Ginsenoside Rh2 Induces Human Hepatoma Cell Apoptosisvia Bax/Bak Triggered Cytochrome C Release and Caspase-9/Caspase-8 Activation

Abstract
:1. Introduction
2. Results
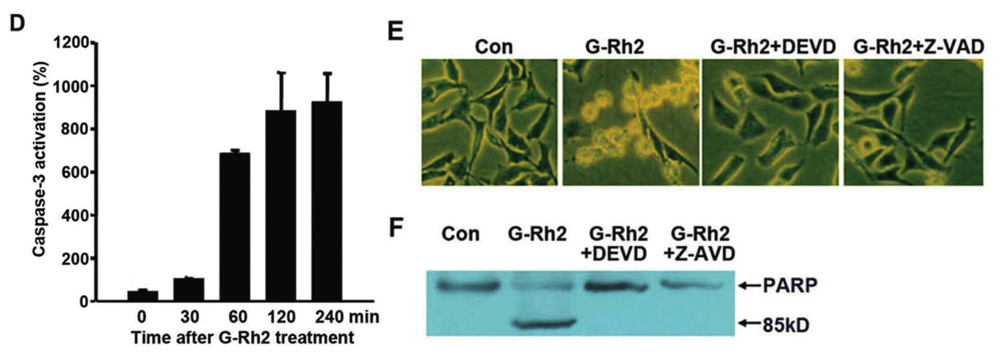
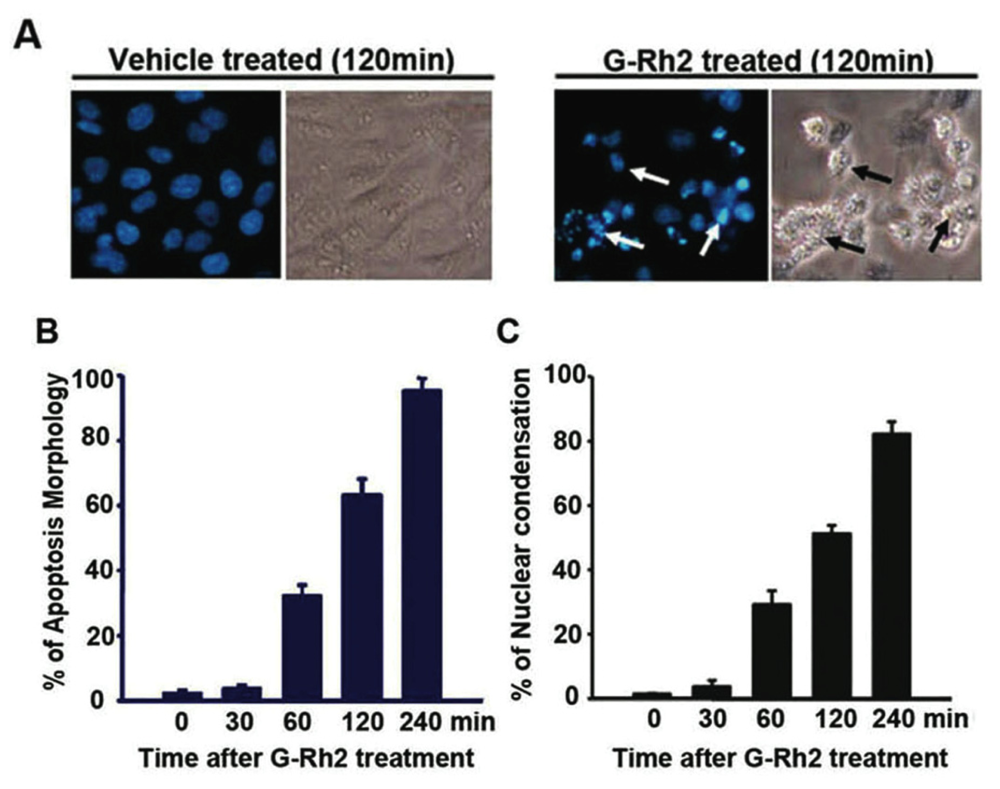
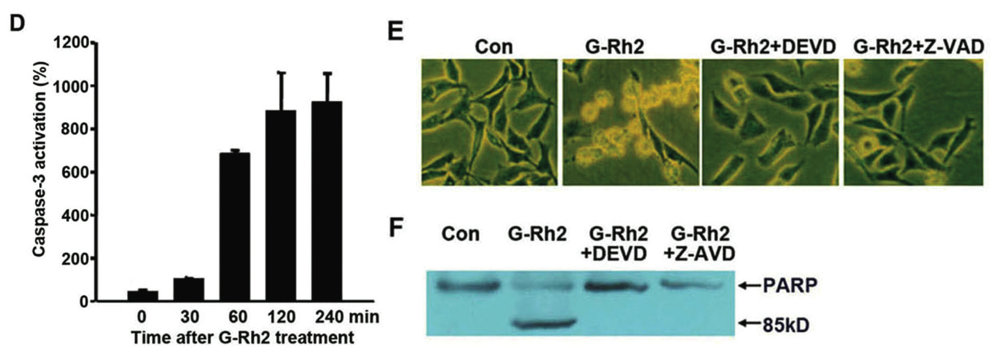
2.1. G-Rh2-Induced Apoptosis Is Mediated Via Caspase Activation
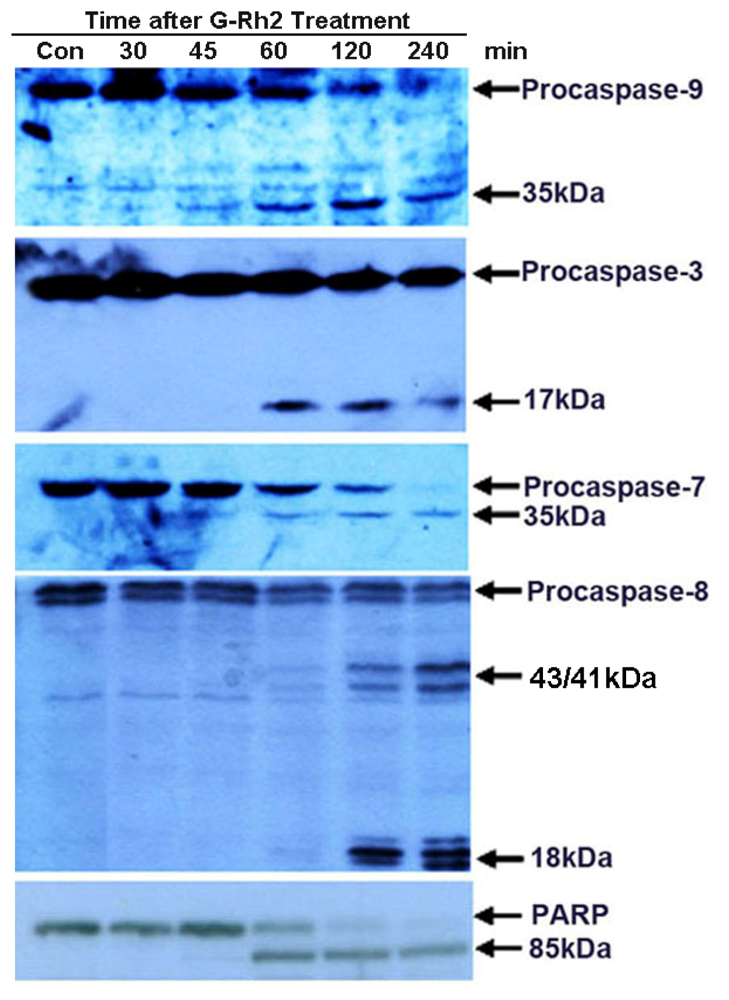
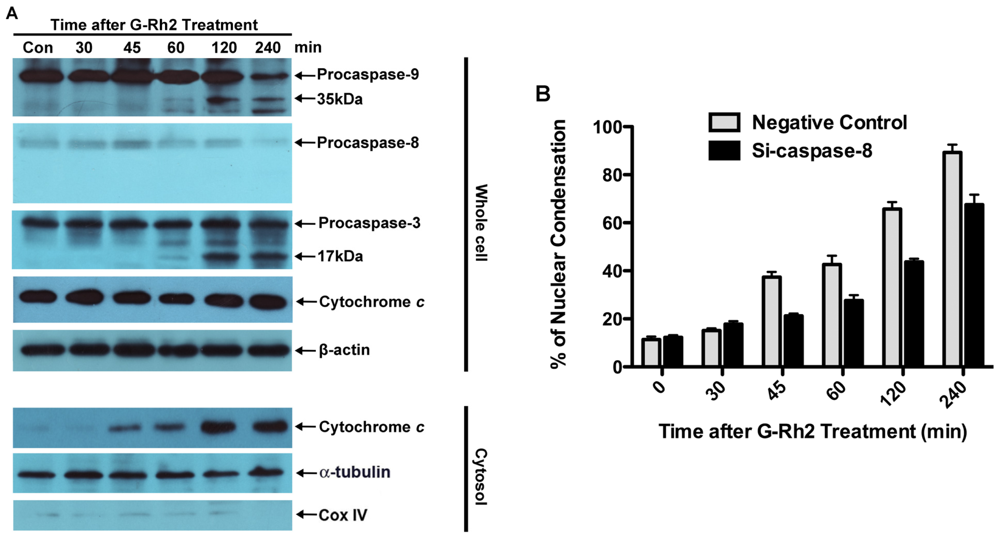
2.2. G-Rh2 Induces Caspase-9,-3,-7, and -8 Cleavages in SK-HEP-1 Cells
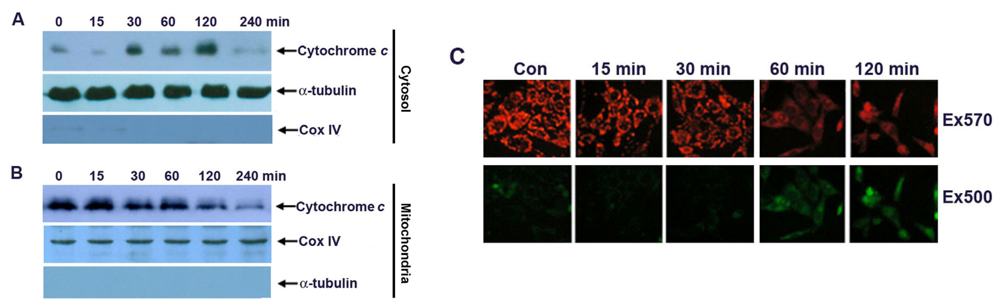
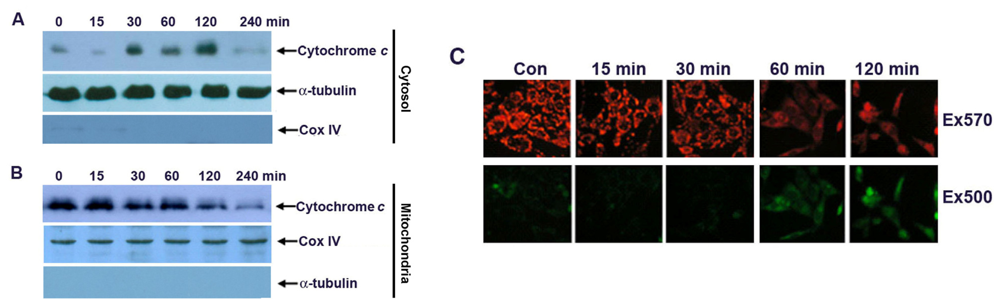
2.3. Apoptosis of G-Rh2-Induced SK-HEP-1 Cells Is Mediated through Cytochrome C Release and Depolarization of Mitochondrial Membrane Potential
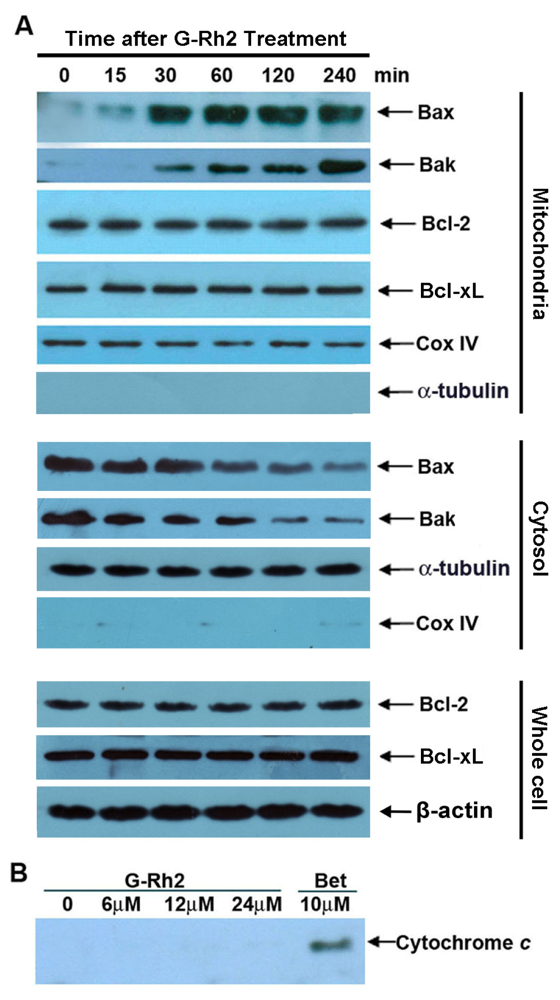
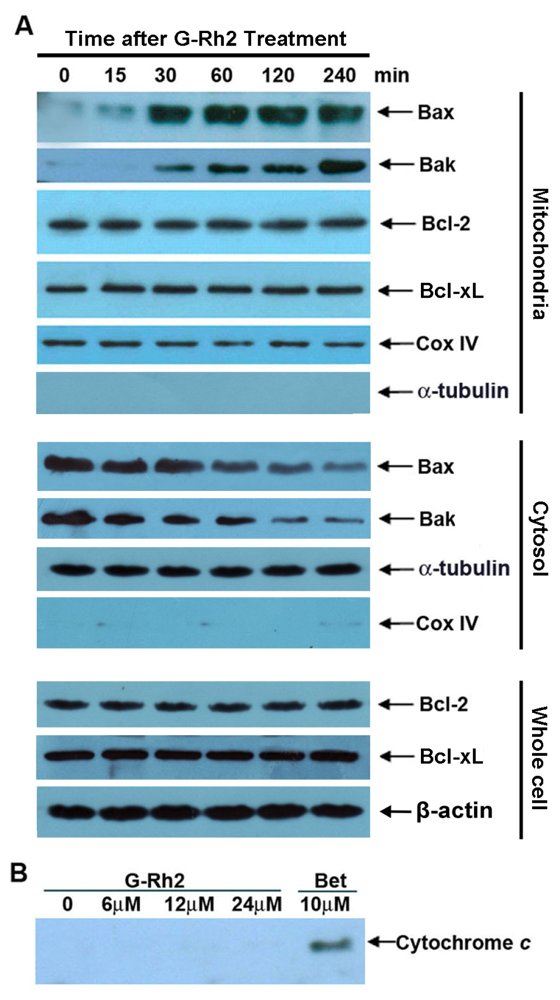
2.4. G-Rh2-Induced Apoptosis in SK-HEP-1 Cells Is Associated with Mitochondrial Translocation of Bax and Bak
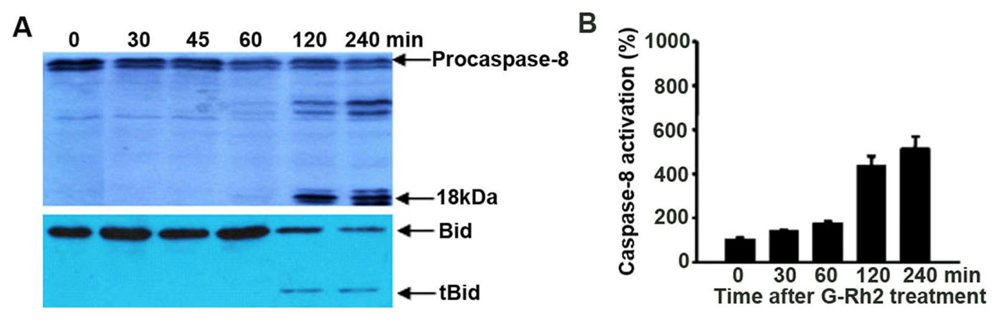
2.5. Caspase-8 Is Also Activated in G-Rh2-Treated Cells
3. Experimental Section
3.1. Materials
3.2. Cell Culture Conditions and Apoptosis Assays
3.3. Immuoblot Analysis
3.4. Caspase Assay
3.5. Preparation of Mitochondrial and Cytosolic Protein Extracts and Cytochrome C Release Assay
