Form Follows Function: Structural and Catalytic Variation in the Class A Flavoprotein Monooxygenases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
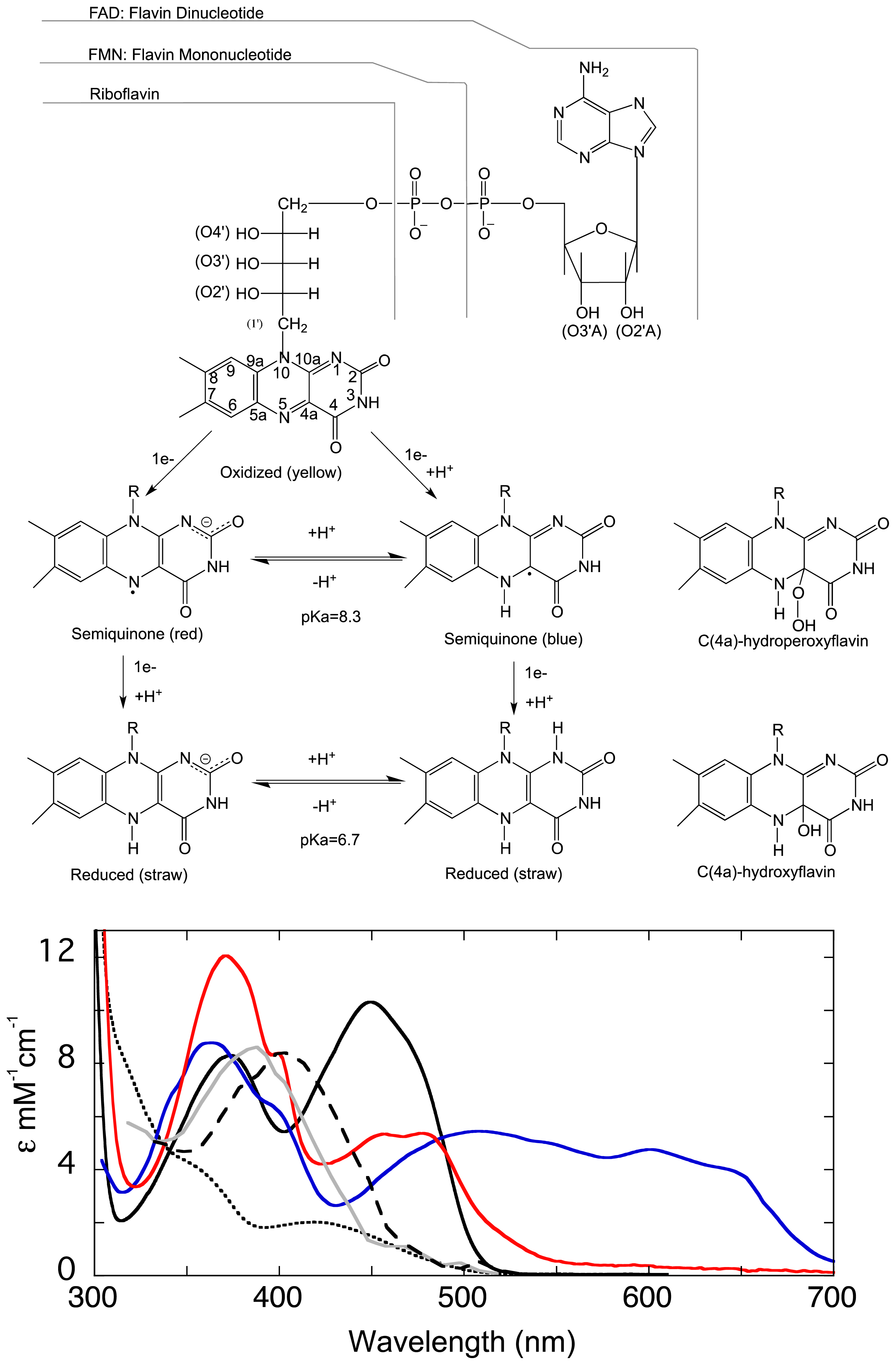
1.1. The Flavin Cofactor
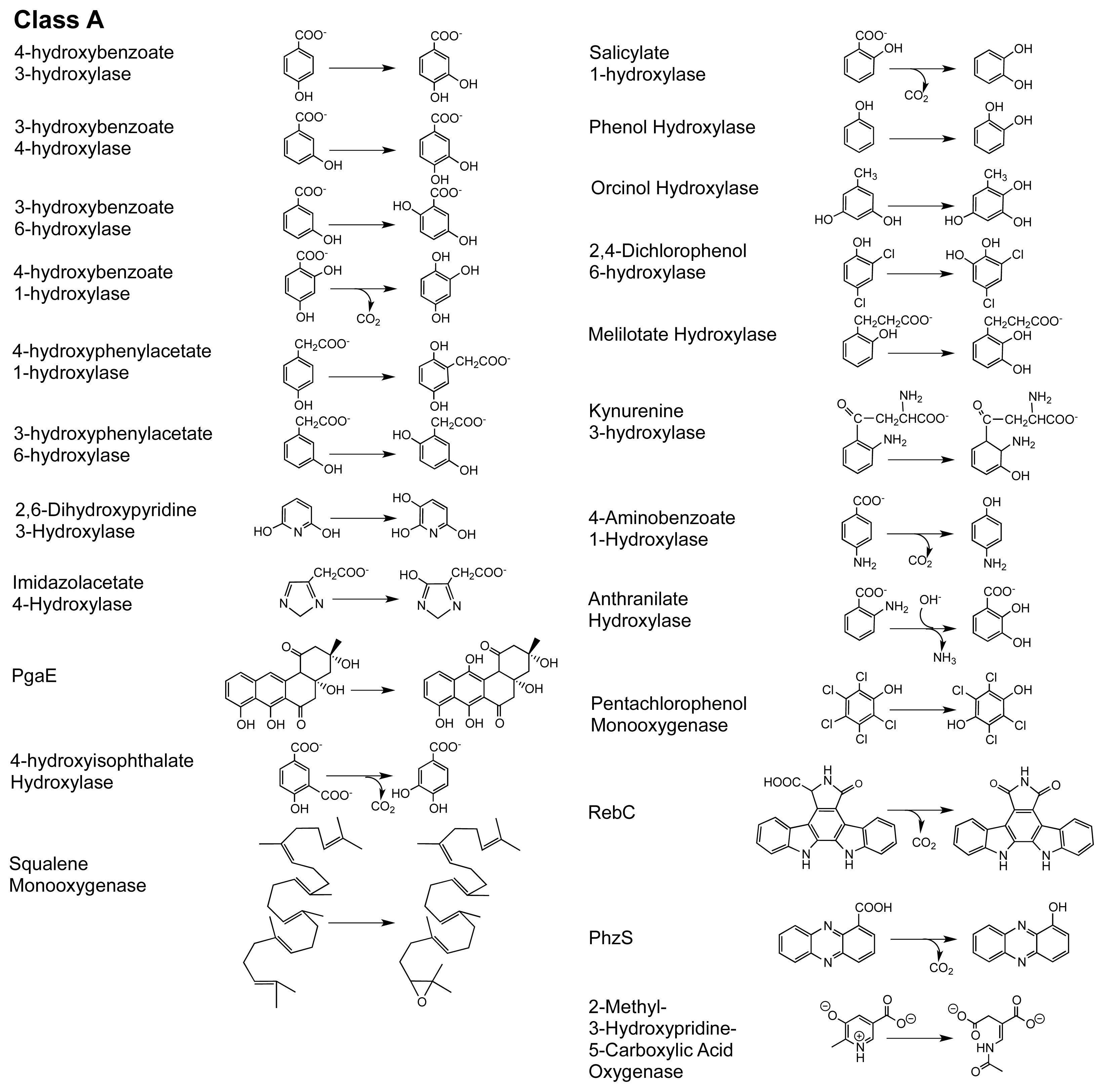
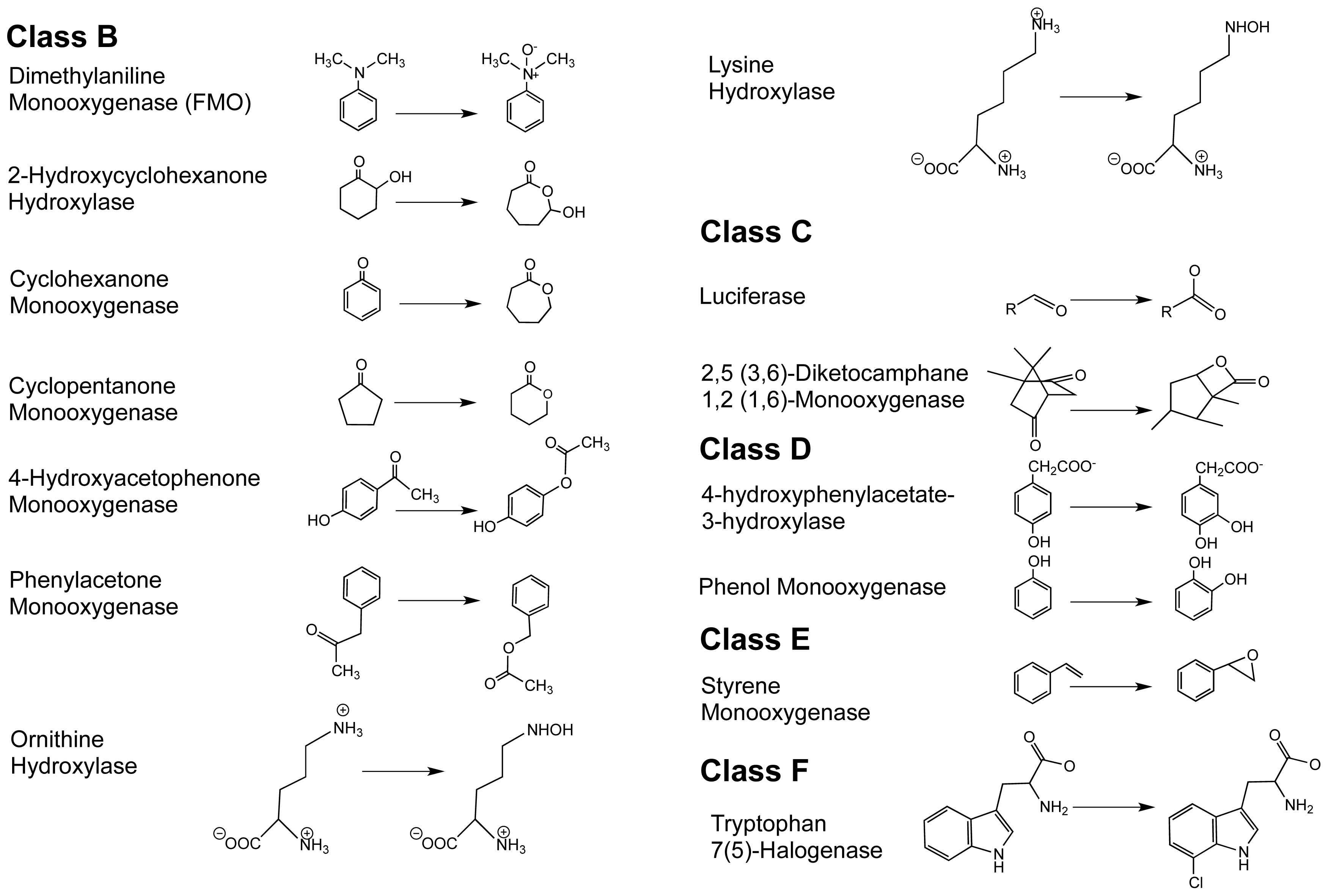
1.2. The FPMO Classes
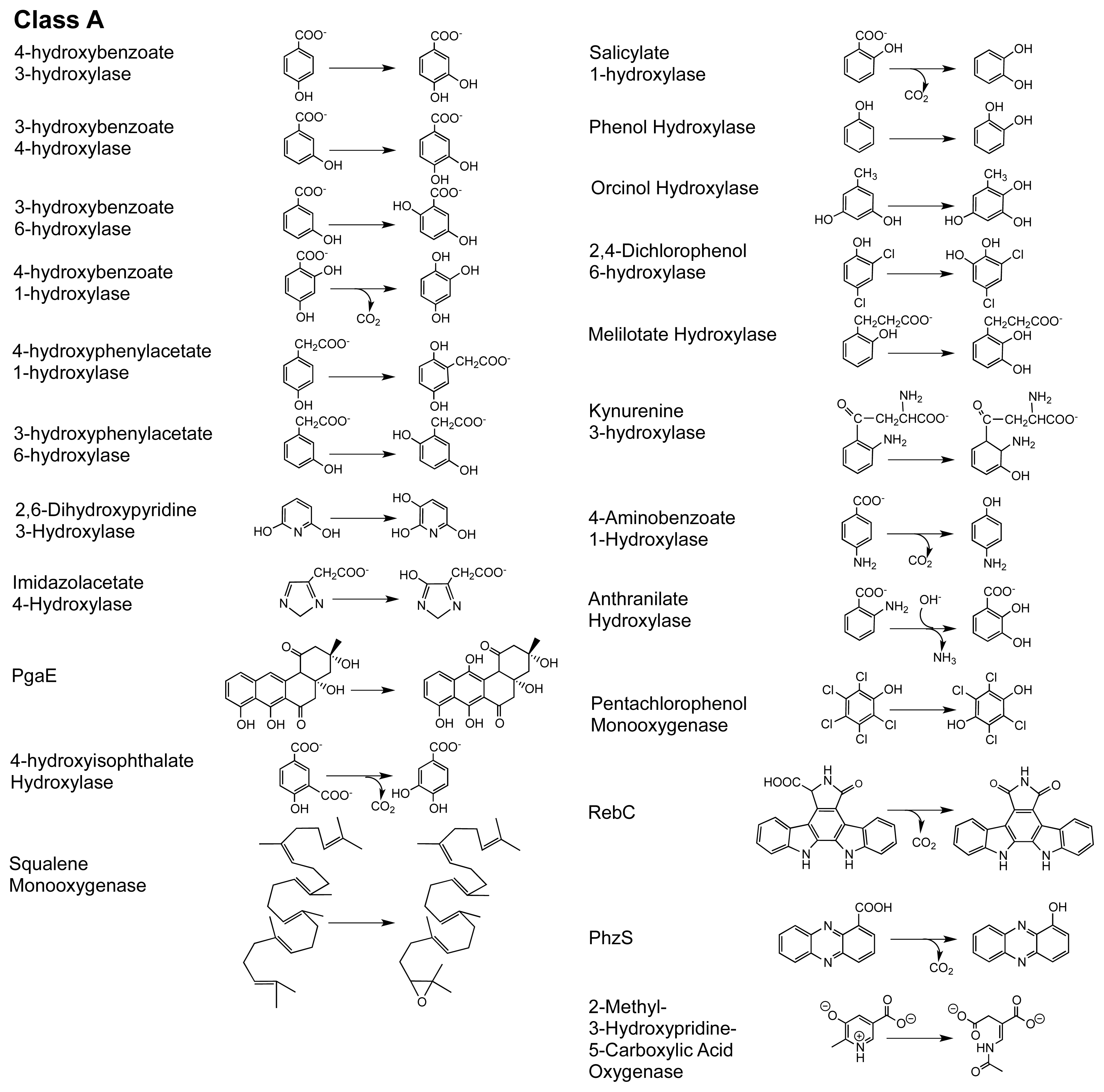
2. The Class A FPMOs
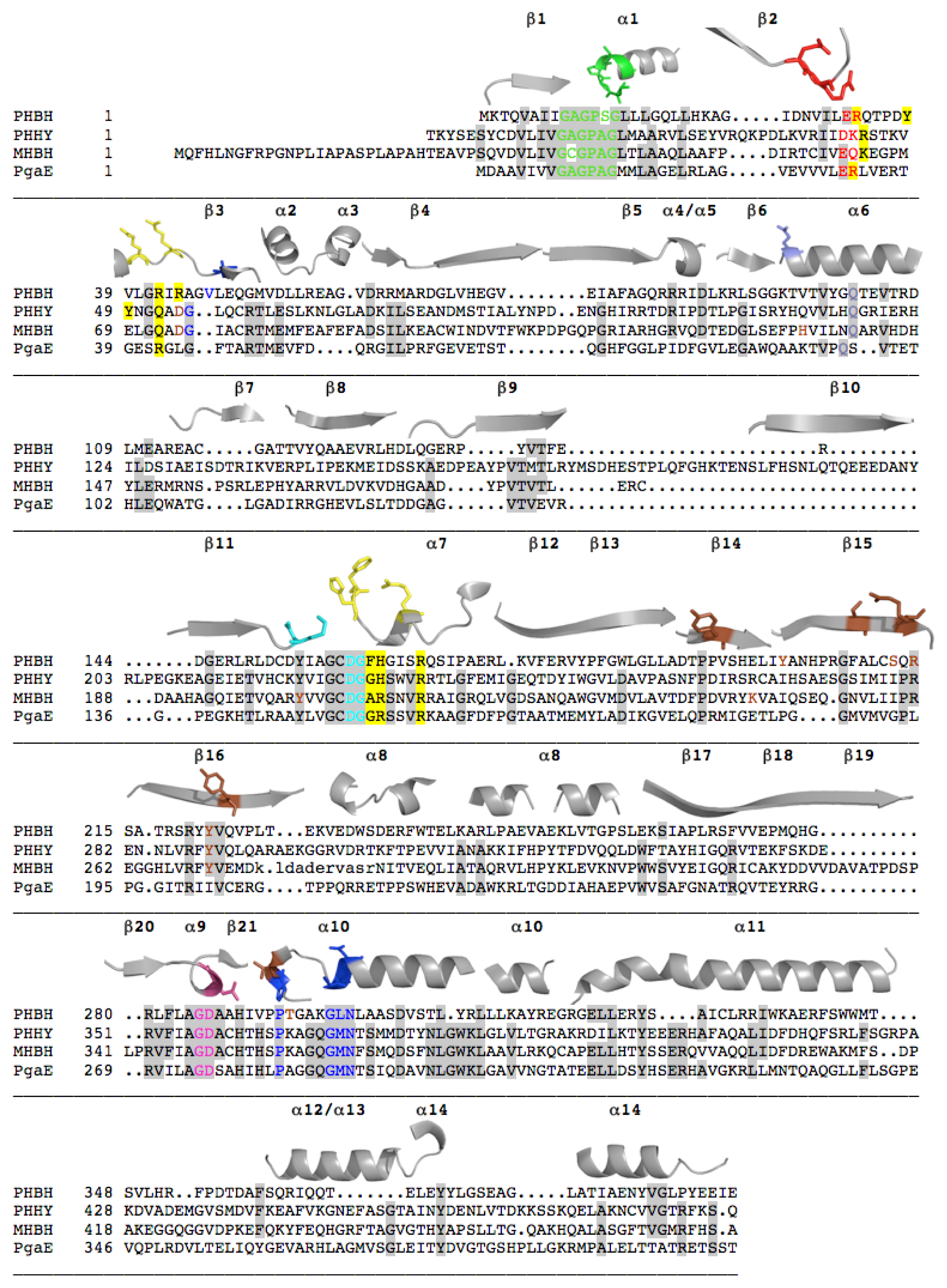
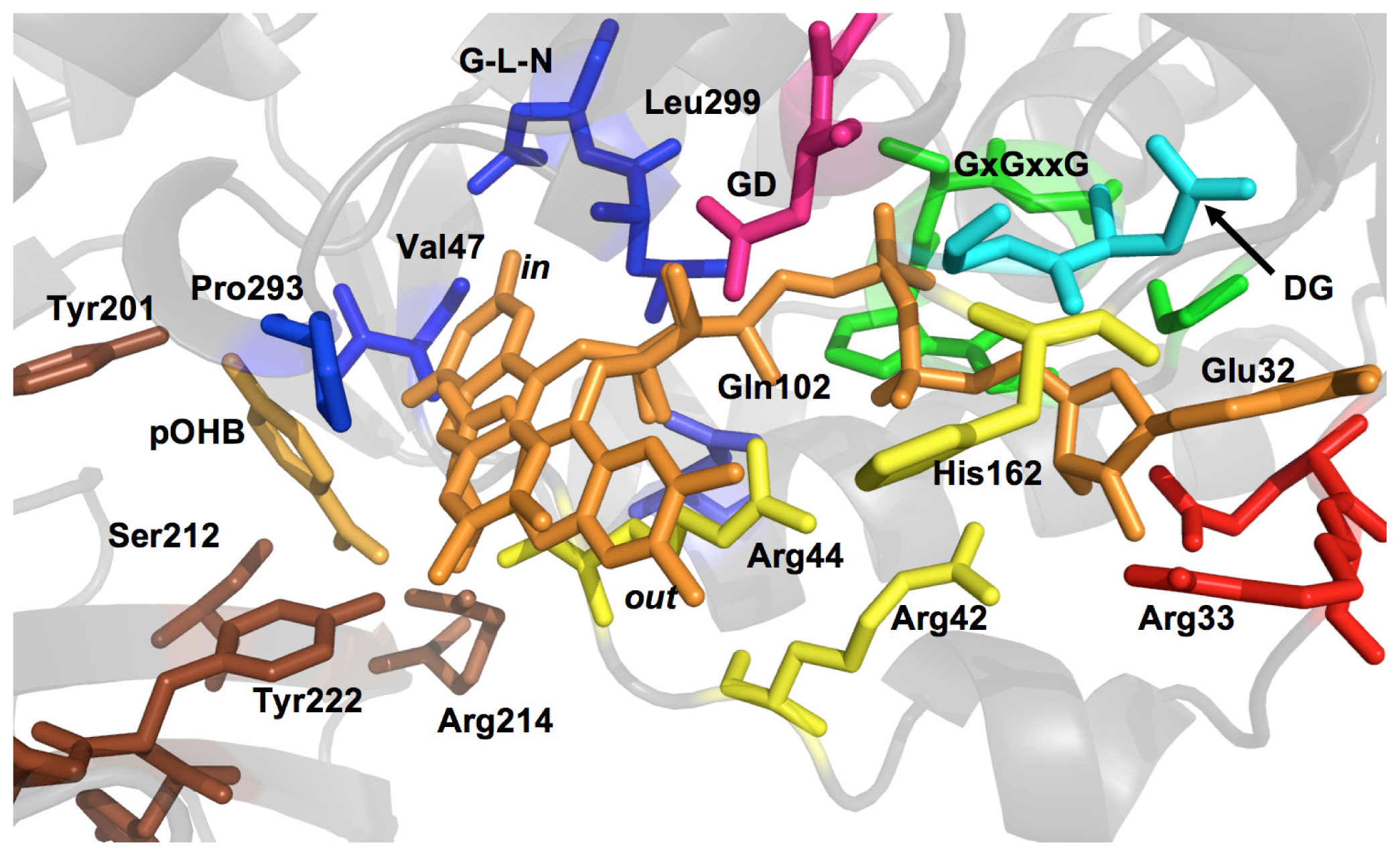
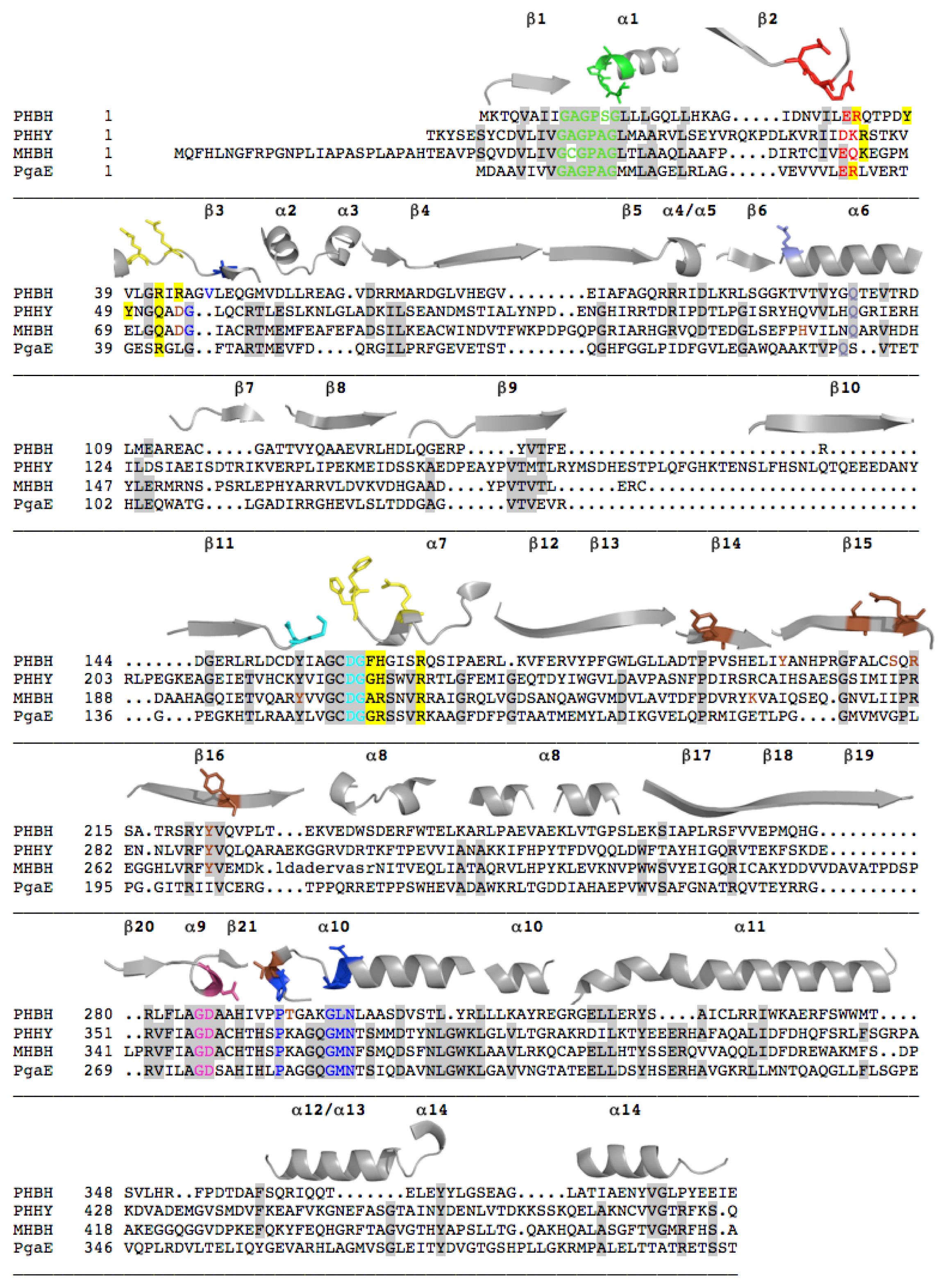
2.1. Functional Assignment of Conserved Residues
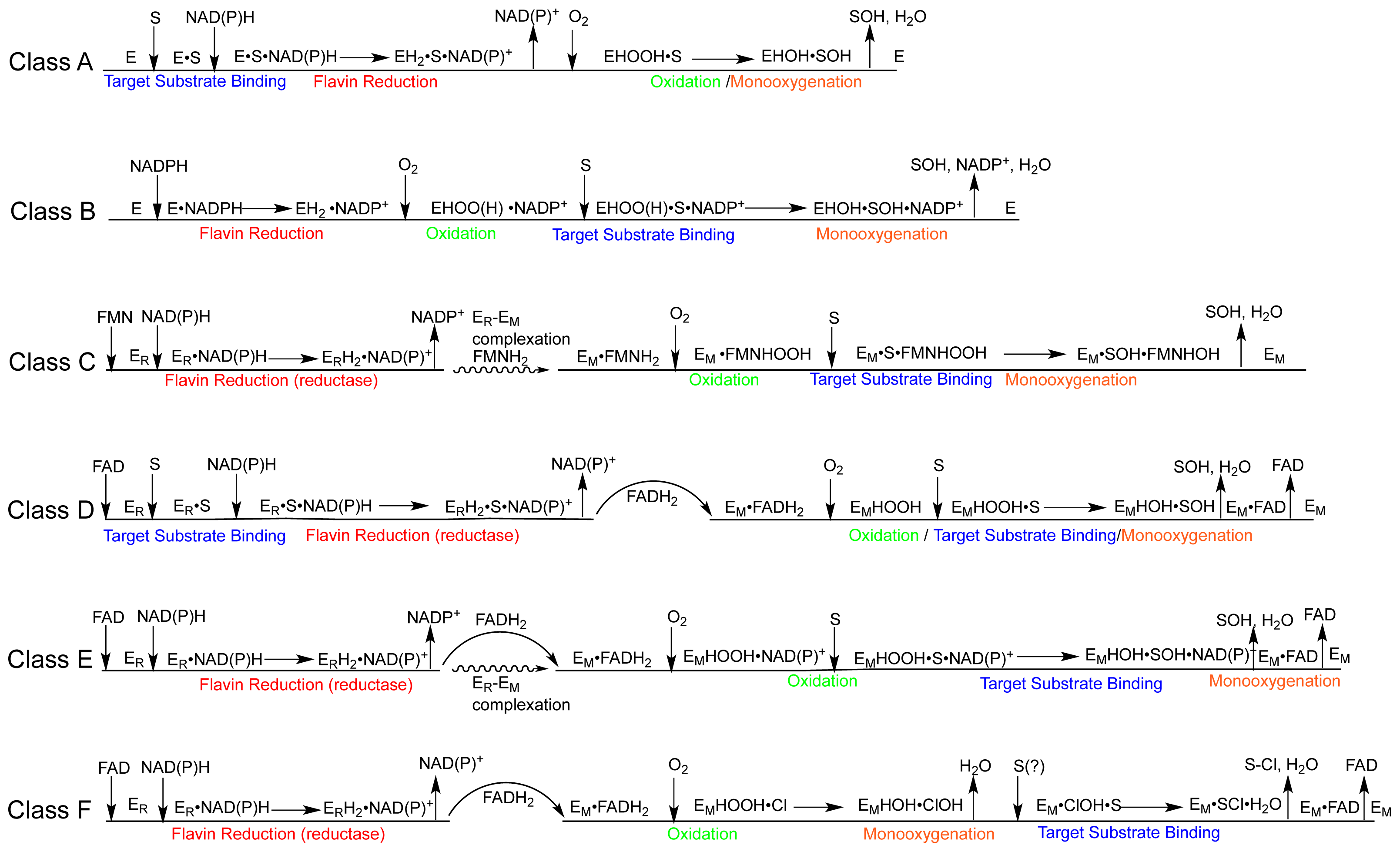
2.2. The Mechanism of the Class A Fpmos
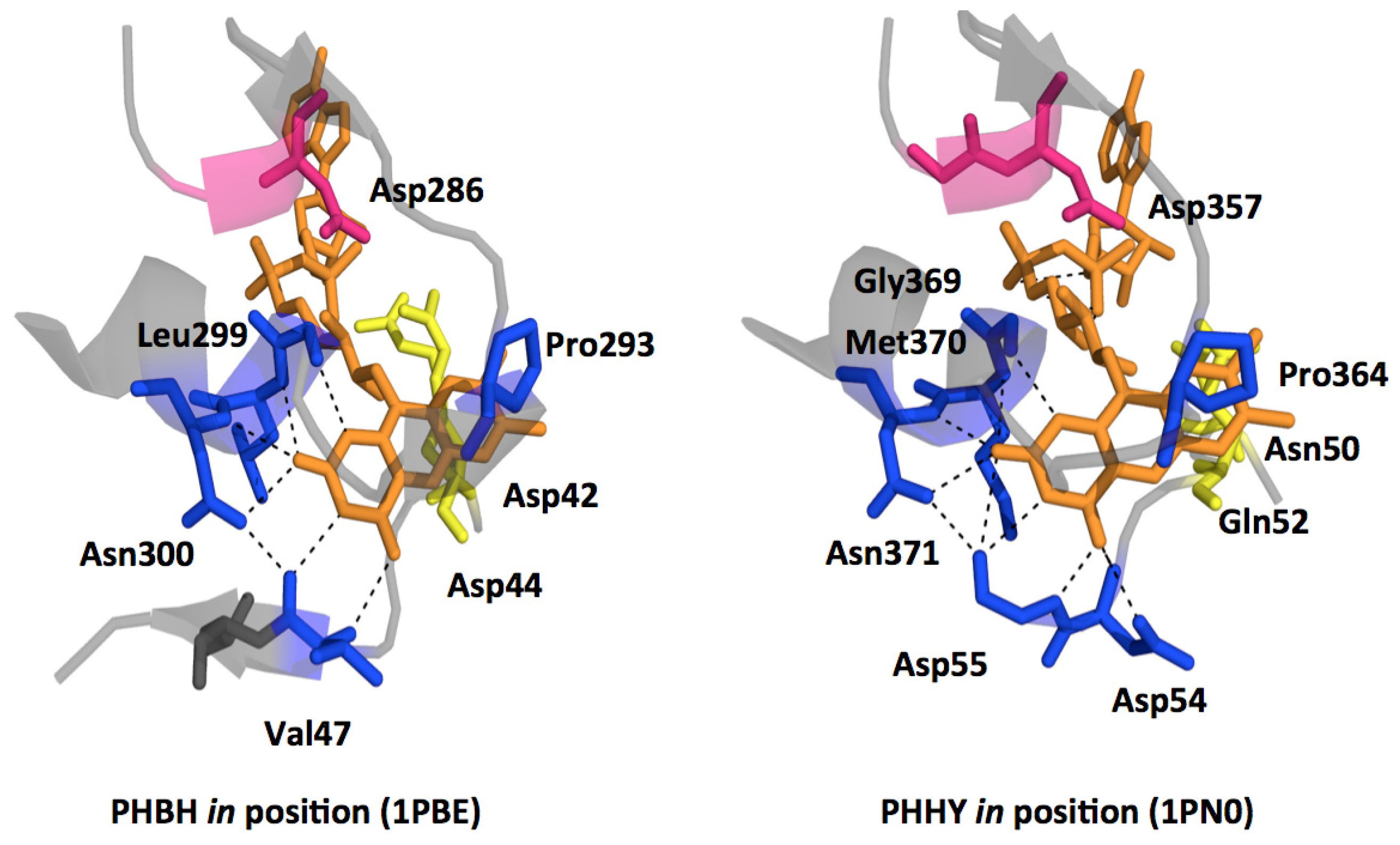
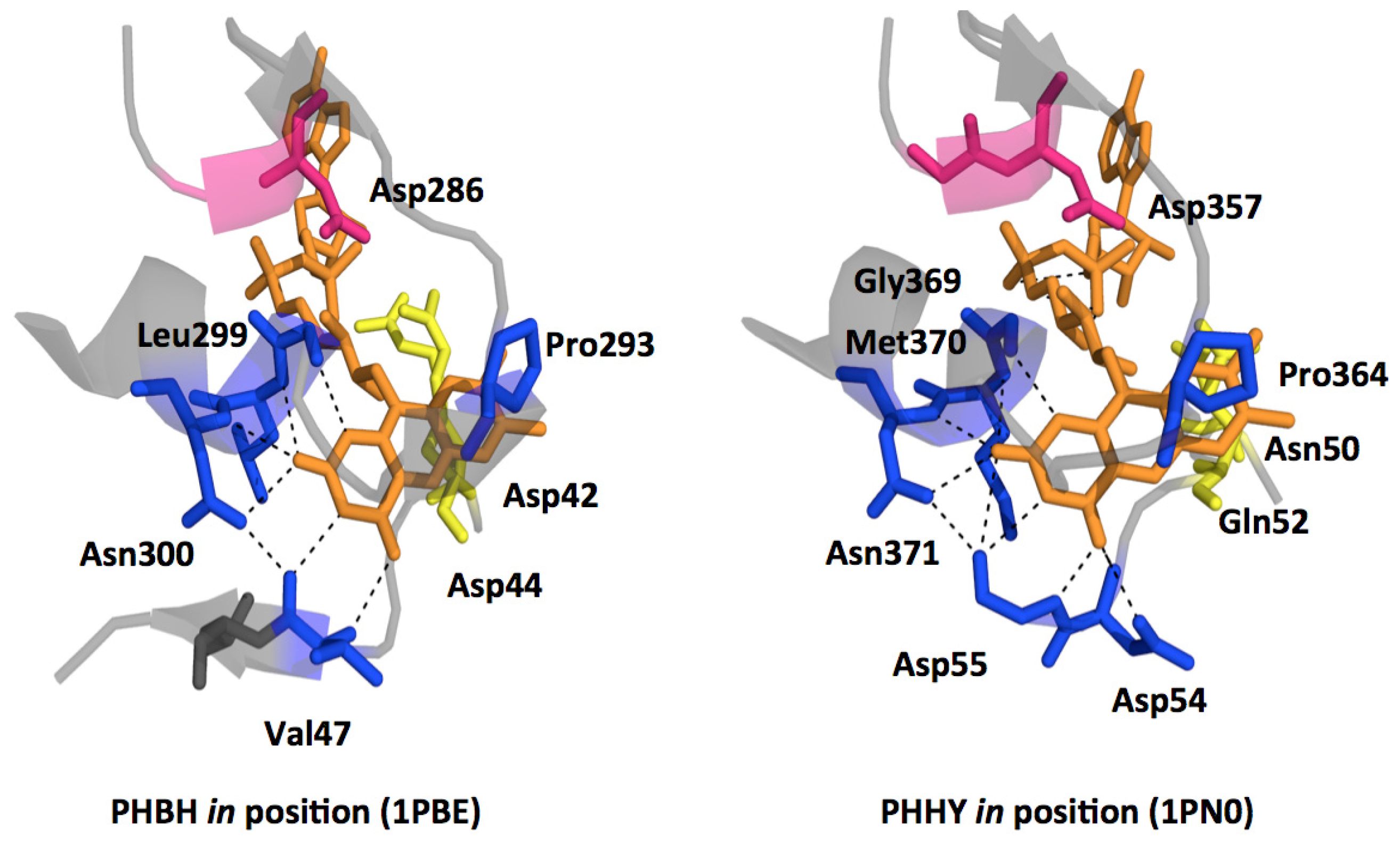
2.2.1. Substrate Binding
2.2.2. Reductive Half Reaction
2.2.3. The Oxidative Half Reaction
2.2.4. Product Release
3. Perspective
Abbreviations
| FPMO | Flavoprotein monooxygenase |
| PHBH | para-hydroxybenzoate hydroxylase |
| MHBH | meta-hydroxybenzoate hydroxylase |
| PHHY | phenol hydroxylase |
| DHPH | dihydroxypyridine hydroxylase |
| PhzS | phenzine decarboxylase |
| CabE and PgaE | Flavoenzymes involved in the biosythesis of angucycline |
| RebC | rebeccamycin |
| FAD | flavin adenine dinucleotide |
| FMN | flavin mononucleotide |
| NADH | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| pOHB | para-hydroxybenzoate |
References
- Goodwin, T.W.; Mercer, E.I. Introduction to Plant Biochemistry, 2nd ed; Pergamon Press: Sydney, Australia, 1983; p. 677. [Google Scholar]
- Daniel, O.; Meier, M.S.; Schlatter, J.; Frischknecht, P. Selected phenolic compounds in cultivated plants: Ecologic functions, health implications, and modulation by pesticides. Environ. Health Persp 1999, 107, S109–S114. [Google Scholar]
- Medvedev, V.I. Migration of polycyclic aromatic hydrocarbons from resins used in the food industry. Vopr. Pitan 1973, 6, 70–73. [Google Scholar]
- Costa, A.S.; Romao, L.P.; Araujo, B.R.; Lucas, S.C.; Maciel, S.T.; Wisniewski, A., Jr; Alexandre, M.R. Environmental strategies to remove volatile aromatic fractions (BTEX) from petroleum industry wastewater using biomass. Bioresour. Technol. 2012, 105, 31–39. [Google Scholar]
- Gottschalk, G. Bacterial Metabolism; Springer-Verlag: New York, NY, USA, 1985; p. 359. [Google Scholar]
- Ryan, K.S.; Howard-Jones, A.R.; Hamill, M.J.; Elliott, S.J.; Walsh, C.T.; Drennan, C.L. Crystallographic trapping in the rebeccamycin biosynthetic enzyme RebC. Proc. Natl. Acad. Sci. USA 2007, 104, 15311–15316. [Google Scholar]
- Koskiniemi, H.; Metsa-Ketela, M.; Dobritzsch, D.; Kallio, P.; Korhonen, H.; Mantsala, P.; Schneider, G.; Niemi, J. Crystal structures of two aromatic hydroxylases involved in the early tailoring steps of angucycline biosynthesis. J. Mol. Biol 2007, 372, 633–648. [Google Scholar]
- Tsuji, H.; Ogawa, T.; Bando, N.; Sasaoka, K. Purification and properties of 4-aminobenzoate hydroxylase, a new monooxygenase from Agaricus bisporus. J. Biol. Chem 1986, 261, 13203–13209. [Google Scholar]
- Subramanian, V.; Vaidyanathan, C.S. Anthranilate hydroxylase from Aspergillus niger: New type of NADPH-linked nonheme iron monooxygenase. J. Bact 1984, 160, 651–655. [Google Scholar]
- Powlowski, J.B.; Dagley, S.; Massey, V.; Ballou, D.P. Properties of anthranilate hydroxylase (deaminating), a flavoprotein from Trichosporon cutaneum. J. Biol. Chem 1987, 262, 69–74. [Google Scholar]
- Dewick, P.M. The biosynthesis of shikimate metabolites. Nat. Prod. Rep 1988, 5, 73–97. [Google Scholar]
- Kurnasov, O.; Goral, V.; Colabroy, K.; Gerdes, S.; Anantha, S.; Osterman, A.; Begley, T.P. NAD biosynthesis: Identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol 2003, 10, 1195–1204. [Google Scholar]
