Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites


Abstract
:1. Introduction
2. Phase I Drug Metabolism
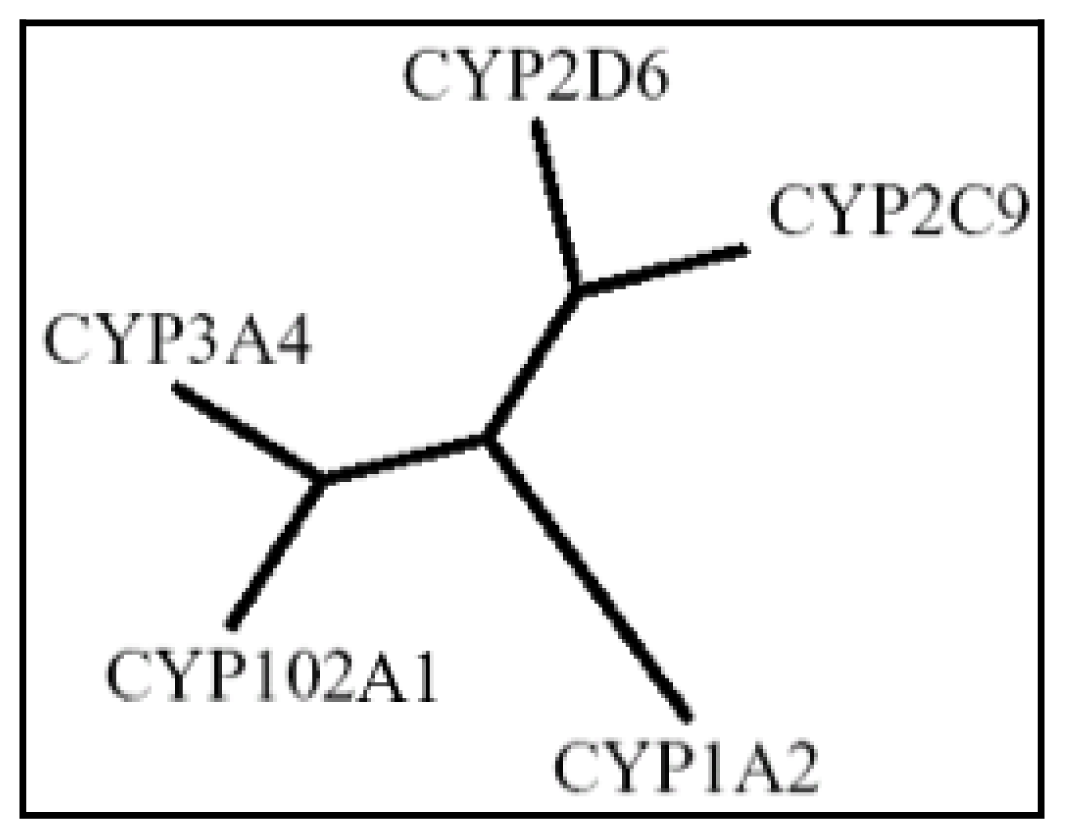
3. P450 BM3 as Biocatalyst
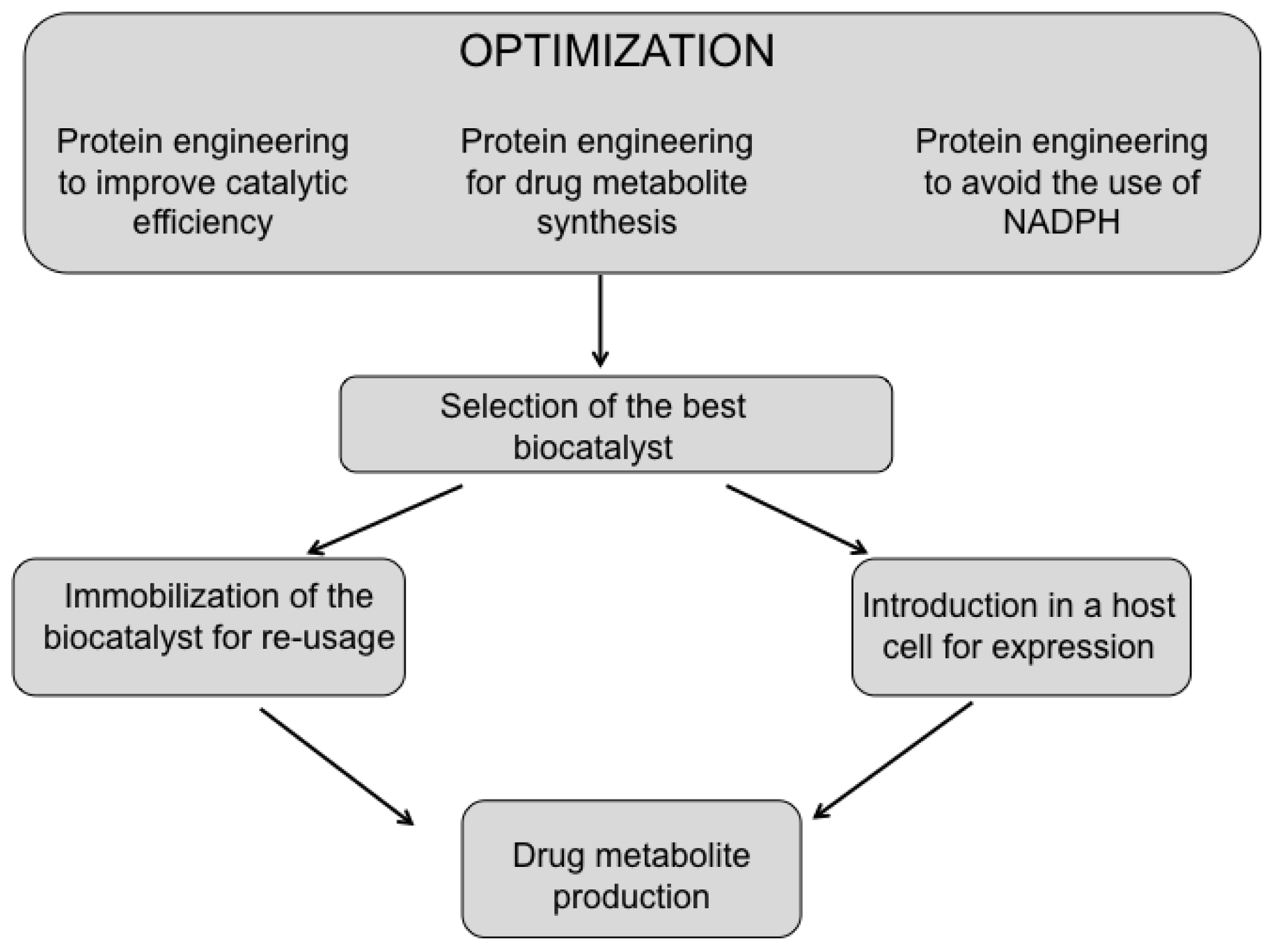
4. Optimization of P450 BM3 as a Biocatalyst for Drug Metabolites Production
- The increase of the substrate specificity by protein engineering;
- The improvement of the catalytic performance (KM, kcat, coupling efficiency) of P450 BM3 toward drugs;
- The substitution of the costly NADPH cofactor;
- The immobilization and scale-up of the process for industrial application.