3.6. Depolarization Assay of Mitochondrial Membrane Potential by Staining with MitoCapture
3.7. RNA Interference
4. Discussion
5. Conclusions
Acknowledgments
References
- Park, H.M.; Kim, S.J.; Kim, J.S.; Kang, H.S. Reactive oxygen species mediated ginsenoside Rg3- and Rh2-induced apoptosis in hepatoma cells through mitochondrial signaling pathways. Food Chem. Toxicol 2012, 50, 2736–2741. [Google Scholar]
- Chan, S.W. Panax ginseng, Rhodiola rosea and Schisandra chinensis. Int. J. Food Sci. Nutr 2012, 63, 75–81. [Google Scholar]
- Jin, Y.H.; Yim, H.; Park, J.H.; Lee, S.K. Cdk2 activity is associated with depolarization of mitochondrial membrane potential during apoptosis. Biochem. Biophys. Res. Commun 2003, 305, 974–980. [Google Scholar]
- Kaneko, H.; Nakanishi, K. Proof of the mysterious efficacy of ginseng: Basic and clinical trials: Clinical effects of medical ginseng, korean red ginseng: Specifically, its anti-stress action for prevention of disease. J. Pharmacol. Sci 2004, 95, 158–162. [Google Scholar]
- Cheng, Y.; Shen, L.H.; Zhang, J.T. Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action. Acta Pharm. Sinica 2005, 26, 143–149. [Google Scholar]