- Crozier, K.R.; Moran, G.R. Heterologous expression and purification of kynurenine-3-monooxygenase from Pseudomonas fluorescens strain 17400. Protein Expr. Purif 2007, 51, 324–333. [Google Scholar]
- Van Berkel, W.J.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotech 2006, 124, 670–689. [Google Scholar]
- Palfey, B.A.; McDonald, C.A. Control of catalysis in flavin-dependent monooxygenases. Arch. Biochem. Biophys 2009, 493, 26–36. [Google Scholar]
- Montersino, S.; Tischler, D.; Gassner, G.T.; van Berkel, W.J.H. Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv. Synth. Catal 2011, 353, 2301–2319. [Google Scholar]
- Flavins and Flavoproteins; Proceedings of the 15th International Symposium of Flavins and Flavoproteins, Tokyo, Japan, 1–22 April 2005, Nishino, T.; Miura, R. (Eds.) ARchiTect Inc: Tokyo, Japan, 2005.
- Mattevi, A. To be or not to be an oxidase: Challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci 2006, 31, 276–283. [Google Scholar]
- Macheroux, P.; Kappes, B.; Ealick, S.E. Flavogenomics—A genomic and structural view of flavin-dependent proteins. FEBS J 2011, 278, 2625–2634. [Google Scholar]
- De Colibus, L.; Mattevi, A. New frontiers in structural flavoenzymology. Curr. Opin. Struct. Biol 2006, 16, 722–728. [Google Scholar]
- Dym, O.; Eisenberg, D. Sequence-structure analysis of FAD-containing proteins. Prot. Sci 2001, 10, 1712–1728. [Google Scholar]
- Massey, V.; Ghisla, S.; Moore, E.G. 8-mercaptoflavins as active site probes of flavoenzymes. J. Biol. Chem 1979, 254, 9640–9650. [Google Scholar]
- Jorns, M.S.; Wang, B.; Jordan, S.P. DNA repair catalyzed by Escherichia coli DNA photolyase containing only reduced flavin: Elimination of the enzyme’s second chromophore by reduction with sodium borohydride. Biochemistry 1987, 26, 6810–6816. [Google Scholar]
- DeRose, V.J.; Woo, J.C.G.; Hawe, W.P.; Hoffman, B.M.; Silverman, R.B.; Yelekci, K. Observation of a flavin semiquinone in the resting state of monoamine oxidase B by electron paramagnetic resonance and electron nuclear double resonance spectroscopy. Biochemistry 1996, 35, 11085–11091. [Google Scholar]
- Mincey, T.; Tayrien, G.; Mildvan, A.S.; Abeles, R.H. Presence of a flavin semiquinone in methanol oxidase. Proc. Natl. Acad. Sci. USA 1980, 77, 7099–7101. [Google Scholar]
- Massey, V.; Gibson, Q.H. Role of semiquinones in flavoprotein catalysis. Fed. Proc 1964, 23, 18–29. [Google Scholar]
- Entsch, B.; Ballou, D.P. Purification, properties, and oxygen reactivity of p-hydroxybenzoate hydroxylase from Pseudomonas aeruginosa. Biochim. Biophys. Acta 1989, 999, 313–322. [Google Scholar]
- Entsch, B.; Ballou, D.P.; Massey, V. Flavin-oxygen derivatives involved in hydroxylation by p-hydroxybenzoate hydroxylase. J. Biol. Chem 1976, 251, 2550–2563. [Google Scholar]
- Palfey, B.A.; Massey, V. Flavin-dependent enzymes. Comp. Biol. Cat 1998, 3, 83–154. [Google Scholar]
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans 2000, 28, 283–296. [Google Scholar]
- Stankovich, M. Chemistry and Biochemistry of Flavoenzyme; Muller, F., Ed.; CRC Press: Boca Raton, FL, USA, 1991; Volume 1, pp. 401–425. [Google Scholar]
- Palfey, B.A.; Ballou, D.; Massey, V. Oxygen activation by flavins and pterins. Act. Oxyg. Biochem 1995, 2, 37–83. [Google Scholar]
- Barman, B.G.; Tollin, G. Flavine-protein interactions in flavoenzymes. Thermodynamics and kinetics of reduction of Azotobacter flavodoxin. Biochemistry 1972, 11, 4755–4759. [Google Scholar]
- Gomez-Moreno, C.; Choy, M.; Edmondson, D.E. Purification and properties of the bacterial flavoprotein: Thiamin dehydrogenase. J. Biol. Chem 1979, 254, 7630–7635. [Google Scholar]
