Role of Oxidative Stress in Hepatocarcinogenesis Induced by Hepatitis C Virus

{kind=link}
Abstract
:1. Introduction
2. Survey of HCV-Positive HCC-Related Host Factors
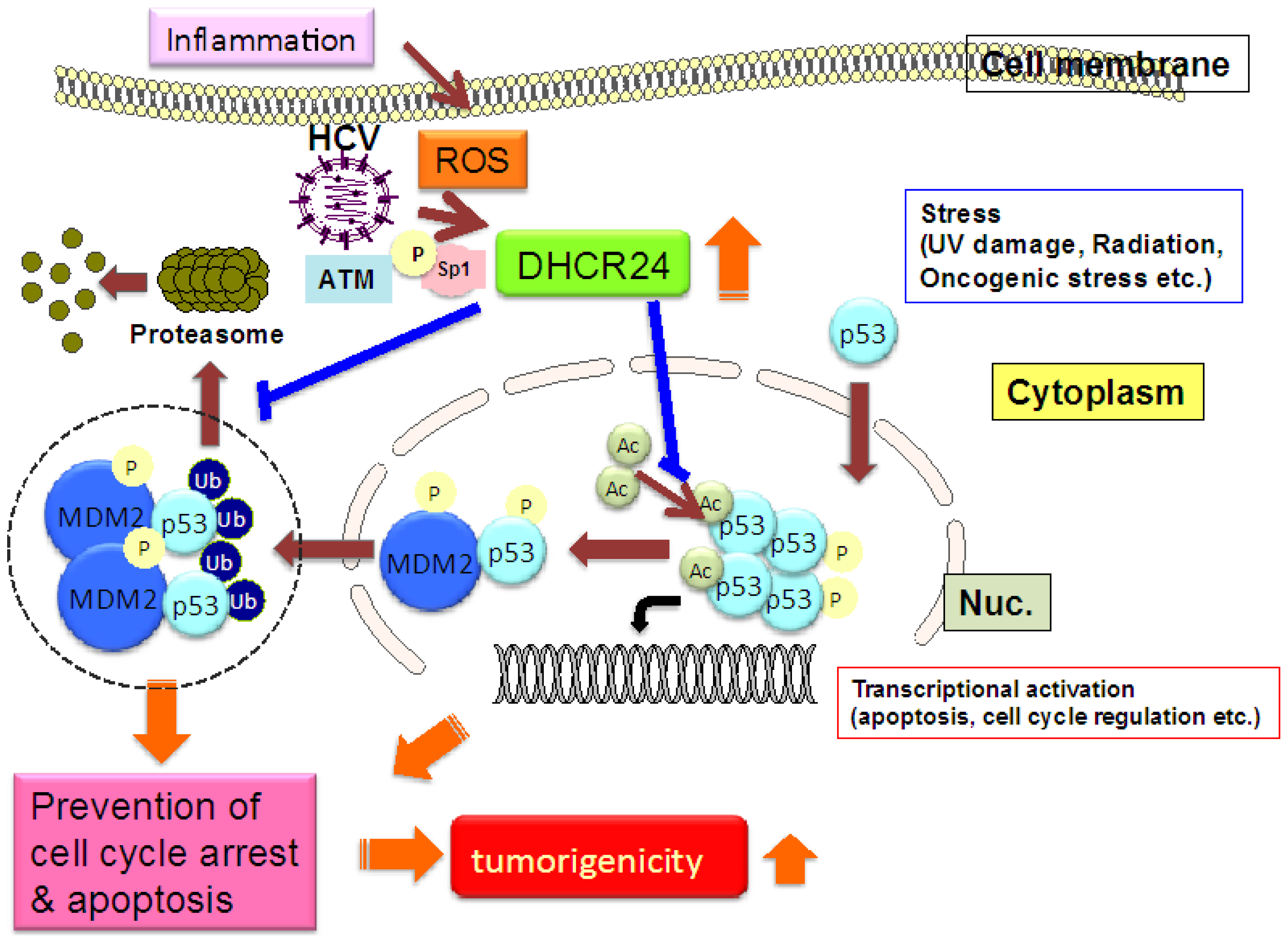
3. HCV Induces DHCR24 Expression through Oxidative Stress
4. Overexpression of DHCR24 Results in Impairment of p53 Activity
5. Conclusion
Acknowledgements
References
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar]
- Jenny-Avital, E.R. Hepatitis C. Curr. Opin. Infect. Dis 1998, 11, 293–299. [Google Scholar]
- Saito, I.; Miyamura, T.; Ohbayashi, A.; Harada, H.; Katayama, T.; Kikuchi, S.; Watanabe, Y.; Koi, S.; Onji, M.; Ohta, Y.; et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 6547–6549. [Google Scholar]
- Lavanchy, D. The global burden of hepatitis C. Liver Int 2009, 29, 74–81. [Google Scholar]
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int 2009, 3, 526–536. [Google Scholar]
- Garcia-Monzon, C.; Majano, P.L.; Zubia, I.; Sanz, P.; Apolinario, A.; Moreno-Otero, R. Intrahepatic accumulation of nitrotyrosine in chronic viral hepatitis is associated with histological severity of liver disease. J. Hepatol 2000, 32, 331–338. [Google Scholar]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol 2007, 22, 108–111. [Google Scholar]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol 2005, 13, 159–163. [Google Scholar]
- Fujinaga, H.; Tsutsumi, T.; Yotsuyanagi, H.; Moriya, K.; Koike, K. Hepatocarcinogenesis in hepatitis C: HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species. Oncology 2011, 81, 11–17. [Google Scholar]
- Zhu, Z.; Wilson, A.T.; Mathahs, M.M.; Wen, F.; Brown, K.E.; Luxon, B.A.; Schmidt, W.N. Heme oxygenase-1 suppresses hepatitis C virus replication and increases resistance of hepatocytes to oxidant injury. Hepatology 2008, 48, 1430–1439. [Google Scholar]
- Jaiswal, M.; LaRusso, N.F.; Shapiro, R.A.; Billiar, T.R.; Gores, G.J. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 2001, 120, 190–199. [Google Scholar]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol 2010, 185, 6985–6998. [Google Scholar]
- Machida, K.; Tsukamoto, H.; Liu, J.C.; Han, Y.P.; Govindarajan, S.; Lai, M.M.; Akira, S.; Ou, J.H. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology 2010, 52, 480–492. [Google Scholar]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med 1998, 4, 1065–1067. [Google Scholar]
