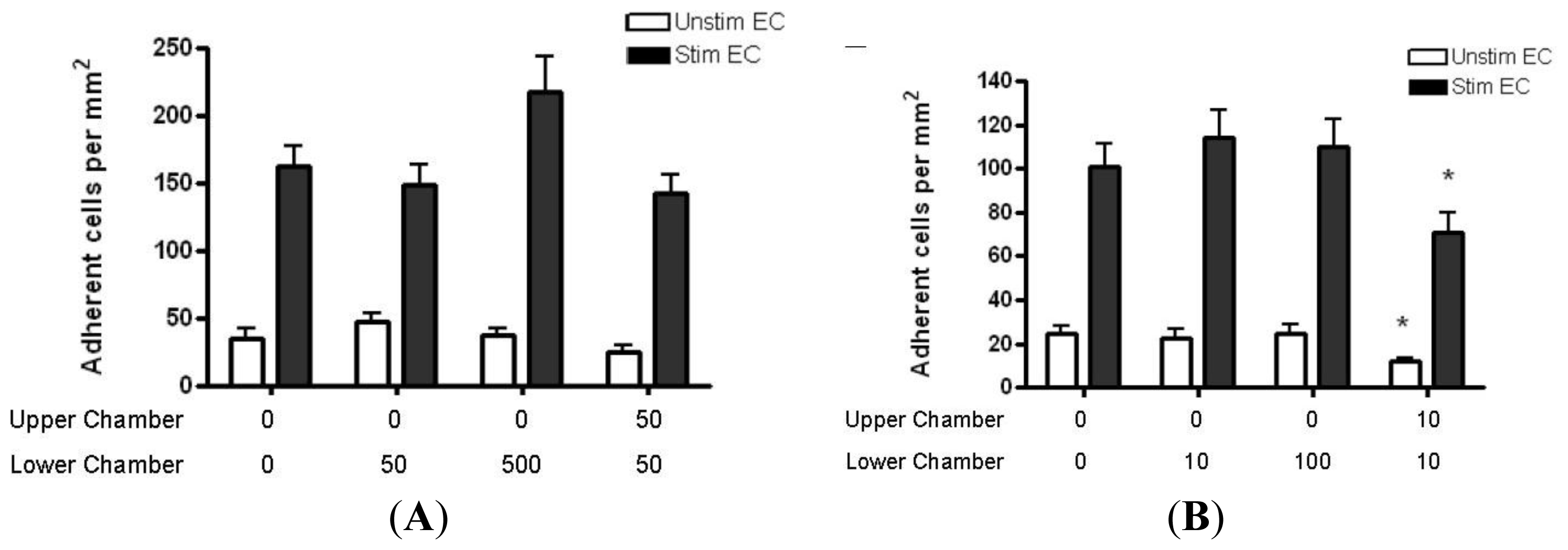
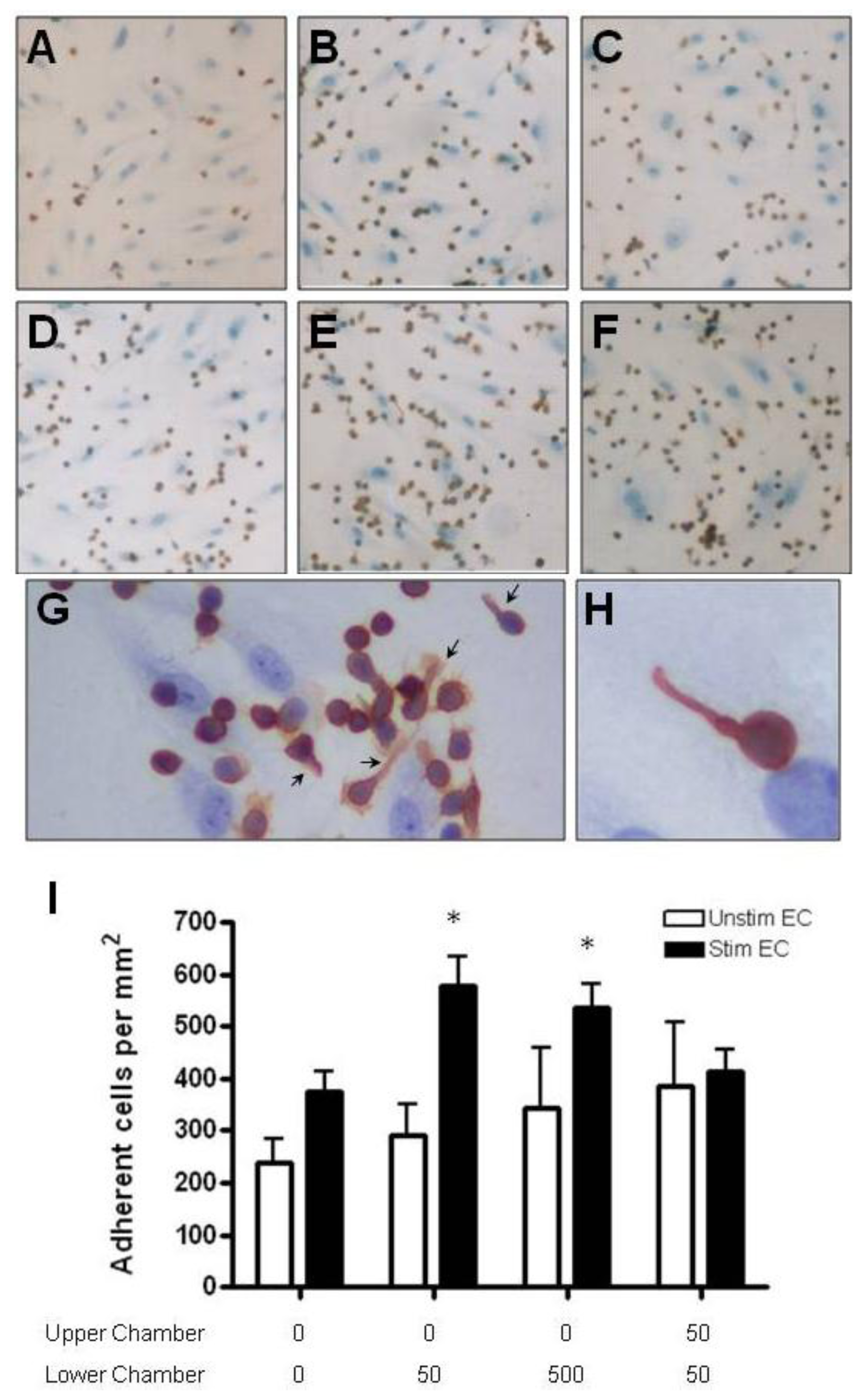
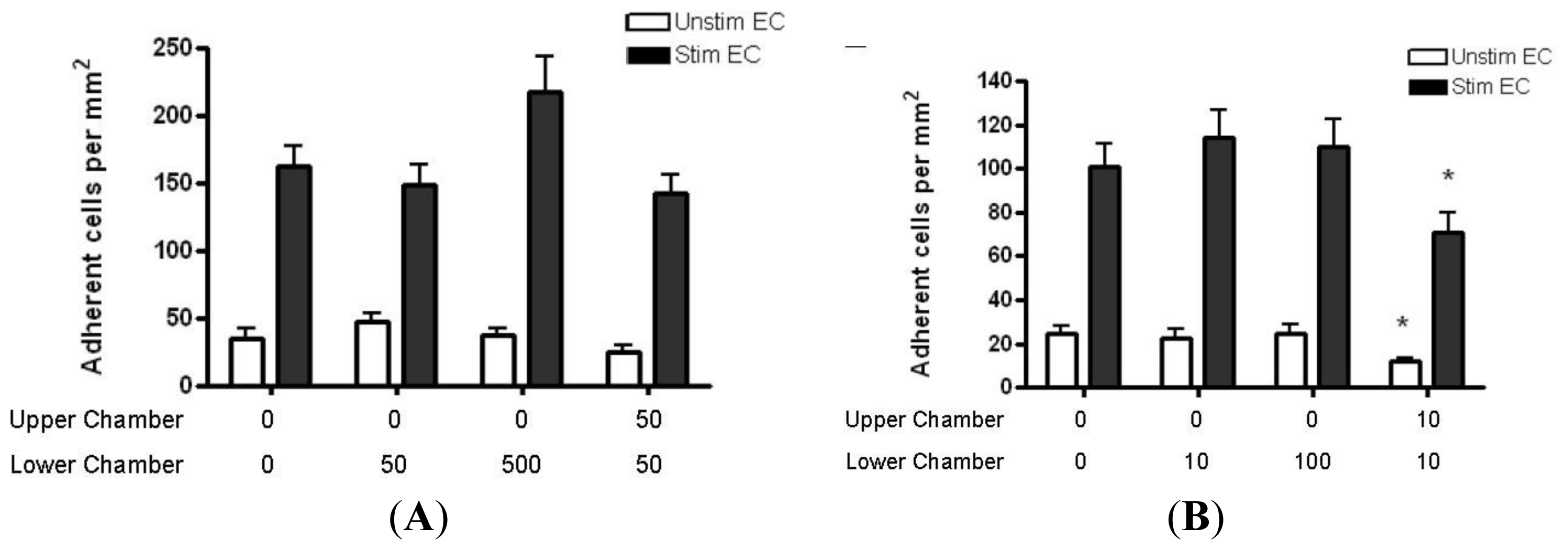
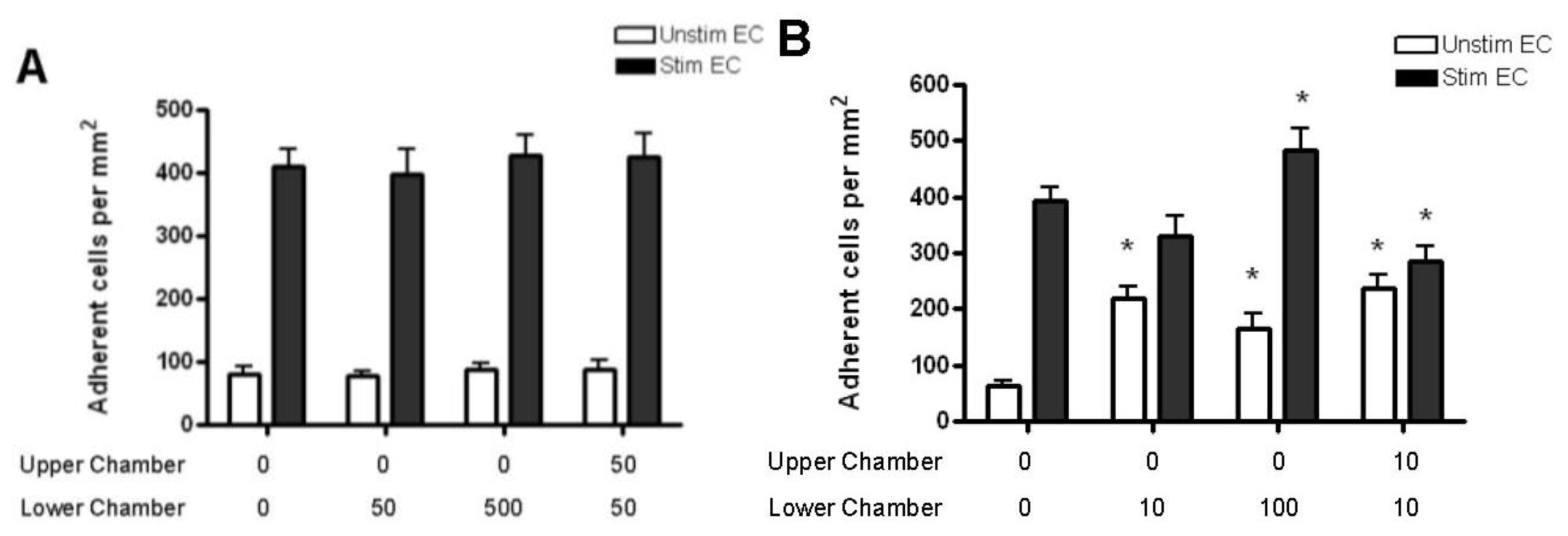
2.4. Adhesion of CD4+ T Cell Subsets to HBMEC in Response to CCL2 and CCL3
3. Experimental Section
3.1. Isolation and Culture of Human Brain Microvessel Endothelial Cells
3.2. Isolation and Characterization of T Cell Subsets
3.3. Antibodies, Cytokines and Chemokines
3.4. CD4+ T Cell Activation
3.5. Chemokine Diffusion Assay
3.6. Immunoelectron Microscopic Localization of Chemokines
3.7. T Cell Adhesion Assay
3.8. Statistics
4. Conclusions
Acknowledgments
References
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hebert, C.A.; Horuk, R.; Matsushima, K.; Miller, L.H.; Oppenheim, J.J.; Power, C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev 2000, 52, 145–176. [Google Scholar]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol 2000, 18, 217–242. [Google Scholar]
- Baggiolini, M.; Dewald, B.; Moser, B. Human chemokines: An update. Annu. Rev. Immunol 1997, 15, 675–705. [Google Scholar]
- Qin, S.; LaRosa, G.; Campbell, J.J.; Smith-Heath, H.; Kassam, N.; Shi, X.; Zeng, L.; Buthcher, E.C.; Mackay, C.R. Expression of monocyte chemoattractant protein-1 and interleukin-8 receptors on subsets of T cells: Correlation with transendothelial chemotactic potential. Eur. J. Immunol 1996, 26, 640–647. [Google Scholar]
- Menten, P.; Wuyts, A.; van Damme, J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev 2002, 13, 455–481. [Google Scholar]
- Simpson, J.E.; Newcombe, J.; Cuzner, M.L.; Woodroofe, M.N. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J. Neuroimmunol 1998, 84, 238–249. [Google Scholar]
- McManus, C.; Berman, J.W.; Brett, F.M.; Staunton, H.; Farrell, M.; Brosnan, C.F. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: An immunohistochemical and in situ hybridization study. J. Neuroimmunol 1998, 86, 20–29. [Google Scholar]