- Massey, V. A simple method for the determination of redox potentials. In Flavins and Flavoproteins; Walter de Gruyter: Berlin, Germany, 1991; pp. 59–66. [Google Scholar]
- Einarsdottir, G.H.; Stankovich, M.T.; Powlowski, J.; Ballou, D.P.; Massey, V. Regulation of oxidation-reduction potentials of anthranilate hydroxylase from Trichosporon cutaneum by substrate and effector binding. Biochemistry 1989, 28, 4161–4168. [Google Scholar]
- Williamson, G.; Edmondson, D.E.; Muller, F. Oxidation-reduction potential studies on p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Biochim. Biophys. Acta 1988, 953, 258–262. [Google Scholar]
- Mason, H.S.; Fowlks, F.W.; Peterson, E. Oxygen transfer and electron transport by the phenolase complex. J. Am. Chem. Soc 1955, 77, 2914–2915. [Google Scholar]
- Hayaishi, O. Mechanism of the pyrocatechase reaction. J. Am. Chem. Soc 1955, 77, 5450–5451. [Google Scholar]
- Maeda-Yorita, K.; Aki, K.; Sagai, H.; Misaki, H.; Massey, V. l-lactate oxidase and l-lactate monooxygenase: Mechanistic variations on a common structural theme. Biochimie 1995, 77, 631–642. [Google Scholar]
- Husain, M.; Massey, V. Kinetic studies on the reaction of p-hydroxybenzoate hydroxylase. Agreement of steady state and rapid reaction data. J. Biol. Chem 1979, 254, 6657–6666. [Google Scholar]
- Ballou, D.P. Flavoprotein Monooxygenases. In Flavins and Flavoproteins; Walter de Gruyter: Berlin, Germany, 1984; pp. 605–618. [Google Scholar]
- Meneely, K.M.; Lamb, A.L. Biochemical characterization of a flavin adenine dinucleotide-dependent monooxygenase, ornithine hydroxylase from Pseudomonas aeruginosa, suggests a novel reaction mechanism. Biochemistry 2007, 46, 11930–11937. [Google Scholar]
- Ge, L.; Seah, S.Y. Heterologous expression, purification, and characterization of an l-ornithine N(5)-hydroxylase involved in pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa. J. Bacteriol 2006, 188, 7205–7210. [Google Scholar]
- Macheroux, P.; Plattner, H.J.; Romaguera, A.; Diekmann, H. FAD and substrate analogs as probes for lysine N6-hydroxylase from Escherichia coli EN 222. Eur. J. Biochem 1993, 213, 995–1002. [Google Scholar]
- Chocklett, S.W.; Sobrado, P. Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry 2010, 49, 6777–6783. [Google Scholar]
- Abu-Soud, H.; Mullins, L.S.; Baldwin, T.O.; Raushel, F.M. Stopped-flow kinetic analysis of the bacterial luciferase reaction. Biochemistry 1992, 31, 3807–3813. [Google Scholar]
- Orru, R.; Dudek, H.M.; Martinoli, C.; Pazmino, D.E.T.; Royant, A.; Weik, M.; Fraaije, M.W.; Mattevi, A. Snapshots of enzymatic Baeyer-Villiger catalysis: Oxygen activation and intermediate stabilization. J. Biol. Chem 2011, 286, 29284–29291. [Google Scholar]
- Sheng, D.; Ballou, D.P.; Massey, V. Mechanistic studies of cyclohexanone monooxygenase: Chemical properties of intermediates involved in catalysis. Biochemistry 2001, 40, 11156–11167. [Google Scholar]
- Olucha, J.; Meneely, K.M.; Chilton, A.S.; Lamb, A.L. Two structures of an N-hydroxylating flavoprotein monooxygenase: Ornithine hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem 2011, 286, 31789–31798. [Google Scholar]
- Franceschini, S.; Fedkenheuer, M.; Vogelaar, N.J.; Robinson, H.H.; Sobrado, P.; Mattevi, A. Structural insight into the mechanism of oxygen activation and substrate selectivity of flavin-dependent N-hydroxylating monooxygenases. Biochemistry 2012, 51, 7043–7045. [Google Scholar]
- Chaiyen, P.; Suadee, C.; Wilairat, P. A novel two-protein component flavoprotein hydroxylase. Eur. J. Biochem 2001, 268, 5550–5561. [Google Scholar]