4.1. Protein Engineering to Improve Substrate Selectivity
4.2. Protein Engineering to Improve Catalytic Efficiency
4.3. Substitution of the Costly NADPH Cofactor
4.4. Immobilization and Scale-Up of the Bioprocess for Industrial Application
5. Conclusions and Future Perspectives
Acknowledgment
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discovery 2004, 3, 711–715. [Google Scholar]
- Singh, S.S. Preclinical pharmacokinetics: An approach towards safer and efficacious drugs. Curr. Drug Metab 2006, 7, 165–182. [Google Scholar]
- Fitzgerald, J.D.; O’Donnell, S.R. Pharmacology of 4-hydroxypropranolol, a metabolite of propranolol. Br. J. Pharmacol 1971, 43, 222–235. [Google Scholar]
- Coltart, D.J.; Shand, D.G. Plasma propranolol levels in the quaniitative assessment of β-adrenergic blockade in man. Br. Med. J 1970, 3, 731–734. [Google Scholar]
- Oatis, J.E., Jr; Russell, M.P.; Knapp, D.R.; Walle, T. Ring-hydroxylated propranolol: Synthesis and beta-receptor antagonist and vasodilating activities of the seven isomers. J. Med. Chem. 1981, 24, 309–314. [Google Scholar]
- Miller, J.A. Carcinogenesis by chemicals: An overview—G. H. A. Clowes memorial lecture. Cancer Res 1970, 30, 559–576. [Google Scholar]
- Miller, J.A. Brief-history of chemical carcinogenesis. Cancer Lett 1994, 83, 9–14. [Google Scholar]
- Guengerich, F.P. Generation of reactive intermediates. J. Biochem. Mol. Toxicol 2005, 19, 173–174. [Google Scholar]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther 1973, 187, 185–194. [Google Scholar]
- Dahlin, D.C.; Miwa, G.T.; Lu, A.Y.; Nelson, S.D. N-acetyl-p-benzoquinone imine: A cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. USA 1984, 81, 1327–1331. [Google Scholar]
- Markham, A.; Wagstaff, A.J. Fexofenadine. Drugs 1998, 55, 269–274. [Google Scholar]
- Ortiz de Montellano, P.R.; DeVoss, J.J. CytochromeP450: Structure, Mechanism, and Biochemistry, 3rd ed; Ortiz de Montellano, P.R., Ed.; Kluwer Academic Publishers: New York, NY, USA, 2005; pp. 183–246. [Google Scholar]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol 2001, 14, 611–650. [Google Scholar]
- Lewis, D.F.; Sheridan, G. Cytochromes P450, oxygen, and evolution. Sci. World J 2001, 1, 151–157. [Google Scholar]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev 1996, 96, 2841–2888. [Google Scholar]
- Sakaguchi, M.; Mihara, K.; Sato, R. A short amino-terminal segment of microsomal cytochrome-P-450 functions both as an insertion signal and as a stop transfer sequence. EMBO J 1987, 6, 2425–2431. [Google Scholar]
- Black, S.D. Membrane topology of the mammalian P450-cytochromes. FASEB J 1992, 6, 680–685. [Google Scholar]
- Porter, T.D.; Kasper, C.B. Coding nucleotide-sequence of rat nadph-cytochrome-P-450 oxidoreductase-cdna and identification of flavin-binding domains. Proc. Natl. Acad. Sci. USA 1985, 82, 973–977. [Google Scholar]
- Bertz, R.J.; Granneman, G.R. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin. Pharmacoket 1997, 32, 210–258. [Google Scholar]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar]
- Baranczewski, P.; Stanczak, A.; Kautiainen, A.; Sandin, P.; Edlund, P.O. Introduction to early in vitro identification of metabolites of new chemical entities in drug discovery and development. Pharmacol. Rep 2006, 58, 341–352. [Google Scholar]
- Fantuzzi, A.; Capria, E.; Mak, L.H.; Dodhia, V.R.; Sadeghi, S.J.; Collins, S.; Somers, G.; Huq, E.; Gilardi, G. An electrochemical microfluidic platform for human p450 drug metabolism profiling. Anal. Chem 2010, 82, 10222–10227. [Google Scholar]
- Parikh, A.; Gillam, E.M.J.; Guengerich, F.P. Drug metabolism by Escherichia coli expressing human cytochromes P450. Nat. Biotechnol 1997, 15, 784–788. [Google Scholar]
- Vail, R.B.; Homann, M.J.; Hanna, I.; Zaks, A. Preparative synthesis of drug metabolites using human cytochrome P450s 3A4, 2C9 and 1A2 with NADPH-P450 reductase expressed in Escherichia coli. J. Ind. Microbiol. Biotechnol 2005, 32, 67–74. [Google Scholar]
- Rushmore, T.H.; Reider, P.J.; Slaughter, D.; Assang, C.; Shou, M. Bioreactor systems in drug metabolism: Synthesis of cytochrome P450-generated metabolites. Metab. Eng 2000, 2, 115–125. [Google Scholar]
- Gillam, E.M.; Guengerich, F.P. Exploiting the versatility of human cytochrome P450 enzymes: The promise of blue roses from biotechnology. IUBMB Life 2001, 52, 271–277. [Google Scholar]
- Kumar, S.; Liu, H.; Halpert, J.R. Engineering of cytochrome P450 3A4 for enhanced peroxide-mediated substrate oxidation using directed evolution and site-directed mutagenesis. Drug Metab. Dispos 2006, 34, 1958–1965. [Google Scholar]