- Nam, M.H.; Kim, S.I.; Liu, J.R.; Yang, D.C.; Lim, Y.P.; Kwon, K.H.; Yoo, J.S.; Park, Y.M. Proteomic analysis of Korean ginseng (Panax ginseng C.A. Meyer). J. Chromatogr. B 2005, 815, 147–155. [Google Scholar]
- Lee, K.Y.; Park, J.A.; Chung, E.; Lee, Y.H.; Kim, S.I.; Lee, S.K. Ginsenoside-Rh2 blocks the cell cycle of SK-HEP-1 cells at the G1/S boundary by selectively inducing the protein expression of p27kip1. Cancer Lett 1996, 110, 193–200. [Google Scholar]
- Oh, M.; Choi, Y.H.; Choi, S.; Chung, H.; Kim, K.; Kim, S.I.; Kim, D.K.; Kim, N.D. Anti-proliferating effects of ginsenoside Rh2 on MCF-7 human breast cancer cells. Int. J. Oncol 1999, 14, 869–875. [Google Scholar]
- Li, B.; Zhao, J.; Wang, C.Z.; Searle, J.; He, T.C.; Yuan, C.S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett 2011, 301, 185–192. [Google Scholar]
- Park, J.A.; Lee, K.Y.; Oh, Y.J.; Kim, K.W.; Lee, S.K. Activation of caspase-3 protease via a Bcl-2-insensitive pathway during the process of ginsenoside Rh2-induced apoptosis. Cancer Lett 1997, 121, 73–81. [Google Scholar]
- Kim, H.E.; Oh, J.H.; Lee, S.K.; Oh, Y.J. Ginsenoside RH-2 induces apoptotic cell death in rat C6 glioma via a reactive oxygen- and caspase-dependent but Bcl-X(L)-independent pathway. Life Sci. 1999, 65, PL33–40. [Google Scholar]