- Kirchner, U.; Westphal, A.H.; Muller, R.; van Berkel, W.J. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem 2003, 278, 47545–47553. [Google Scholar]
- Sucharitakul, J.; Chaiyen, P.; Entsch, B.; Ballou, D.P. The reductase of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii requires p-hydroxyphenylacetate for effective catalysis. Biochemistry 2005, 44, 10434–10442. [Google Scholar]
- Ukaegbu, U.E.; Kantz, A.; Beaton, M.; Gassner, G.T.; Rosenzweig, A.C. Structure and ligand binding properties of the epoxidase component of styrene monooxygenase. Biochemistry 2010, 49, 1678–1688. [Google Scholar]
- Kantz, A.; Gassner, G.T. Nature of the reaction intermediates in the flavin adenine dinucleotide-dependent epoxidation mechanism of styrene monooxygenase. Biochemistry 2011, 50, 523–532. [Google Scholar]
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys 2010, 497, 1–12. [Google Scholar]
- Ballou, D.P.; Entsch, B.; Cole, L.J. Dynamics involved in catalysis by single-component and two-component flavin-dependent aromatic hydroxylases. Biochem. Biophys. Res. Commun 2005, 338, 590–598. [Google Scholar]
- Sucharitakul, J.; Chaiyen, P.; Entsch, B.; Ballou, D.P. Kinetic mechanisms of the oxygenase from a two-component enzyme, p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii. J. Biol. Chem 2006, 281, 17044–17053. [Google Scholar]
- Dong, C.; Flecks, S.; Unversucht, S.; Haupt, C.; van Pee, K.H.; Naismith, J.H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar]
- Flecks, S.; Patallo, E.P.; Zhu, X.; Ernyei, A.J.; Seifert, G.; Schneider, A.; Dong, C.; Naismith, J.H.; van Pee, K.H. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chem. Int. Ed. Engl 2008, 47, 9533–9536. [Google Scholar]
- Zhu, X.; de Laurentis, W.; Leang, K.; Herrmann, J.; Ihlefeld, K.; van Pee, K.H.; Naismith, J.H. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J. Mol. Biol 2009, 391, 74–85. [Google Scholar]
- Yeh, E.; Cole, L.J.; Barr, E.W.; Bollinger, J.M., Jr; Ballou, D.P.; Walsh, C.T. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry 2006, 45, 7904–7912. [Google Scholar]
- Yeh, E.; Blasiak, L.C.; Koglin, A.; Drennan, C.L.; Walsh, C.T. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 2007, 46, 1284–1292. [Google Scholar]
- Chaiyen, P. Flavoenzymes catalyzing oxidative aromatic ring-cleavage reactions. Arch. Biochem. Biophys 2010, 493, 62–70. [Google Scholar]
- Gatti, D.L.; Palfey, B.A.; Lah, M.S.; Entsch, B.; Massey, V.; Ballou, D.P.; Ludwig, M.L. The mobile flavin of 4-OH benzoate hydroxylase. Science 1994, 266, 110–114. [Google Scholar]
- Cole, L.J.; Entsch, B.; Ortiz-Maldonado, M.; Ballou, D.P. Properties of p-hydroxybenzoate hydroxylase when stabilized in its open conformation. Biochemistry 2005, 44, 14807–14817. [Google Scholar]
- Van der Bolt, F.J.; Vervoort, J.; van Berkel, W.J. Flavin motion in p-hydroxybenzoate hydroxylase. Substrate and effector specificity of the Tyr22-->Ala mutant. Eur. J. Biochem 1996, 237, 592–600. [Google Scholar]
- Schreuder, H.A.; Mattevi, A.; Obmolova, G.; Kalk, K.H.; Hol, W.G.; van der Bolt, F.J.; van Berkel, W.J. Crystal structures of wild-type p-hydroxybenzoate hydroxylase complexed with 4-aminobenzoate-2,4-dihydroxybenzoate, and 2-hydroxy-4-aminobenzoate and of the Tyr222Ala mutant complexed with 2-hydroxy-4-aminobenzoate. Evidence for a proton channel and a new binding mode of the flavin ring. Biochemistry 1994, 33, 10161–10170. [Google Scholar]
- Brender, J.R.; Dertouzos, J.; Ballou, D.P.; Massey, V.; Palfey, B.A.; Entsch, B.; Steel, D.G.; Gafni, A. Conformational dynamics of the isoalloxazine in substrate-free p-hydroxybenzoate hydroxylase: Single-molecule studies. J. Am. Chem. Soc 2005, 127, 18171–18178. [Google Scholar]