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol 2006, 124, 128–145. [Google Scholar]
- Lamb, D.C.; Waterman, M.R.; Kelly, S.L.; Guengerich, F.P. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol 2007, 18, 504–512. [Google Scholar]
- Gillam, E.M. Extending the capabilities of nature’s most versatile catalysts: directed evolution of mammalian xenobiotic-metabolizing P450s. Arch. Biochem. Biophys 2007, 464, 176–186. [Google Scholar]
- Kumar, S. Engineering cytochrome P450 biocatalysts for biotechnology, medicine and bioremediation. Expert Opin. Drug Metab. Toxicol 2010, 6, 115–131. [Google Scholar]
- Guengerich, F.P. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discovery 2002, 1, 359–366. [Google Scholar]
- Julsing, M.K.; Cornelissen, S.; Buehler, B.; Schmid, A. Heme-iron oxygenases: Powerful industrial biocatalysts? Curr. Opin. Chem. Biol 2008, 12, 177–186. [Google Scholar]
- Nahri, L.O.; Fulco, A.J. Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem 1986, 261, 7160–7169. [Google Scholar]
- Noble, M.A.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Mackay, A.C.; Reid, G.A.; Hanzlik, R.P.; Munro, A.W. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J 1999, 339, 371–379. [Google Scholar]
- Munro, A.W.; Daff, S.; Coggins, J.R.; Lindsay, J.G.; Chapman, S.K. Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur. J. Biochem 1996, 239, 403–409. [Google Scholar]
- Di Nardo, G.; Fantuzzi, A.; Sideri, A.; Panicco, P.; Sassone, C.; Giunta, C.; Gilardi, G. Wild-type CYP102A1 as a biocatalyst: Turnover of drugs usually metabolised by human liver enzymes. J. Biol. Inorg. Chem 2007, 12, 313–323. [Google Scholar]
- Tsotsou, G.E.; Cass, A.E.G.; Gilardi, G. High throughput assay for cytochrome P450BM3 for screening libraries of substrates and combinatorial mutants. Biosens. Bioelectron 2002, 17, 119–131. [Google Scholar]
- Tsotsou, G.E.; di Nardo, G.; Sadeghi, S.J.; Fruttero, R.; Lazzarato, L.; Bertinaria, M.; Gilardi, G. A rapid screening for cytochrome P450 catalysis on new chemical entities: Cytochrome P450 BM3 and 1,2,5-oxadiazole derivatives. J. Biomol. Screening 2012. [Google Scholar] [CrossRef]
- Yun, C.H.; Kim, K.H.; Kim, D.H.; Jung, H.C.; Pan, J.G. The bacterial P450BM3: A prototype for a biocatalyst with human P450 activities. Trends Biotechnol 2007, 25, 289–298. [Google Scholar]
- Kim, D.H.; Kim, K.H.; Kim, D.H.; Liu, K.H.; Jung, H.C.; Pan, J.G.; Yun, C.H. Generation of Human Metabolites of 7-Ethoxycoumarin by Bacterial Cytochrome P450BM3. Drug Metab. Dispos 2008, 36, 2166–2170. [Google Scholar]
- Kim, D.H.; Ahn, T.; Jung, H.C.; Pan, J.G.; Yun, C.H. Generation of the human metabolite piceatannol from the anticancer-preventive agent resveratrol by bacterial cytochrome P450 BM3. Drug Metab. Dispos 2008, 37, 932–936. [Google Scholar]
- Warman, A.J.; Roitel, O.; Neeli, R.; Girvan, H.M.; Seward, H.E.; Murray, S.A.; McLean, K.J.; Joyce, M.G.; Toogood, H.; Holt, R.A.; et al. Flavocytochrome P450BM3: an update on structure and mechanism of a biotechnologically important enzyme. Biochem. Soc. Trans 2005, 33, 747–753. [Google Scholar]
- McLean, K.J.; Girvan, H.M.; Munro, A.W. Cytochrome P450/redox partner fusion enzymes: Biotechnological and toxicological prospects. Expert Opin. Drug Metab. Toxicol 2007, 3, 847–863. [Google Scholar]
- Ravichandran, K.G.; Boddupalli, S.S.; Hasemann, C.A.; Peterson, J.A.; Deisenhofer, J. Crystal-structure of hemoprotein domain of p450bm-3, a prototype for microsomal P450’s. Science 1993, 261, 731–736. [Google Scholar]
- Li, H.Y.; Poulos, T.L. The structure of the cytochrome P450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Biol 1997, 4, 140–146. [Google Scholar]
- Rentmeister, A.; Brown, T.R.; Snow, C.D.; Carbone, M.N.; Arnold, F.H. Engineered bacterial mimics of human drug metabolizing enzyme CYP2C9. ChemCatChem 2011, 3, 1065–1071. [Google Scholar]
- Van Vugt-Lussenburg, B.M.A.; Babel, L.C.; Vermeulen, N.P.E.; Commandeur, J.N.M. Evaluation of alkoxyresorufins as fluorescent substrates for cytochrome P450BM3 and site-directed mutants. Anal. Biochem 2005, 341, 148–155. [Google Scholar]
- Van Vugt-Lussenburg, B.M.A.; Damsten, M.C.; Maasdijk, D.M.; Vermeulen, N.P.E.; Commandeur, J.N.M. Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450BM3. Biochem. Biophys. Res. Commun 2006, 346, 810–818. [Google Scholar]
- Patten, C.J.; Thomas, P.E.; Guy, R.L.; Lee, M.; Gonzalez, F.J.; Guengerich, F.P.; Yang, C.S. Cytochrome-P450 enzymes involved in acetaminophen activation by rat and human liver-microsomes and their kinetics. Chem. Res. Toxicol 1993, 6, 511–518. [Google Scholar]