- Tsukiyama-Kohara, K.; Tone, S.; Maruyama, I.; Inoue, K.; Katsume, A.; Nuriya, H.; Ohmori, H.; Ohkawa, J.; Taira, K.; Hoshikawa, Y.; et al. Activation of the CKI-CDK-Rb-E2F pathway in full genome hepatitis C virus-expressing cells. J. Biol. Chem 2004, 279, 14531–14541. [Google Scholar]
- Nishimura, T.; Kohara, M.; Izumi, K.; Kasama, Y.; Hirata, Y.; Huang, Y.; Shuda, M.; Mukaidani, C.; Takano, T.; Tokunaga, Y.; et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol Delta24-reductase. J. Biol. Chem 2009, 284, 36442–36452. [Google Scholar]
- Crameri, A.; Biondi, E.; Kuehnle, K.; Lutjohann, D.; Thelen, K.M.; Perga, S.; Dotti, C.G.; Nitsch, R.M.; Ledesma, M.D.; Mohajeri, M.H. The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J 2006, 25, 432–443. [Google Scholar]
- Kedjouar, B.; De Medina, P.; Oulad-Abdelghani, M.; Payre, B.; Silvente-Poirot, S.; Favre, G.; Faye, J.C.; Poirot, M. Molecular characterization of the microsomal tamoxifen binding site. J. Biol. Chem 2004, 279, 34048–34061. [Google Scholar]
- Waterham, H.R.; Koster, J.; Romeijn, G.J.; Hennekam, R.C.; Vreken, P.; Andersson, H.C.; FitzPatrick, D.R.; Kelley, R.I.; Wanders, R.J. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet 2001, 69, 685–694. [Google Scholar]
- Greeve, I.; Hermans-Borgmeyer, I.; Brellinger, C.; Kasper, D.; Gomez-Isla, T.; Behl, C.; Levkau, B.; Nitsch, R.M. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J. Neurosci 2000, 20, 7345–7352. [Google Scholar]
- Benvenuti, S.; Saccardi, R.; Luciani, P.; Urbani, S.; Deledda, C.; Cellai, I.; Francini, F.; Squecco, R.; Rosati, F.; Danza, G.; et al. Neuronal differentiation of human mesenchymal stem cells: Changes in the expression of the Alzheimer’s disease-related gene seladin-1. Exp. Cell Res 2006, 312, 2592–2604. [Google Scholar]
- Kuehnle, K.; Crameri, A.; Kalin, R.E.; Luciani, P.; Benvenuti, S.; Peri, A.; Ratti, F.; Rodolfo, M.; Kulic, L.; Heppner, F.L.; et al. Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress. Mol. Cell. Biol 2008, 28, 539–550. [Google Scholar]
- Luciani, P.; Gelmini, S.; Ferrante, E.; Lania, A.; Benvenuti, S.; Baglioni, S.; Mantovani, G.; Cellai, I.; Ammannati, F.; Spada, A.; et al. Expression of the antiapoptotic gene seladin-1 and octreotide-induced apoptosis in growth hormone-secreting and nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab 2005, 90, 6156–6161. [Google Scholar]