- Van Der Voorn, P.; Tekstra, J.; Beelen, R.H.; Tensen, C.P.; van Der Valk, P.; De Groot, C.J. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol 1999, 154, 45–51. [Google Scholar]
- Mahad, D.J.; Ransohoff, R.M. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol 2003, 15, 23–32. [Google Scholar]
- Lukacs, N.W.; Strieter, R.M.; Elner, V.M.; Evanoff, H.L.; Burdick, M.; Kunkel, S.L. Intercellular adhesion molecule-1 mediates the expression of monocyte-derived MIP-1 alpha during monocyte-endothelial cell interactions. Blood 1994, 83, 1174–1178. [Google Scholar]
- Weiss, J.M.; Downie, S.A.; Lyman, W.D.; Berman, J.W. Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J. Immunol 1998, 161, 6896–6903. [Google Scholar]
- Bajetto, A.; Bonavia, R.; Barbero, S.; Schettini, G. Characterization of chemokines and their receptors in the central nervous system: Physiopathological implications. J. Neurochem 2002, 82, 1311–1329. [Google Scholar]
- Chui, R.; Dorovini-Zis, K. Regulation of CCL2 and CCL3 expression in human brain endothelial cells by cytokines and lipopolysaccharide. J. Neuroinflamm 2010, 7, 1. [Google Scholar]
- Gay, F.W.; Drye, T.J.; Dick, G.W.; Esiri, M.M. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain 1997, 120, 1461–1483. [Google Scholar]
- Henderson, A.P.; Barnett, M.H.; Parratt, J.D.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol 2009, 66, 739–753. [Google Scholar]
- Quandt, J.; Dorovini-Zis, K. The beta chemokines CCL4 and CCL5 enhance adhesion of specific CD4+ T cell subsets to human brain endothelial cells. J. Neuropathol. Exp. Neurol 2004, 63, 350–362. [Google Scholar]
- Liu, K.K.; Dorovini-Zis, K. Regulation of CXCL12 and CXCR4 expression by human brain endothelial cells and their role in CD4+ and CD8+ T cell adhesion and transendothelial migration. J. Neuroimmunol 2009, 215, 49–64. [Google Scholar]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp. Neurol 2004, 190, 446–455. [Google Scholar]
- Huynh, H.K.; Dorovini-Zis, K. Effects of interferon-gamma on primary cultures of human brain microvessel endothelial cells. Am. J. Pathol 1993, 142, 1265–1278. [Google Scholar]