- Fei, X.F.; Wang, B.X.; Tashiro, S.; Li, T.J.; Ma, J.S.; Ikejima, T. Apoptotic effects of ginsenoside Rh2 on human malignant melanoma A375-S2 cells. Acta Pharmacol. Sinica 2002, 23, 315–322. [Google Scholar]
- Jin, Y.H.; Yoo, K.J.; Lee, Y.H.; Lee, S.K. Caspase 3-mediated cleavage of p21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J. Biol. Chem 2000, 275, 30256–30263. [Google Scholar]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res 2012, 318, 1213–1220. [Google Scholar]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar]
- Ricci, J.E.; Munoz-Pinedo, C.; Fitzgerald, P.; Bailly-Maitre, B.; Perkins, G.A.; Yadava, N.; Scheffler, I.E.; Ellisman, M.H.; Green, D.R. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2004, 117, 773–786. [Google Scholar]
- Dewson, G.; Kluck, R.M. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci 2009, 122, 2801–2808. [Google Scholar]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem 1998, 273, 33942–33948. [Google Scholar]
- Thorburn, A. Death receptor-induced cell killing. Cell. Signal 2004, 16, 139–144. [Google Scholar]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar]
- Degli Esposti, M.; Ferry, G.; Masdehors, P.; Boutin, J.A.; Hickman, J.A.; Dive, C. Post-translational modification of Bid has differential effects on its susceptibility to cleavage by caspase 8 or caspase 3. J. Biol. Chem 2003, 278, 15749–15757. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Ham, Y.M.; Choi, J.S.; Chun, K.H.; Joo, S.H.; Lee, S.K. The c-Jun N-terminal kinase 1 activity is differentially regulated by specific mechanisms during apoptosis. J. Biol. Chem 2003, 278, 50330–50337. [Google Scholar]
- Ham, Y.M.; Lim, J.H.; Na, H.K.; Choi, J.S.; Park, B.D.; Yim, H.; Lee, S.K. Ginsenoside-Rh2-induced mitochondrial depolarization and apoptosis are associated with reactive oxygen species- and Ca2+-mediated c-Jun NH2-terminal kinase 1 activation in HeLa cells. J. Pharmacol. Exp. Therapeut 2006, 319, 1276–1285. [Google Scholar]
- Oh, J.I.; Chun, K.H.; Joo, S.H.; Oh, Y.T.; Lee, S.K. Caspase-3-dependent protein kinase C delta activity is required for the progression of Ginsenoside-Rh2-induced apoptosis in SK-HEP-1 cells. Cancer Lett 2005, 230, 228–238. [Google Scholar]
- Kirkin, V.; Joos, S.; Zornig, M. The role of Bcl-2 family members in tumorigenesis. Biochim. Biophys. Acta 2004, 1644, 229–249. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence number | siRNA Duplex | |