- Reinen, J.; van Leeuwen, J.S.; Li, Y.; Sun, L.; Grootenhuis, P.D.; Decker, C.J.; Saunders, J.; Vermeulen, N.P.; Commandeur, J.N. Efficient screening of cytochrome P450 BM3 mutants for their metabolic activity and diversity toward a wide set of drug-like molecules in chemical space. Drug Metab. Dispos 2011, 39, 1568–1576. [Google Scholar]
- Wen, B.; Ma, L.; Zhu, M. Bioactivation of the tricyclic antidepressant amitriptyline and its metabolite nortriptyline to arene oxide intermediates in human liver microsomes and recombinant P450s. Chemico-Biol. Interact 2008, 173, 59–67. [Google Scholar]
- Van Vugt-Lussenburg, B.M.A.; Stjernschantz, E.; Lastdrager, J.; Oostenbrink, C.; Vermeulen, N.P.E.; Commandeur, J.N.M. Identification of critical residues in novel drug metabolizing mutants of cytochrome P450BM3 using random mutagenesis. J. Med. Chem 2007, 50, 455–461. [Google Scholar]
- Li, X.Q.; Bjorkman, A.; Andersson, T.B.; Ridderstrom, M.; Masimirembwa, C.M. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: A new high affinity and turnover enzyme-specific probe substrate. J. Pharmacol. Exp. Ther 2002, 300, 399–407. [Google Scholar]
- Sawayama, A.M.; Chen, M.M.Y.; Kulanthaivel, P.; Kuo, M.S.; Hemmerle, H.; Arnold, F.H. A panel of cytochrome P450 BM3 variants to produce drug metabolites and diversify lead compounds. Chemistry 2009, 15, 11723–11729. [Google Scholar]
- Matsumoto, S.; Yamazoe, Y. Involvement of multiple human cytochromes P450 in the liver microsomal metabolism of astemizole and a comparison with terfenadine. Br. J. Clin. Pharmacol 2001, 51, 133–142. [Google Scholar]
- Landwehr, M.; Hochrein, L.; Otey, C.R.; Kasrayan, A.; Backvall, J.E.; Arnold, F.H. Enantioselective alpha-hydroxylation of 2-arylacetic acid derivatives and buspirone catalyzed by engineered cytochrome P450BM-3. J. Am. Chem. Soc 2006, 128, 6058–6059. [Google Scholar]
- Zhu, M.S.; Zhao, W.P.; Jimenez, H.; Zhang, D.L.; Yeola, S.; Dai, R.K.; Vachharajani, N.; Mitroka, J. Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes. Drug Metab. Dispos 2005, 33, 500–507. [Google Scholar]
- Peter, R.; Bocker, R.; Beaune, P.H.; Iwasaki, M.; Guengerich, F.P.; Yang, C.S. Hydroxylation of chlorzoxazone as a specific probe for human liver cytochromeP-450IIE1. Chem. Res. Toxicol 1990, 3, 566–573. [Google Scholar]
- Hiratsuka, M.; Hinai, Y.; Sasaki, T.; Konno, Y.; Imagawa, K.; Ishikawa, M.; Mizugaki, M. Characterization of human cytochrome P450 enzymes involved in the metabolism of cilostazol. Drug Metab. Dispos 2007, 35, 1730–1732. [Google Scholar]
- Brosen, K.; Naranjo, C.A. Review of pharmacokinetic and pharmacodynamic interaction studies with citalopram. Eur. Neuropsychopharmacol 2001, 11, 275–283. [Google Scholar]
- Damsten, M.C.; van Vugt-Lussenburg, B.M.A.; Zeldenthuis, T.; de Vlieger, J.S.B.; Commandeur, J.N.M.; Vermeulen, N.P.E. Application of drug metabolising mutants of cytochrome P450BM3 (CYP102A1) as biocatalysts for the generation of reactive metabolites. Chemico-Biol. Interact 2008, 171, 96–107. [Google Scholar]
- Urichuk, L.; Prior, T.I.; Dursun, S.; Baker, G. Metabolism of atypical antipsychotics: involvement of cytochrome P450 enzymes and relevance for drug-drug interactions. Curr. Drug Metab 2008, 9, 410–418. [Google Scholar]
- Yu, A.M.; Haining, R.L. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: Can dextromethorphan be used as a dual probe for both CYP2D6 and CYP3A activities? Drug Metab. Dispos 2001, 29, 1514–1520. [Google Scholar]
- Van Leeuwen, J.S.; Vredenburg, G.; Dragovic, S.; Tjong, T.F.J.; Vos, J.C.; Vermeulen, N.P.E. Metabolism related toxicity of diclofenac in yeast as model system. Toxicol. Lett 2011, 200, 162–168. [Google Scholar]
- Tsotsou, G.E.; Sideri, A.; Goyal, A.; di Nardo, G.; Gilardi, G. Identification of mutant Asp251Gly/Gln307His of cytochrome P450 BM3 for the generation of metabolites of diclofenac, ibuprofen and tolbutamide. Chemistry 2012, 18, 3582–3588. [Google Scholar]
- Mancy, A.; Antignac, M.; Minoletti, C.; Dijols, S.; Mouries, V.; Duong, N.T.H.; Battioni, P.; Dansette, P.M.; Mansuy, D. Diclofenac and its derivatives as tools for studying human cytochromes P450 active sites: Particular efficiency and regioselectivity of P4502Cs. Biochemistry 1999, 38, 14264–14270. [Google Scholar]
- Pinto, A.G.; Horlander, J.; Chalasani, N.; Hamman, M.; Asghar, A.; Kolwankar, D.; Hall, S.D. Diltiazem inhibits human intestinal cytochrome P450 3A (CYP3A) activity in vivo without altering the expression of intestinal mRNA or protein. Br. J. Clin. Pharmacol 2005, 59, 440–446. [Google Scholar]