- Selmaj, K.; Raine, C.S.; Cannella, B.; Brosnan, C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Invest 1991, 87, 949–954. [Google Scholar]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; Merrill, J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med 1989, 170, 607–612. [Google Scholar]
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol 1995, 37, 424–435. [Google Scholar]
- Woodroofe, M.N.; Cuzner, M.L. Cytokine mRNA expression in inflammatory multiple sclerosis lesions: Detection by non-radioactive in situ hybridization. Cytokine 1993, 5, 583–588. [Google Scholar]
- Song, L.; Pachter, J.S. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc. Res 2004, 67, 78–89. [Google Scholar]
- Roberts, T.K.; Eugenin, E.A.; Lopez, L.; Romero, I.A.; Weksler, B.B.; Couraud, P.-O.; Berman, J.W. CCL2 disrupts the adherens junction: implications for neuroinflammation. Lab. Invest 2012, 92, 1213–1233. [Google Scholar]
- Ge, S.; Song, L.; Serwanski, D.R.; Kuziel, W.A.; Pachter, J.S. Transcellular transport of CCL2 across brain microvascular endothelial cells. J. Neurochem 2008, 104, 1219–1232. [Google Scholar]
- Man, S.; Ubogu, E.E.; Ransohoff, R.M. Inflammatory cell migration into the central nervous system: A few new twists on an old tale. Brain Pathol 2007, 17, 243–250. [Google Scholar]
- Crola Da Silva, C.; Lamerant-Fayel, N.; Paprocka, M.; Mitterrand, M.; Gosset, D.; Dus, D.; Kieda, C. Selective human endothelial cell activation by chemokines as a guide to cell homing. Immunology 2009, 126, 394–404. [Google Scholar]
- Parish, C.R. The role of heparan sulphate in inflammation. Nat. Rev. Immunol 2006, 6, 633–643. [Google Scholar]
- Klein, N.J.; Shennan, G.I.; Heyderman, R.S.; Levin, M. Alteration in glycosaminoglycan metabolism and surface charge on human umbilical vein endothelial cells induced by cytokines, endotoxin and neutrophils. J. Cell Sci 1992, 102, 821–832. [Google Scholar]