- Entsch, B.; Cole, L.J.; Ballou, D.P. Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch. Biochem. Biophys 2005, 433, 297–311. [Google Scholar]
- Wang, J.; Ortiz-Maldonado, M.; Entsch, B.; Massey, V.; Ballou, D.; Gatti, D.L. Protein and ligand dynamics in 4-hydroxybenzoate hydroxylase. Proc. Natl. Acad. Sci. USA 2002, 99, 608–613. [Google Scholar]
- Enroth, C.; Neujahr, H.; Schneider, G.; Lindqvist, Y. The crystal structure of phenol hydroxylase in complex with FAD and phenol provides evidence for a concerted conformational change in the enzyme and its cofactor during catalysis. Structure 1998, 6, 605–617. [Google Scholar]
- Gatti, D.L.; Entsch, B.; Ballou, D.P.; Ludwig, M.L. pH-dependent structural changes in the active site of p-hydroxybenzoate hydroxylase point to the importance of proton and water movements during catalysis. Biochemistry 1996, 35, 567–578. [Google Scholar]
- Moran, G.R.; Entsch, B.; Palfey, B.A.; Ballou, D.P. Evidence for flavin movement in the function of p-hydroxybenzoate hydroxylase from studies of the mutant Arg220Lys. Biochemistry 1996, 35, 9278–9285. [Google Scholar]
- Moran, G.R.; Entsch, B.; Palfey, B.A.; Ballou, D.P. Mechanistic insights into p-hydroxybenzoate hydroxylase from studies of the mutant Ser212Ala. Biochemistry 1999, 38, 6292–6299. [Google Scholar]
- Frederick, K.K.; Palfey, B.A. Kinetics of proton-linked flavin conformational changes in p-hydroxybenzoate hydroxylase. Biochemistry 2005, 44, 13304–13314. [Google Scholar]
- Ortiz-Maldonado, M.; Cole, L.J.; Dumas, S.M.; Entsch, B.; Ballou, D.P. Increased positive electrostatic potential in p-hydroxybenzoate hydroxylase accelerates hydroxylation but slows turnover. Biochemistry 2004, 43, 1569–1579. [Google Scholar]
- Schreuder, H.A.; Hol, W.G.; Drenth, J. Analysis of the active site of the flavoprotein p-hydroxybenzoate hydroxylase and some ideas with respect to its reaction mechanism. Biochemistry 1990, 29, 3101–3108. [Google Scholar]
- Entsch, B.; Palfey, B.A.; Ballou, D.P.; Massey, V. Catalytic function of tyrosine residues in para-hydroxybenzoate hydroxylase as determined by the study of site-directed mutants. J. Biol. Chem 1991, 266, 17341–17349. [Google Scholar]
- Lah, M.S.; Palfey, B.A.; Schreuder, H.A.; Ludwig, M.L. Crystal structures of mutant Pseudomonas aeruginosa p-hydroxybenzoate hydroxylases: The Tyr201Phe, Tyr385Phe, and Asn300Asp variants. Biochemistry 1994, 33, 1555–1564. [Google Scholar]
- Crozier-Reabe, K.R.; Phillips, R.S.; Moran, G.R. Kynurenine 3-monooxygenase from Pseudomonas fluorescens: Substrate-like inhibitors both stimulate flavin reduction and stabilize the flavin-peroxo intermediate yet result in the production of hydrogen peroxide. Biochemistry 2008, 47, 12420–12433. [Google Scholar]
- Palfey, B.A.; Federick, K.K.; Basu, R.; Xu, D.; Ballou, D.P.; Moran, G.R.; Entsch, B. Wavin’ Flavins and Passwords: Dynamics and Control in the Reactions of p-Hydroxybenzoate Hydroxylase. In Flavins and Flavoproteins; Ghisla, S., Kroneck, P., Eds.; Agency for Scientific Publications: Berlin, Germay, 1999; pp. 351–358. [Google Scholar]
- Palfey, B.A.; Moran, G.R.; Entsch, B.; Ballou, D.P.; Massey, V. Substrate recognition by “password” in p-hydroxybenzoate hydroxylase. Biochemistry 1999, 38, 1153–1158. [Google Scholar]
- Treiber, N.; Schulz, G.E. Structure of 2,6-dihydroxypyridine 3-hydroxylase from a nicotine-degrading pathway. J. Mol. Biol 2008, 379, 94–104. [Google Scholar]
- Hiromoto, T.; Fujiwara, S.; Hosokawa, K.; Yamaguchi, H. Crystal structure of 3-hydroxybenzoate hydroxylase from Comamonas testosteroni has a large tunnel for substrate and oxygen access to the active site. J. Mol. Biol 2006, 364, 878–896. [Google Scholar]