|---|---|---|
| Sense (5′-3′) | Antisense (5′-3′) | |
| 1 | GCUGCUCUUCCGAAUUAAU | AUUAAUUCGGAAGAGCAGC |
| 2 | CCUGCUGGAUAUUUUCAUA | UAUGAAAAUAUCCAGCAGG |
| 3 | CAGAUCAGAAUUGAGGUCU | AGACCUCAAUUCUGAUCUG |
| Negative control | CCUACGCCACCAAUUUCGU | ACGAAAUUGGUGGCGUAGG |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, X.-X.; Guo, Q.; Li, Y.; Lee, S.K.; Wei, X.-N.; Jin, Y.-H. Ginsenoside Rh2 Induces Human Hepatoma Cell Apoptosisvia Bax/Bak Triggered Cytochrome C Release and Caspase-9/Caspase-8 Activation. Int. J. Mol. Sci. 2012, 13, 15523-15535. https://doi.org/10.3390/ijms131215523
Guo X-X, Guo Q, Li Y, Lee SK, Wei X-N, Jin Y-H. Ginsenoside Rh2 Induces Human Hepatoma Cell Apoptosisvia Bax/Bak Triggered Cytochrome C Release and Caspase-9/Caspase-8 Activation. International Journal of Molecular Sciences. 2012; 13(12):15523-15535. https://doi.org/10.3390/ijms131215523
Chicago/Turabian StyleGuo, Xiao-Xi, Qiao Guo, Yang Li, Seung Ki Lee, Xue-Ning Wei, and Ying-Hua Jin. 2012. "Ginsenoside Rh2 Induces Human Hepatoma Cell Apoptosisvia Bax/Bak Triggered Cytochrome C Release and Caspase-9/Caspase-8 Activation" International Journal of Molecular Sciences 13, no. 12: 15523-15535. https://doi.org/10.3390/ijms131215523