- Sallusto, F.; Lanzavecchia, A.; Mackay, C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 1998, 19, 568–574. [Google Scholar]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar]
- Sorensen, T.L.; Tani, M.; Jensen, J.; Pierce, V.; Lucchinetti, C.; Folcik, V.A.; Qin, S.; Rottman, J.; Sellebjerg, F.; Strieter, R.M.; et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Invest 1999, 103, 807–815. [Google Scholar]
- Szczucinski, A.; Losy, J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol. Scand 2007, 115, 137–146. [Google Scholar]
- Simpson, J.; Rezaie, P.; Newcombe, J.; Cuzner, M.L.; Male, D.; Woodroofe, M.N. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J. Neuroimmunol 2000, 108, 192–200. [Google Scholar]
- Wong, D.; Dorovini-Zis, K. Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J. Neuroimmunol 1992, 39, 11–21. [Google Scholar]
- Wong, D.; Dorovini-Zis, K. Expression of vascular cell adhesion molecule-1 (VCAM-1) by human brain microvessel endothelial cells in primary culture. Microvasc. Res 1995, 49, 325–339. [Google Scholar]
- Wong, D.; Dorovini-Zis, K. Platelet/endothelial cell adhesion molecule-1 (PECAM-1) expression by human brain microvessel endothelial cells in primary culture. Brain Res 1996, 731, 217–220. [Google Scholar]
- Wong, D.; Dorovini-Zis, K. Regualtion by cytokines and lipopolysaccharide of E-selectin expression by human brain microvessel endothelial cells in primary culture. J. Neuropathol. Exp. Neurol 1996, 55, 225–235. [Google Scholar]
- Wong, D.; Prameya, R.; Dorovini-Zis, K. In vitro adhesion and migration of T lymphocytes across monolayers of human brain microvessel endothelial cells: regulation by ICAM-1, VCAM-1, E-selectin and PECAM-1. J. Neuropathol. Exp. Neurol 1999, 58, 138–152. [Google Scholar]
- Krummel, M.F.; Macara, I. Maintenance and modulation of T cell polarity. Nat. Immunol 2006, 7, 1143–1149. [Google Scholar]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol 2007, 7, 678–689. [Google Scholar]
- Thelen, M.; Stein, J.V. How chemokines invite leukocytes to dance. Nat. Immunol 2008, 9, 953–959. [Google Scholar]