- Schreuder, H.A.; Prick, P.A.; Wierenga, R.K.; Vriend, G.; Wilson, K.S.; Hol, W.G.; Drenth, J. Crystal structure of the p-hydroxybenzoate hydroxylase-substrate complex refined at 1.9 A resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J. Mol. Biol 1989, 208, 679–696. [Google Scholar]
- Kalin, M.; Neujahr, H.Y.; Weissmahr, R.N.; Sejlitz, T.; Johl, R.; Fiechter, A.; Reiser, J. Phenol hydroxylase from Trichosporon cutaneum: Gene cloning, sequence analysis, and functional expression in Escherichia coli. J. Bacteriol 1992, 174, 7112–7120. [Google Scholar]
- Eppink, M.H.; Schreuder, H.A.; van Berkel, W.J. Identification of a novel conserved sequence motif in flavoprotein hydroxylases with a putative dual function in FAD/NAD(P)H binding. Protein Sci 1997, 6, 2454–2458. [Google Scholar]
- Eppink, M.H.; Schreuder, H.A.; van Berkel, W.J. Interdomain binding of NADPH in p-hydroxybenzoate hydroxylase as suggested by kinetic, crystallographic and modeling studies of histidine 162 and arginine 269 variants. J. Biol. Chem 1998, 273, 21031–21039. [Google Scholar]
- Eppink, M.H.; Schreuder, H.A.; van Berkel, W.J. Structure and function of mutant Arg44Lys of 4-hydroxybenzoate hydroxylase implications for NADPH binding. Eur. J. Biochem 1995, 231, 157–165. [Google Scholar]
- Eppink, M.H.; Schreuder, H.A.; van Berkel, W.J. Lys42 and Ser42 variants of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens reveal that Arg42 is essential for NADPH binding. Eur. J. Biochem 1998, 253, 194–201. [Google Scholar]
- Xu, D.; Ballou, D.P.; Massey, V. Studies of the mechanism of phenol hydroxylase: Mutants Tyr289Phe, Asp54Asn, and Arg281Met. Biochemistry 2001, 40, 12369–12378. [Google Scholar]
- Palfey, B.A.; Basu, R.; Frederick, K.K.; Entsch, B.; Ballou, D.P. Role of protein flexibility in the catalytic cycle of p-hydroxybenzoate hydroxylase elucidated by the Pro293Ser mutant. Biochemistry 2002, 41, 8438–8446. [Google Scholar]
- Gohain, N.; Thomashow, L.S.; Mavrodi, D.V.; Blankenfeldt, W. The purification, crystallization and preliminary structural characterization of FAD-dependent monooxygenase PhzS, a phenazine-modifying enzyme from Pseudomonas aeruginosa. Acta Cryst 2006, 62, 989–992. [Google Scholar]
- White-Stevens, R.H.; Kamin, H. Studies of a flavoprotein, salicylate hydroxylase. I. Preparation, properties, and the uncoupling of oxygen reduction from hydroxylation. J. Biol. Chem 1972, 247, 2358–2370. [Google Scholar]
- Schreuder, H.A.; van der Laan, J.M.; Swarte, M.B.; Kalk, K.H.; Hol, W.G.; Drenth, J. Crystal structure of the reduced form of p-hydroxybenzoate hydroxylase refined at 2.3 A resolution. Proteins 1992, 14, 178–190. [Google Scholar]
- Ryan, K.S.; Chakraborty, S.; Howard-Jones, A.R.; Walsh, C.T.; Ballou, D.P.; Drennan, C.L. The FAD cofactor of RebC shifts to an IN conformation upon flavin reduction. Biochemistry 2008, 47, 13506–13513. [Google Scholar]
- Wessiak, A.; Schopfer, L.M.; Massey, V. pH Dependence of the reoxidation of p-hydroxybenzoate hydroxylase 2,4-dihydroxybenzoate complex. J. Biol. Chem 1984, 259, 12547–12556. [Google Scholar]
- Clarkson, J.; Palfey, B.A.; Carey, P.R. Probing the chemistries of the substrate and flavin ring system of p-hydroxybenzoate hydroxylase by raman difference spectroscopy. Biochemistry 1997, 36, 12560–12566. [Google Scholar]
- Palfey, B.A.; Entsch, B.; Ballou, D.P.; Massey, V. Changes in the catalytic properties of p-hydroxybenzoate hydroxylase caused by the mutation Asn300Asp. Biochemistry 1994, 33, 1545–1554. [Google Scholar]
- Moran, G.R.; Entsch, B.; Palfey, B.A.; Ballou, D.P. Electrostatic effects on substrate activation in para-hydroxybenzoate hydroxylase: Studies of the mutant lysine 297 methionine. Biochemistry 1997, 36, 7548–7556. [Google Scholar]