- Bourrie, M.; Meunier, V.; Berger, Y.; Fabre, G. Role of cytochrome P-4502C9 in irbesartan oxidation by human liver microsomes. Drug Metab. Dispos 1999, 27, 288–296. [Google Scholar]
- Kim, K.H.; Kang, J.Y.; Kim, D.H.; Park, S.H.; Park, S.H.; Kim, D.; Park, K.D.; Lee, Y.J.; Jung, H.C.; Pan, J.G.; et al. Generation of human chiral metabolites of simvastatin and lovastatin by bacterial CYP102A1 mutants. Drug Metab. Dispos 2011, 39, 140–150. [Google Scholar]
- Garcia, M.J.; Reinoso, R.F.; Navarro, A.S.; Prous, J.R. Clinical pharmacokinetics of statins. Methods Findings Exp. Clin. Pharmacol 2003, 25, 457–481. [Google Scholar]
- Miners, J.O.; Coulter, S.; Tukey, R.H.; Veronese, M.E.; Birkett, D.J. Cytochromes P450, 1A2, and 2C9 are responsible for the human hepatic O-demethylation of R- and S-naproxen. Biochem. Pharmacol 1996, 51, 1003–1008. [Google Scholar]
- Guengerich, F.P.; Martin, M.V.; Beaune, P.H.; Kremers, P.; Wolff, T.; Waxman, D.J. Characterization of rat and human-liver microsomal cytochrome-P-450 forms involved in nifedipine oxidation, a prototype for genetic-polymorphism in oxidative drug-metabolism. J. Biol. Chem 1986, 261, 5051–5060. [Google Scholar]
- Dixon, C.M.; Colthup, P.V.; Serabjit-Singh, C.J.; Kerr, B.M.; Boehlert, C.C.; Park, G.R.; Tarbit, M.H. Multiple forms of cytochrome-P450 are involved in the metabolism of ondansetron in humans. Drug Metab. Dispos 1995, 23, 1225–1230. [Google Scholar]
- Park, S.H.; Kim, D.H.; Kim, D.; Jung, H.C.; Pan, J.G.; Ahn, T.; Yun, C.H. Engineering bacterial cytochrome P450 (P450) BM3 into a prototype with human P450 enzyme activity using indigo formation. Drug Metab. Dispos 2010, 38, 732–739. [Google Scholar]
- Yun, C.H.; Miller, G.P.; Guengerich, F.P. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry 2000, 39, 11319–11329. [Google Scholar]
- Botsch, S.; Gautier, J.C.; Beaune, P.; Eichelbaum, M.; Kroemer, H.K. Identification and characterization of the cytochrome-P450 enzymes involved in n-dealkylation of propafenone-molecular-base for interaction potential and variable disposition of active metabolites. Mol. Pharmacol 1993, 43, 120–126. [Google Scholar]
- Otey, C.R.; Bandara, G.; Lalonde, J.; Takahashi, K.; Arnold, F.H. Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol. Bioeng 2006, 93, 494–499. [Google Scholar]
- McGinnity, D.F.; Parker, A.J.; Soars, M.; Riley, R.J. Automated definition of the enzymology of drug oxidation by the major human drug metabolizing cytochrome P450s. Drug Metab. Dispos 2000, 28, 1327–1334. [Google Scholar]
- Meuldermans, W.; Hendrickx, J.; Lauwers, W.; Hurkmans, R.; Swysen, E.; Heykants, J. Excretion and biotransformation of astemizole in rats, guinea-pigs, dogs, and man. Drug Dev. Res 1986, 8, 37–51. [Google Scholar]
- Bidstrup, T.B.; Bjornsdottir, I.; Sidelmann, U.G.; Thomsen, M.S.; Hansen, K.T. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br. J. Clin. Pharmacol 2003, 56, 305–314. [Google Scholar]
- Transon, C.; Leemann, T.; Dayer, P. In vitro comparative inhibition profiles of major human drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6 and CYP3A4) by HMG-CoA reductase inhibitors. Eur. J. Clin. Pharmacol 1996, 50, 209–215. [Google Scholar]
- Tracy, T.S.; Korzekwa, K.R.; Gonzalez, F.J.; Wainer, I.W. Cytochrome P450 isoforms involved in metabolism of the enantiomers of verapamil and norverapamil. Br. J. Clin. Pharmacol 1999, 47, 545–552. [Google Scholar]
- Rea, V.; Kolkman, A.J.; Vottero, E.; Stronks, E.J.; Ampt, K.A.M.; Honing, M.; Vermeulen, N.P.E.; Wijmenga, S.S.; Commandeur, J.N.M. Active site substitution A82W improves the regioselectivity of steroid hydroxylation by cytochrome P450 BM3 mutants as rationalized by spin relaxation nuclear magnetic resonance studies. Biochemistry 2012, 51, 750–760. [Google Scholar]
- Venkataraman, H.; de Beer, S.B.A.; van Bergen, L.A.H.; van Essen, N.; Geerke, D.P.; Vermeulen, N.P.E.; Commandeur, J.N.M. A single active site mutation inverts stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450 BM3. ChemBioChem 2012, 13, 520–523. [Google Scholar]
- Laine, L.; Goldkind, L.; Curtis, S.P.; Connors, L.G.; Zhang, Y.; Cannon, C.P. How common is diclofenac-associated liver injury? Analysis of 17,289 arthritis patients in a long-term prospective clinical trial. Am. J. Gastroenterol 2009, 104, 356–362. [Google Scholar]
- McTavish, D.; Sorkin, E.M. Verapamil—An updated review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension. Drugs 1989, 38, 19–76. [Google Scholar]
- Peters, M.W.; Meinhold, P.; Glieder, A.; Arnold, F.H. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc 2003, 125, 13442–13450. [Google Scholar]