- Del Pozo, M.A.; Cabanas, C.; Montoya, M.C.; Ager, A.; Sanchez-Mateos, P.; Sanchez-Madrid, F. ICAMs redistributed by chemokines to cellular uropods as a mechanism for recruitment of T lymphocytes. J. Cell Biol 1997, 137, 493–508. [Google Scholar]
- Taub, D.D.; Conlon, K.; Lloyd, A.R.; Oppenheim, J.J.; Kelvin, D.J. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science 1993, 260, 355–358. [Google Scholar]
- Nakajima, H.; Fukuda, K.; Doi, Y.; Sugino, M.; Kimura, F.; Hanafusa, T.; Ikemoto, T.; Shimizu, A. Expression of TH1/TH2-related chemokine receptors on peripheral T cells and correlation with clinical disease activity in patients with multiple sclerosis. Eur. Neurol 2004, 52, 162–168. [Google Scholar]
- Schall, T.J.; Bacon, K.; Camp, R.D.; Kaspari, J.W.; Goeddel, D.V. Human macrophage inflammatory protein alpha (MIP-1 alpha) and MIP-1 beta chemokines attract distinct populations of lymphocytes. J. Exp. Med 1993, 177, 1821–1826. [Google Scholar]
- Callahan, M.K.; Williams, K.A.; Kivisakk, P.; Pearce, D.; Stins, M.F.; Ransohoff, R.M. CXCR3 marks CD4+ memory T lymphocytes that are competent to migrate across a human brain microvascular endothelial cell layer. J. Neuroimmunol 2004, 153, 150–157. [Google Scholar]
- Sato, K.; Kawasaki, H.; Morimoto, C.; Yamashima, N.; Matsuyama, T. An abortive ligand-induced activation of CCR1-mediated downstream signaling event and a deficiency of CCR5 expression are associated with the hyporesponsiveness of human naive CD4+ T cells to CCL3 and CCL5. J. Immunol 2002, 168, 6263–6272. [Google Scholar]
- Longden, J.; Cooke, E.L.; Hill, S.J. Effect of CCR5 receptor antagonists on endocytosis of the human CCR5 receptor in CHO-K1 cells. Br. J. Pharmacol 2008, 153, 1513–1527. [Google Scholar]
- Carr, M.W.; Roth, S.J.; Luther, E.; Rose, S.S.; Springer, T.A. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. USA 1994, 91, 3652–3656. [Google Scholar]