- Van Berkel, W.; Westphal, A.; Eschrich, K.; Eppink, M.; de Kok, A. Substitution of Arg214 at the substrate-binding site of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Eur. J. Biochem 1992, 210, 411–419. [Google Scholar]
- Eschrich, K.; van der Bolt, F.J.; de Kok, A.; van Berkel, W.J. Role of Tyr201 and Tyr385 in substrate activation by p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Eur. J. Biochem 1993, 216, 137–146. [Google Scholar]
- Neujahr, H.Y.; Kjellen, K.G. Phenol hydroxylase from yeast. Reaction with phenol derivatives. J. Biol. Chem 1978, 253, 8835–8841. [Google Scholar]
- Neujahr, H.Y.; Gaal, A. Phenol hydroxylase from yeast. Purification and properties of the enzyme from Trichosporon cutaneum. Eur. J. Biochem 1973, 35, 386–400. [Google Scholar]
- Mortberg, M.; Neujahr, H.Y. In situ and in vitro kinetics of phenol hydroxylase. Biochem. Biophys. Res. Commun 1987, 146, 41–46. [Google Scholar]
- Mortberg, M.; Neujahr, H.Y. Activation enthalpies and pH dependence of phenol hydroxylase from Trichosporon cutaneum, in vitro and in situ. FEBS Lett 1988, 242, 75–78. [Google Scholar]
- Müller, F. Phenol Hydroxylase. In Chemistry and Biochemistry of Flavoenzymes; CRC Press: Boca Raton, FL, USA, 1991; pp. 65–85. [Google Scholar]
- Detmer, K.; Massey, V. Effect of substrate and pH on the oxidative half-reaction of phenol hydroxylase. J. Biol. Chem 1985, 260, 5998–6005. [Google Scholar]
- Peelen, S.; Rietjens, I.M.; van Berkel, W.J.; van Workum, W.A.; Vervoort, J. 19F-NMR study on the pH-dependent regioselectivity and rate of the ortho-hydroxylation of 3-fluorophenol by phenol hydroxylase from Trichosporon cutaneum. Implications for the reaction mechanism. Eur. J. Biochem 1993, 218, 345–353. [Google Scholar]
- Jones, K.C.; Ballou, D.P. Reactions of the 4a-hydroperoxide of liver microsomal flavin-containing monooxygenase with nucleophilic and electrophilic substrates. J. Biol. Chem 1986, 261, 2553–2559. [Google Scholar]
- Frederick, K.K.; Ballou, D.P.; Palfey, B.A. Protein dynamics control proton transfers to the substrate on the His72Asn mutant of p-hydroxybenzoate hydroxylase. Biochemistry 2001, 40, 3891–3899. [Google Scholar]
- Ortiz-Maldonado, M.; Entsch, B.; Ballou, D.P. Conformational changes combined with charge-transfer interactions are essential for reduction in catalysis by p-hydroxybenzoate hydroxylase. Biochemistry 2003, 42, 11234–11242. [Google Scholar]
- Eppink, M.H.; Overkamp, K.M.; Schreuder, H.A.; van Berkel, W.J. Switch of coenzyme specificity of p-hydroxybenzoate hydroxylase. J. Mol. Biol 1999, 292, 87–96. [Google Scholar]
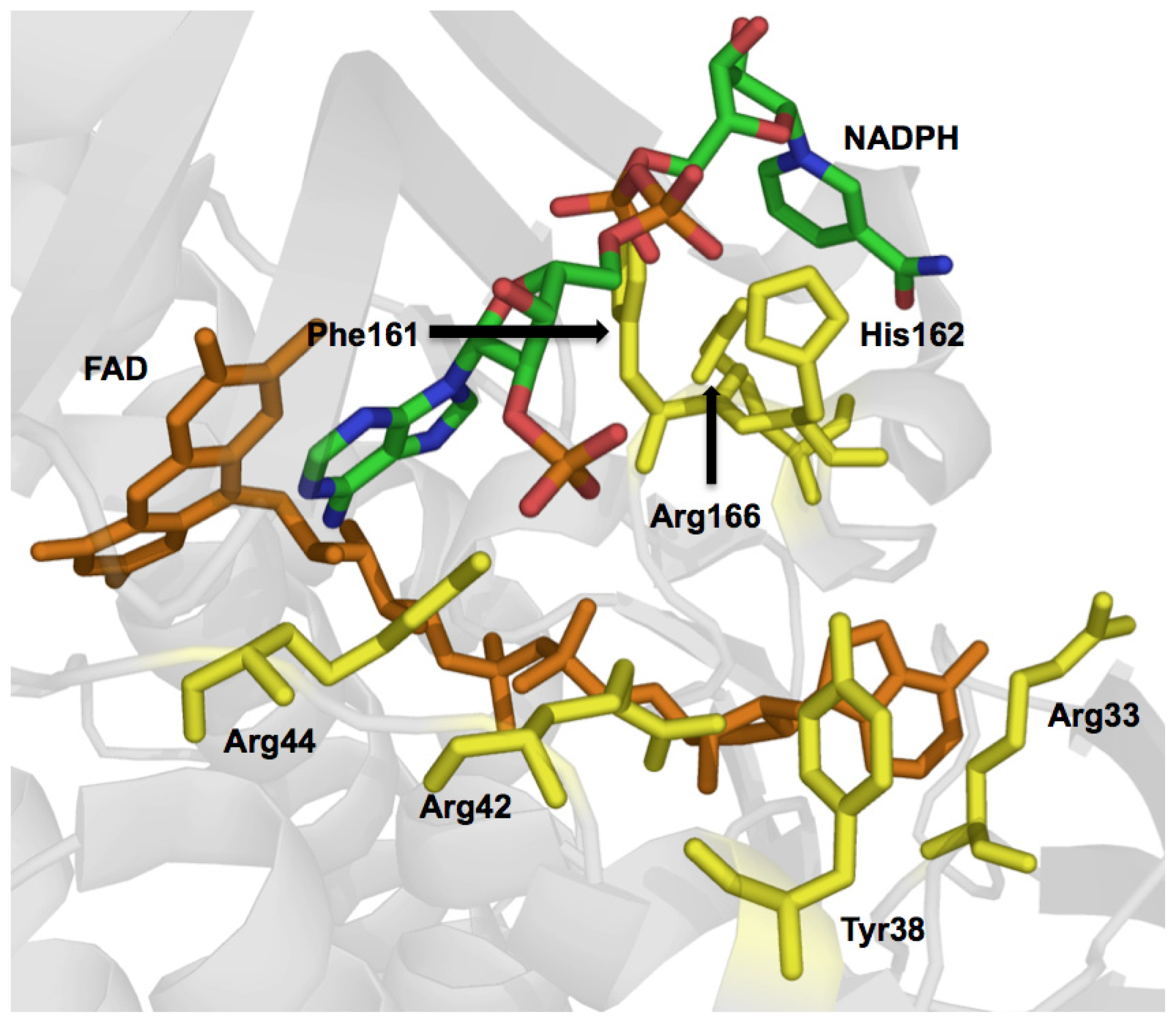
- Eppink, M.H.; Bunthol, C.; Schreuder, H.A.; van Berkel, W.J. Phe161 and Arg166 variants of p-hydroxybenzoate hydroxylase. Implications for NADPH recognition and structural stability. FEBS Lett 1999, 443, 251–255. [Google Scholar]
- Howell, L.G.; Spector, T.; Massey, V. Purification and properties of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. J. Biol. Chem 1972, 247, 4340–4350. [Google Scholar]
- Entsch, B. Hydroxybenzoate hydroxylase. Methods Enzymol 1990, 188, 138–147. [Google Scholar]
- Pai, E.F.; Schulz, G.E. The catalytic mechanism of glutathione reductase as derived from x-ray diffraction analyses of reaction intermediates. J. Biol. Chem 1983, 258, 1752–1757. [Google Scholar]
- Xu, D.; Enroth, C.; Lindqvist, Y.; Ballou, D.P.; Massey, V. Studies of the mechanism of phenol hydroxylase: Effect of mutation of proline 364 to serine. Biochemistry 2002, 41, 13627–13636. [Google Scholar]