- Meinhold, P.; Peters, M.W.; Hartwick, A.; Hernandez, A.R.; Arnold, F.H. Engineering cytochrome P450BM3 for terminal alkane hydroxylation. Adv. Synth. Catal 2006, 348, 763–772. [Google Scholar]
- Kroemer, H.K.; Gautier, J.C.; Beaune, P.; Henderson, C.; Wolf, C.R.; Eichelbaum, M. Identification of P450 enzymes involved in metabolism of verapamil in humans. Naunyn-Schmiedebergs Arch. Pharm 1993, 348, 332–337. [Google Scholar]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylnositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem 1994, 269, 4241–5248. [Google Scholar]
- Van der Weide, J.; Steijns, L.S.W.; van Weelden, M.J.M. The effect of smoking and cytochrome P450CYP1A2 genetic polymorphism on clozapine clearance and dose requirement. Pharmacogenetics 2003, 13, 169–172. [Google Scholar]
- Cowart, L.A.; Falck, J.R.; Capdevila, J.H. Structural determinants of active site binding affinity and metabolism by cytochrome P450BM-3. Arch. Biochem. Biophys 2001, 387, 117–124. [Google Scholar]
- Haines, D.C.; Hegde, A.; Chen, B.; Zhao, W.; Bondlela, M.; Humphreys, J.M.; Mullin, D.A.; Tomchick, D.R.; Machius, M.; Peterson, J.A. A single active-site mutation of P450BM-3 dramatically enhances substrate binding and rate of product formation. Biochemistry 2011, 50, 8333–8341. [Google Scholar]
- Hunter, D.J.B.; Behrendorff, J.B.Y.H.; Johnston, W.A.; Hayes, P.Y.; Huang, W.; Bonn, B.; Hayes, M.A.; de Voss, J.J.; Gillam, E.M.J. Facile production of minor metabolites for drug development using a CYP3A shuffled library. Metab. Eng 2011, 13, 682–693. [Google Scholar]
- Rosic, N.N.; Huang, W.; Johnston, W.A.; DeVoss, J.J.; Gillam, E.M.J. Extending the diversity of cytochrome P450 enzymes by DNA family shuffling. Gene 2007, 395, 40–48. [Google Scholar]
- Johnston, W.A.; Huang, W.; de Voss, J.J.; Hayes, M.A.; Gillam, E.M.J. A shuffled CYP1A library shows both structural integrity and functional diversity. Drug Metab. Dispos 2007, 35, 2177–2185. [Google Scholar]
- Huang, W.; Johnston, W.A.; Hayes, M.A.; de Voss, J.J.; Gillam, E.M.J. A shuffled CYP2C library with a high degree of structural integrity and functional versatility. Arch. Biochem. Biophys 2007, 467, 193–205. [Google Scholar]
- Rosic, N.N. Versatile capacity of shuffled cytochrome P450s for dye production. Appl. Microbiol. Biotechnol 2009, 82, 203–210. [Google Scholar]
- Weis, R.; Winkler, M.; Schittmayer, M.; Kambourakis, S.; Vink, M.; Rozzell, J.D.; Glieder, A. A diversified library of bacterial and fungal bifunctional cytochrome P450 enzymes for drug metabolite synthesis. Adv. Synth. Catal 2009, 351, 2140–2146. [Google Scholar]
- Otey, C.R.; Silberg, J.J.; Voigt, C.A.; Endelman, J.B.; Bandara, G.; Arnold, F.H. Functional evolution and structural conservation in chimeric cytochromes P450: Calibrating a structure-guided approach. Chem. Biol 2004, 11, 309–318. [Google Scholar]
- Gilardi, G.; Meharenna, Y.T.; Tsotsou, G.E.; Sadeghi, S.J.; Fairhead, M.; Giannini, S. Molecular Lego: Design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosens. Bioelectron 2002, 17, 133–145. [Google Scholar]
- Bernhardt, R. Optimized chimeragenesis: Creating diverse P450 functions. Chem. Biol 2004, 11, 287–288. [Google Scholar]
- Sadeghi, S.J.; Ferrero, S.; di Nardo, G.; Gilardi, G. Drug-drug interactions and cooperative effects detected in electrochemically driven human cytochrome P450 3A4. Bioelectrochemistry 2012, 86, 87–91. [Google Scholar]
- Denisov, I.G.; Sligar, S.G. A novel type of allosteric regulation: Functional cooperativity in monomeric proteins. Arch. Biochem. Biophys 2012, 519, 91–102. [Google Scholar]
- Roberts, A.G.; Yang, J.; Halpert, J.R.; Nelson, S.D.; Thummel, K.T.; Atkins, W.M. The structural basis for homotropic and heterotropic cooperativity of midazolam metabolism by human cytochrome P450 3A4. Biochemistry 2011, 50, 10804–10818. [Google Scholar]
- Woods, C.M.; Fernandez, C.; Kunze, K.L.; Atkins, W.M. Allosteric activation of cytochrome P450 3A4 by alpha-naphthoflavone: Branch point regulation revealed by isotope dilution analysis. Biochemistry 2011, 50, 10041–10051. [Google Scholar]
- Fernando, H.; Rumfeldt, J.A.O.; Davydova, N.Y.; Halpert, J.R.; Davydov, D.R. Multiple substrate-binding sites are retained in cytochrome P450 3A4 mutants with decreased cooperativity. Xenobiotica 2011, 41, 281–289. [Google Scholar]
- Frank, D.J.; Denisov, I.G.; Sligar, S.G. Mixing apples and oranges: Analysis of heterotropic cooperativity in cytochrome P450 3A4. Arch. Biochem. Biophys 2009, 488, 146–152. [Google Scholar]
- Frank, D.J.; Denisov, I.G.; Sligar, S.G. Analysis of heterotropic cooperativity in cytochrome P450 3A4 using alpha-naphthoflavone and testosterone. J. Biol. Chem 2011, 286, 5540–5545. [Google Scholar]