- Misu, T.; Onodera, H.; Fujihara, K.; Matsushima, K.; Yoshie, O.; Okita, N.; Takase, S.; Itoyama, Y. Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: Imbalance of Th1/Th2-associated chemokine signaling. J. Neuroimmunol 2001, 114, 207–212. [Google Scholar]
- Giunti, D.; Borsellino, G.; Benelli, R.; Marchese, M.; Capello, E.; Valle, M.T.; Pedemonte, E.; Noonan, D.; Albini, A.; Bernardi, G.; et al. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J Leukocyte Biol 2003, 73, 584–590. [Google Scholar]
- Ding, Z.; Xiong, K.; Issekutz, T.B. Regulation of chemokine-induced transendothelial migration of T lymphocytes by endothelial activation: Differential effects on naive and memory T cells. J. Leukocyte Biol 2000, 67, 825–833. [Google Scholar]
- Dorovini-Zis, K.; Prameya, R.; Bowman, P.D. Culture and characterization of microvascular endothelial cells derived from human brain. Lab. Invest 1991, 64, 425–436. [Google Scholar]
- Santambrogio, L.; Pakaski, M.; Wong, M.L.; Cipriani, B.; Brosnan, C.F.; Lees, M.B.; Dorf, M.E. Antigen presenting capacity of brain microvasculature in altered peptide ligand modulation of experimental allergic encephalomyelitis. J. Neuroimmunol 1999, 93, 81–91. [Google Scholar]





© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, K.K.; Dorovini-Zis, K. Differential Regulation of CD4+ T Cell Adhesion to Cerebral Microvascular Endothelium by the β-Chemokines CCL2 and CCL3. Int. J. Mol. Sci. 2012, 13, 16119-16140. https://doi.org/10.3390/ijms131216119
Liu KK, Dorovini-Zis K. Differential Regulation of CD4+ T Cell Adhesion to Cerebral Microvascular Endothelium by the β-Chemokines CCL2 and CCL3. International Journal of Molecular Sciences. 2012; 13(12):16119-16140. https://doi.org/10.3390/ijms131216119
Chicago/Turabian StyleLiu, Kenneth KY, and Katerina Dorovini-Zis. 2012. "Differential Regulation of CD4+ T Cell Adhesion to Cerebral Microvascular Endothelium by the β-Chemokines CCL2 and CCL3" International Journal of Molecular Sciences 13, no. 12: 16119-16140. https://doi.org/10.3390/ijms131216119