- Bruice, T.C. Mechanisms of flavin catalysis. Acc. Chem. Res 1980, 13, 256–262. [Google Scholar]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem 1994, 269, 22459–22462. [Google Scholar]
- Schreuder, H.A.; van der Laan, J.M.; Hol, W.G.; Drenth, J. Crystal structure of p-hydroxybenzoate hydroxylase complexed with its reaction product 3,4-dihydroxybenzoate. J. Mol. Biol 1988, 199, 637–648. [Google Scholar]
- Ortiz-Maldonado, M.; Ballou, D.P.; Massey, V. Use of free energy relationships to probe the individual steps of hydroxylation of p-hydroxybenzoate hydroxylase: Studies with a series of 8-substituted flavins. Biochemistry 1999, 38, 8124–8137. [Google Scholar]
- Ortiz-Maldonado, M.; Entsch, B.; Ballou, D.P. Oxygen reactions in p-hydroxybenzoate hydroxylase utilize the H-bond network during catalysis. Biochemistry 2004, 43, 15246–15257. [Google Scholar]
- Vervoort, J.; Rietjens, I.M.; van Berkel, W.J.; Veeger, C. Frontier orbital study on the 4-hydroxybenzoate-3-hydroxylase-dependent activity with benzoate derivatives. Eur. J. Biochem 1992, 206, 479–484. [Google Scholar]
- Ridder, L.; Mulholland, A.J.; Rietjens, I.M.; Vervoort, J. Combined quantum mechanical and molecular mechanical reaction pathway calculation for aromatic hydroxylation by p-hydroxybenzoate-3-hydroxylase. J. Mol. Graph. Mod. 1999, 17. [Google Scholar]
- Schopfer, L.M.; Massey, V. Kinetic and mechanistic studies on the oxidation of the melilotate hydroxylase 2-OH-cinnamate complex by molecular oxygen. J. Biol. Chem 1980, 255, 5355–5363. [Google Scholar]
- Ortiz-Maldonado, M.; Gatti, D.; Ballou, D.P.; Massey, V. Structure-function correlations of the reaction of reduced nicotinamide analogues with p-hydroxybenzoate hydroxylase substituted with a series of 8-substituted flavins. Biochemistry 1999, 38, 16636–16647. [Google Scholar]
- Bruice, T.C.; Noar, J.B.; Ball, S.S.; Venkatara, U.V. Monooxygen donation potential of 4a-hydroperoxyflavins as compared with those of a percarboxylic acid and other hydroperoxides. Monooxygen donation to olefin, tertiary amine, alkyl sulfide, and iodide ion. J. Am. Chem. Soc 1983, 105, 2452–2465. [Google Scholar]
- Ridder, L.; Mulholland, A.J.; Rietjens, I.M.C.M.; Vervoort, J. A quantum mechanical/molecular mechanical study of the hydroxylation of phenol and halogenated derivatives by phenol hydroxylase. J. Am. Chem. Soc 2000, 122, 8728–8738. [Google Scholar]
- Enroth, C. High-resolution structure of phenol hydroxylase and correction of sequence errors. Acta Crystallogr. D 2003, 59, 1597–1602. [Google Scholar]
- Peelen, S.; Rietjens, I.M.C.M.; Boersma, M.G.; Vervoort, J. Conversion of phenol derivatives to hydroxylated products by phenol hydroxylase from Trichosporon cutaneum a comparison of regioselectivity and rate of conversion with calculated molecular orbital substrate characteristics. Eur. J. Biochem 1995, 227, 284–291. [Google Scholar]








© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Crozier-Reabe, K.; Moran, G.R. Form Follows Function: Structural and Catalytic Variation in the Class A Flavoprotein Monooxygenases. Int. J. Mol. Sci. 2012, 13, 15601-15639. https://doi.org/10.3390/ijms131215601
Crozier-Reabe K, Moran GR. Form Follows Function: Structural and Catalytic Variation in the Class A Flavoprotein Monooxygenases. International Journal of Molecular Sciences. 2012; 13(12):15601-15639. https://doi.org/10.3390/ijms131215601
Chicago/Turabian StyleCrozier-Reabe, Karen, and Graham R. Moran. 2012. "Form Follows Function: Structural and Catalytic Variation in the Class A Flavoprotein Monooxygenases" International Journal of Molecular Sciences 13, no. 12: 15601-15639. https://doi.org/10.3390/ijms131215601