- Perret, A.; Pompon, D. Electron shuttle between membrane-bound cytochrome P450 3A4 and b(5) rules uncoupling mechanisms. Biochemistry 1998, 37, 11412–11424. [Google Scholar]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. P450(BM3) (CYP102A1): Connecting the dots. Chem. Soc. Rev 2012, 41, 1218–12160. [Google Scholar]
- Joo, H.; Lin, Z.L.; Arnold, F.H. Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 1999, 399, 670–673. [Google Scholar]
- Cirino, P.C.; Arnold, F.H. Regioselectivity and activity of cytochrome P450 BM-3 and mutant F87A in reactions driven by hydrogen peroxide. Adv. Synth. Catal 2002, 344, 932–937. [Google Scholar]
- Cirino, P.C.; Arnold, F.H. A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew. Chem. Int. Ed. Engl 2003, 42, 3299–3301. [Google Scholar]
- Ryan, J.D.; Fish, R.H.; Clark, D.S. Engineering cytochrome p450 enzymes for improved activity towards biomimetic 1,4-NADH cofactors. ChemBioChem 2008, 9, 2579–2582. [Google Scholar]
- Fleming, B.D.; Tian, Y.; Bell, S.G.; Wong, L.L.; Urlacher, V.; Hill, H.A.O. Redox properties of cytochrome P450(BM3) measured by direct methods. Eur. J. Biochem 2003, 270, 4082–4088. [Google Scholar]
- Udit, A.K.; Hill, M.G.; Bittner, V.G.; Arnold, F.H.; Gray, H.B. Reduction of dioxygen catalyzed by pyrene-wired heme domain cytochrome P450BM3 electrodes. J. Am. Chem. Soc 2004, 126, 10218–10219. [Google Scholar]
- Udit, A.K.; Hindoyan, N.; Hill, M.G.; Arnold, F.H.; Gray, H.B. Protein-surfactant film voltammetry of wild-type and mutant cytochrome P450BM3. Inorg. Chem 2005, 44, 4109–4111. [Google Scholar]
- Fantuzzi, A.; Meharenna, Y.T.; Briscoe, P.B.; Sassone, C.; Borgia, B.; Gilardi, G. Improving catalytic properties of P450BM3 haem domain electrodes by molecular Lego. Chem. Commun 2006, 12, 1289–1291. [Google Scholar]
- Fantuzzi, A.; Fairhead, M.; Gilardi, G. Direct electrochemistry of immobilized human cytochrome P450 2E1. J. Am. Chem. Soc 2004, 126, 5040–5041. [Google Scholar]
- Sadeghi, S.J.; Fantuzzi, A.; Gilardi, G. Breakthrough in P450 bioelectrochemistry and future perspectives. Biochim. Biophys. Acta 2011, 1814, 237–248. [Google Scholar]
- Dodhia, V.R.; Sassone, C.; Fantuzzi, A.; di Nardo, G.; Sadeghi, S.J.; Gilardi, G. Modulating the coupling efficiency of human cytochrome P450 CYP3A4 at electrode surfaces through protein engineering. Electrochem. Commun 2008, 10, 1744–1747. [Google Scholar]
- Holtmann, D.; Mangold, K.-M.; Schrader, J. Entrapment of cytochrome P450 BM-3 in polypyrrole for electrochemically-driven biocatalysis. Biotechnol. Lett 2009, 31, 765–770. [Google Scholar]
- Schwaneberg, U.; Appel, D.; Schmitt, J.; Schmid, R.D. P450 in biotechnology: zinc driven omega-hydroxylation of p-nitrophenoxydodecanoic acid using P450BM-3 F87A as a catalyst. J. Biotechnol 2000, 84, 249–257. [Google Scholar]
- Hollmann, F.; Hofstetter, K.; Schmid, A. Non-enzymatic regeneration of nicotinamide and flavin cofactors for monooxygenase catalysis. Trends Biotechnol 2006, 24, 163–171. [Google Scholar]
- Salazar, O.; Cirino, P.C.; Arnold, F.H. Thermostabilization of a cytochrome P450 peroxygenase. ChemBioChem 2003, 4, 891–893. [Google Scholar]
- Li, Y.; Drummond, D.A.; Sawayama, A.M.; Snow, C.D.; Bloom, J.D.; Arnold, F.H. A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nat. Biotechnol 2007, 25, 1051–1056. [Google Scholar]
- Maurer, S.C.; Schulze, H.; Schmid, R.D.; Urlacher, V. Immobilisation of P450BM-3 and an NADP(+) cofactor recycling system: Towards a technical application of heme-containing monooxygenases in fine chemical synthesis. Adv. Synth. Catal 2003, 345, 802–810. [Google Scholar]
- Weber, E.; Sirim, D.; Schreiber, T.; Thomas, B.; Pleiss, J.; Hunger, M.; Glaeser, R.; Urlacher, V.B. Immobilization of P450 BM-3 monooxygenase on mesoporous molecular sieves with different pore diameters. J. Mol. Catal. B 2010, 64, 29–37. [Google Scholar]
- Zhao, L.; Gueven, G.; Li, Y.; Schwaneberg, U. First steps towards a Zn/Co(III)sep-driven P450 BM3 reactor. Appl. Microbiol. Biotechnol 2011, 91, 989–999. [Google Scholar]
- Yim, S.K.; Kim, D.H.; Jung, H.C.; Pan, J.G.; Kang, H.S.; Ahn, T.; Yun, C.H. Surface display of heme- and diflavin-containing cytochrome P450 BM3 in Escherichia coli: A whole-cell biocatalyst for oxidation. J. Microbiol. Biotechnol 2010, 20, 712–717. [Google Scholar]
- Schewe, H.; Holtmann, D.; Schrader, J. P450(BM-3)-catalyzed whole-cell biotransformation of alpha-pinene with recombinant Escherichia coli in an aqueous-organic two-phase system. Appl. Microbiol. Biotechnol 2009, 83, 849–857. [Google Scholar]
- Lu, Y.; Mei, L.H. Optimization of fermentation conditions for P450 BM-3 monooxygenase production by hybrid design methodology. J. Zhejiang Univ. Sci 2007, 8, 27–32. [Google Scholar]
- Pflug, S.; Richter, S.M.; Urlacher, V.B. Development of a fed-batch process for the production of the cytochrome P450 monooxygenase CYP102A1 from Bacillus megaterium in E. coli. J. Biotechnol 2007, 129, 481–488. [Google Scholar]
- Park, J.W.; Lee, J.K.; Kwon, T.J.; Yi, D.H.; Kim, Y.J.; Moon, S.H.; Suh, H.H.; Kang, S.M.; Park, Y.I. Bioconversion of compactin into pravastatin by Streptomyces sp. Biotechnol. Lett 2003, 25, 1827–1831. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
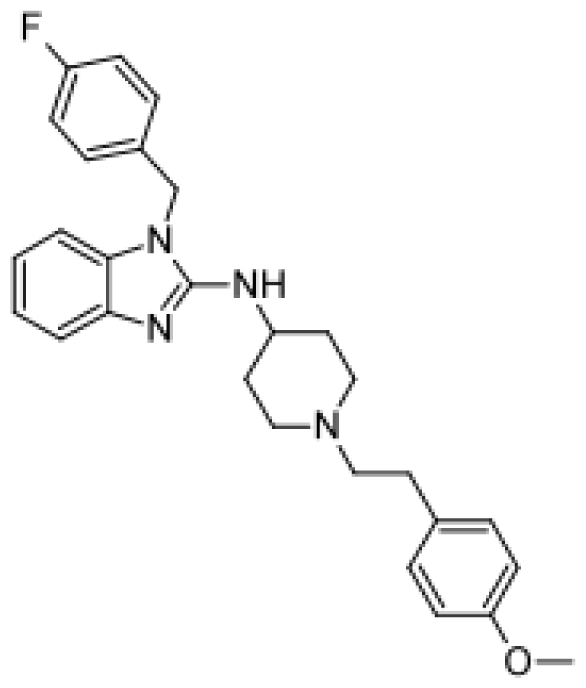{kind=link}
{kind=link}
{kind=link}
| Drug | Structure | Metabolite produced by P450 BM3 WT or variants | Main human P450 involved | Ref. |
|---|---|---|---|---|
| Acetaminophen | 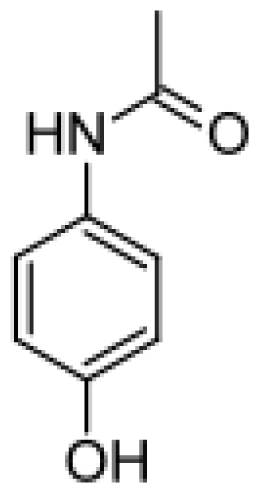 | N-acetyl-p-benzo-quinone imine [49] | 2E1 | [50] |
| Amitriptyline | 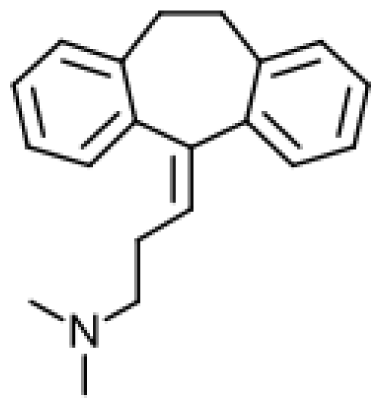 | Nortriptyline [51] | 2C19, 3A4, 1A2 | [52] |
| Amodiaquine | 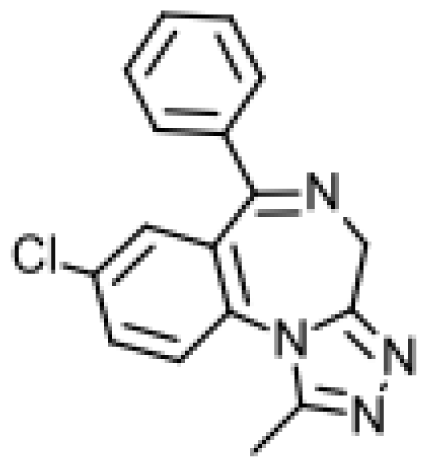 | N-desethylamodiaquine [53] | 2C8 | [54] |
| Astemizole | 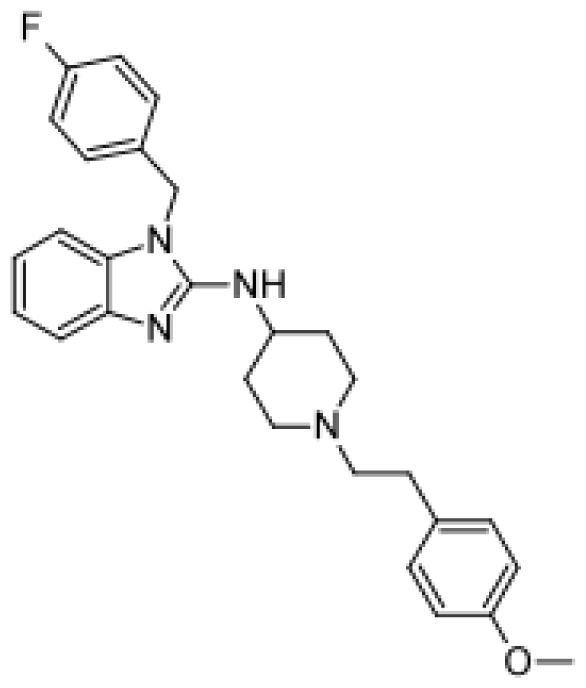 | 6-hydroxyastemizole [55] | 2D6 | [56] |
| 6-hydroxydesmethylastemizole [55] | 2D6 | |||
| Desmethylastemizole [55] | 2D6 | |||
| Norastemizle [ 55] | 3A4 | |||
| Buspirone | 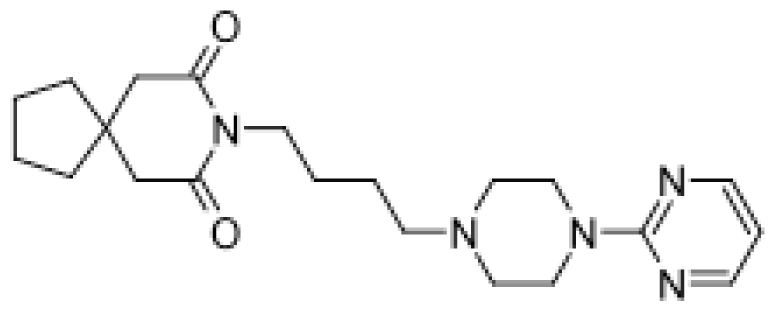 | (R)-6-hydroxybuspirone [57] | 3A4 | [58] |
| Mono- and dihydroxylated metabolites [51] | 3A4 | |||
| Chlorzoxazone | 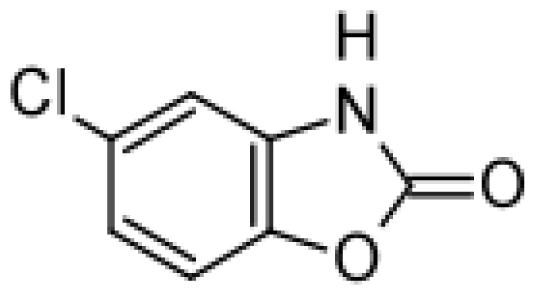 | 6-hydroxychlorzoxazone [37] | 2E1 | [59] |
| Cilostazol |  | Mono- and di-hydroxymetabolites [51] | 3A4/5 | [60] |
| Citalopram | 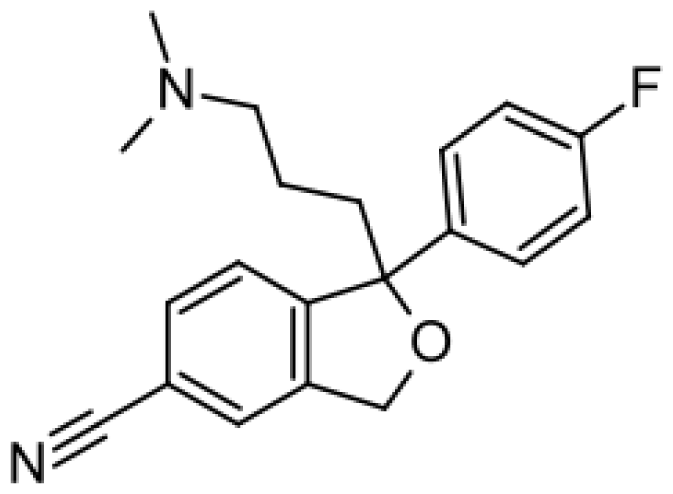 | Demethylcitalopram [51] | 3A4, 2C19 | [61] |
| di-demethylcitalopram [51] | 2D6 | |||
| Clozapine | 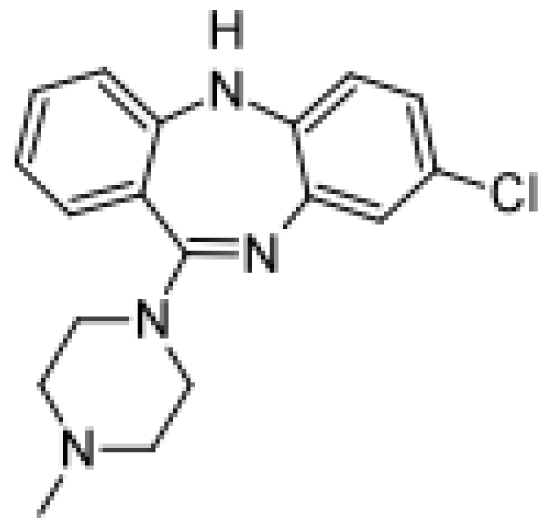 | N-demthylclozapine [62] | 1A2 | [63] |
| Clozapine N-oxide [62] | 1A2 | |||
| Dextromethorphan | 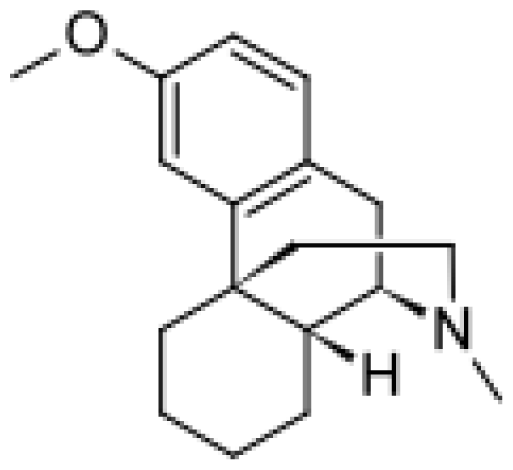 | 3-methoxymorphinan [49] | 2D6, 3A4 | [ 64] |
| Diclofenac | 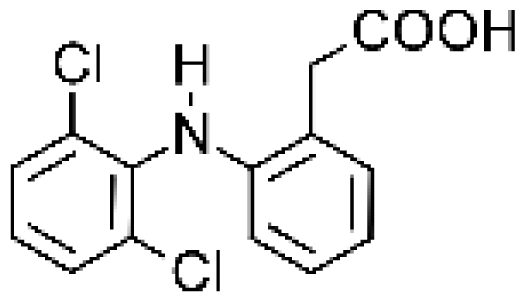 | 4′-hydroxydiclofenac [65, 66] | 2C9/2C19 | [67] |
| 5-hydroxydiclofenac [65] | 2C9/2C19 | |||
| Diltiazem | 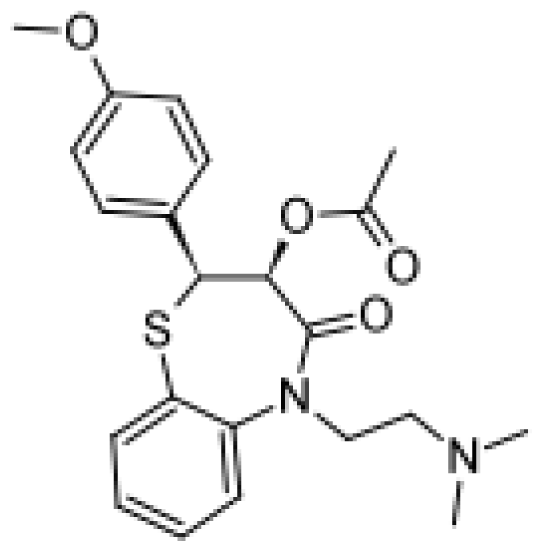 | Demethyldiltiazem [51] | 3A4 | [68] |
| Di-demethyldiltiazem [51] | 3A4 | |||
| Irbesartan |  | Hydroxyirbesartan [51] | 2C9 | [69] |
| Di-hydroxyirbesartan [51] | 2C9 | |||
| Lovastatin |  | 6b-hydroxylovastatin [70] | 3A4/5 | [71] |
| 6′-exomethylenelovastatin [70] | 3A4/5 | |||
| Naproxen | 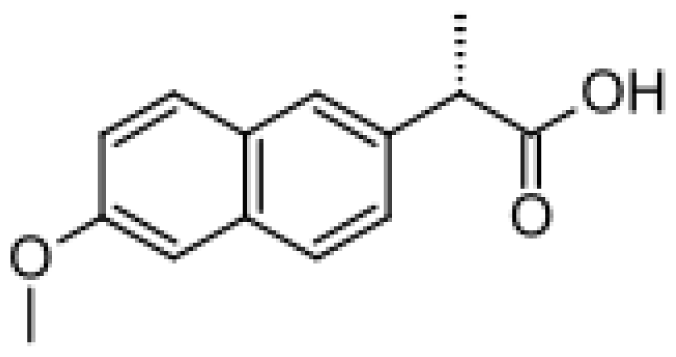 | Desmethylnaproxen [47] | 2C9, 1A2 | [72] |
| Nifedipine |  | Oxidized nifedipine [37] | 3A4 | [73] |
| Ondansetron | 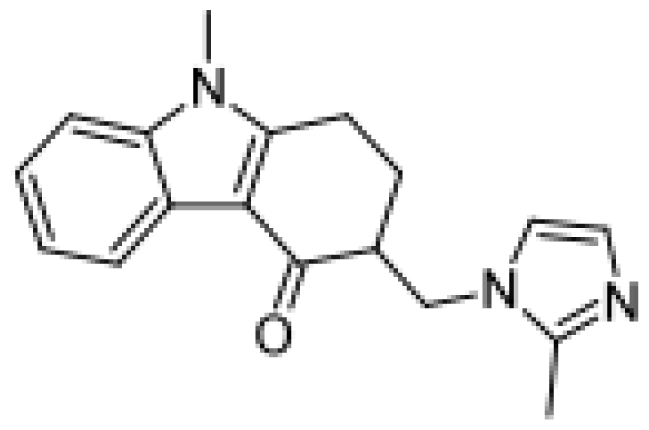 | Hydroxylondansetron [51] | 3A4, 2D6, 1A2 | [74] |
| Dihydroxyondansetron [51] | 3A4, 2D6, 1A2 | |||
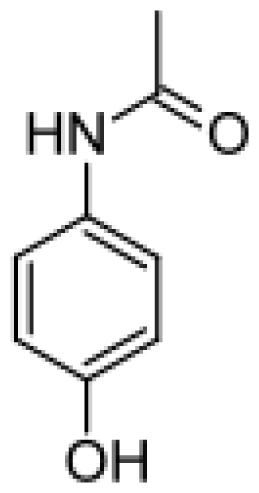
| Phenacetin | 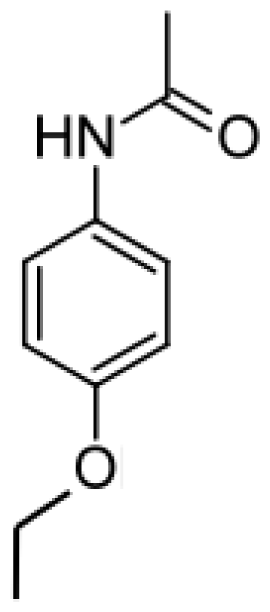 | Acetaminophen [75] | 1A2 | [ 76] |
| Propafenone | 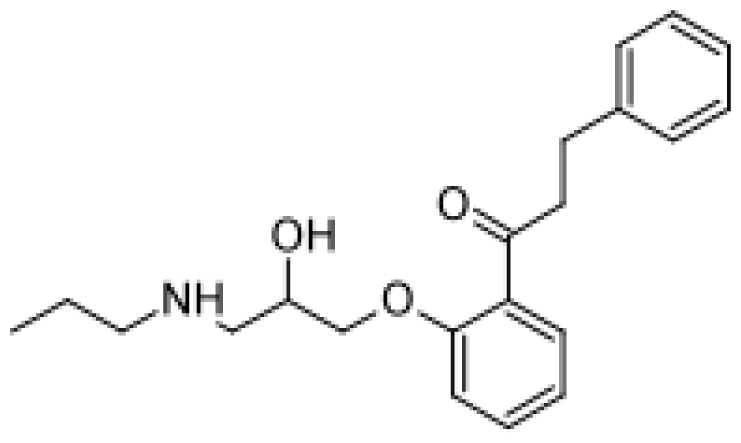 | Hydroxylated metabolites [51] | 2D6 | [77] |
| N-despropylpropafenone [51] | 3A4/1A2 | |||
| Propranolol | 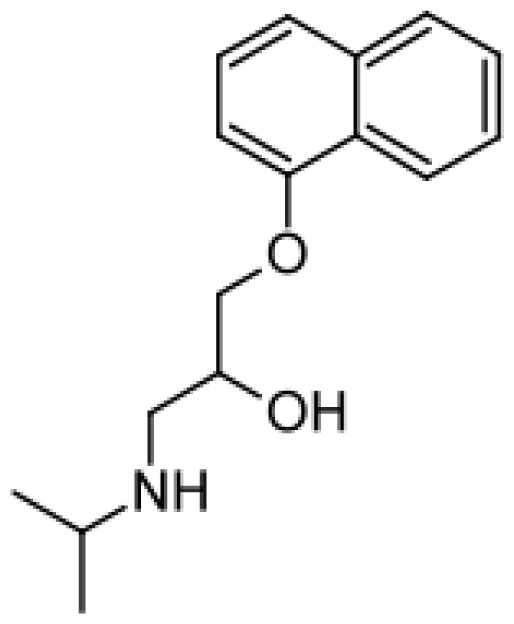 | 4′-hydroxypropranolol [78] | 2D6 | [79] |
| 5′-hydroxypropranolol [78] | 2D6 | |||
| N-despropylpropranolol [37, 78] | 1A2 | |||
| Repaglinide | 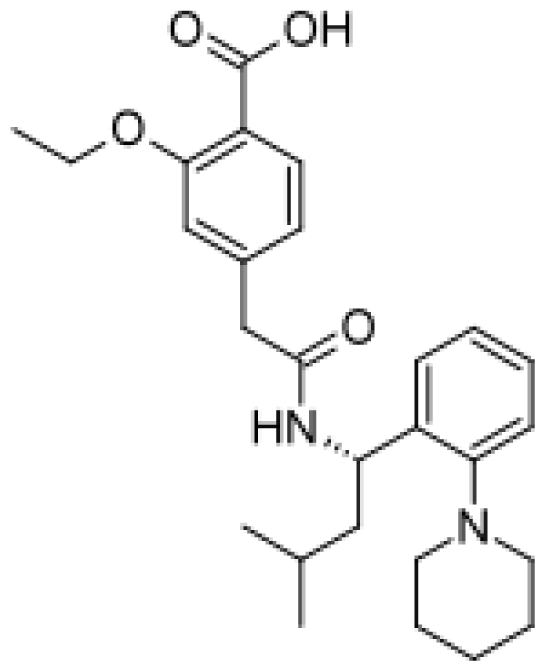 | Hydroxylated metabolites [80] | 3A4, 2C8 | [81] |
| Simvastatin |  | 6b-hydroxysimvastatin [70] | 3A4 | [71] |
| 6′-exomethylenesimvastatin [70] | 3A4 | |||
| Tolbutamide |  | 4-hydroxytolbutamide [37] | 2C | [82] |
| Verapamil | 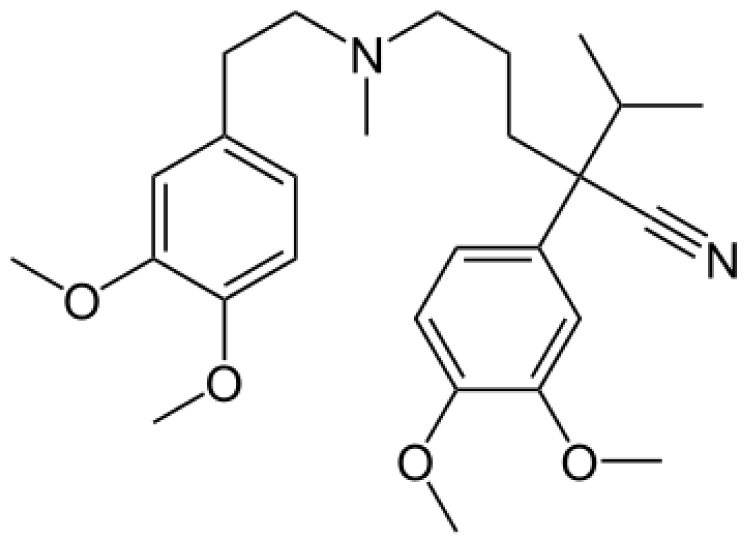 | Norverapamil [ 55] | 3A4 | [83] |
| D-617 [55] | 3A4/3A5, 2C8 | |||
| D-620 [55] | 3A4/3A5, 2C8 | |||
| D-702 [55] | 2C9/2C18 | |||
| PR-22 [55] | 2C8 | |||
| PR-25 [55] | 2C8 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Di Nardo, G.; Gilardi, G. Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites. Int. J. Mol. Sci. 2012, 13, 15901-15924. https://doi.org/10.3390/ijms131215901
Di Nardo G, Gilardi G. Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites. International Journal of Molecular Sciences. 2012; 13(12):15901-15924. https://doi.org/10.3390/ijms131215901
Chicago/Turabian StyleDi Nardo, Giovanna, and Gianfranco Gilardi. 2012. "Optimization of the Bacterial Cytochrome P450 BM3 System for the Production of Human Drug Metabolites" International Journal of Molecular Sciences 13, no. 12: 15901-15924. https://doi.org/10.3390/ijms131215901