- Lu, X.; Kambe, F.; Cao, X.; Kozaki, Y.; Kaji, T.; Ishii, T.; Seo, H. 3Beta-Hydroxysteroid-delta24 reductase is a hydrogen peroxide scavenger, protecting cells from oxidative stress-induced apoptosis. Endocrinology 2008, 149, 3267–3273. [Google Scholar]
- Wu, C.; Miloslavskaya, I.; Demontis, S.; Maestro, R.; Galaktionov, K. Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature 2004, 432, 640–645. [Google Scholar]
- Di Stasi, D.; Vallacchi, V.; Campi, V.; Ranzani, T.; Daniotti, M.; Chiodini, E.; Fiorentini, S.; Greeve, I.; Prinetti, A.; Rivoltini, L.; et al. DHCR24 gene expression is upregulated in melanoma metastases and associated to resistance to oxidative stress-induced apoptosis. Int. J. Cancer 2005, 115, 224–230. [Google Scholar]
- Saito, M.; Kohara, M.; Tsukiyama-Kohara, K. Hepatitis C virus promotes expression of the 3beta-hydroxysterol delta24-reductase through Sp1. J. Med. Virol 2012, 84, 733–746. [Google Scholar]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; et al. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med 2001, 7, 927–933. [Google Scholar]
- Olofsson, B.A.; Kelly, C.M.; Kim, J.; Hornsby, S.M.; Azizkhan-Clifford, J. Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol. Cancer Res 2007, 5, 1319–1330. [Google Scholar]
- Iwahori, S.; Shirata, N.; Kawaguchi, Y.; Weller, S.K.; Sato, Y.; Kudoh, A.; Nakayama, S.; Isomura, H.; Tsurumi, T. Enhanced phosphorylation of transcription factor sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol 2007, 81, 9653–9664. [Google Scholar]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem 2005, 280, 37481–37488. [Google Scholar]
- De Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 2001, 20, 1331–1340. [Google Scholar]
- Bonaccorsi, L.; Luciani, P.; Nesi, G.; Mannucci, E.; Deledda, C.; Dichiara, F.; Paglierani, M.; Rosati, F.; Masieri, L.; Serni, S.; et al. Androgen receptor regulation of the seladin-1/DHCR24 gene: Altered expression in prostate cancer. Lab. Invest 2008, 88, 1049–1056. [Google Scholar]
- Malmlof, M.; Roudier, E.; Hogberg, J.; Stenius, U. MEK-ERK-Mediated phosphorylation of Mdm2 at Ser-166 in hepatocytes. Mdm2 is activated in response to inhibited Akt signaling. J. Biol. Chem 2007, 282, 2288–2296. [Google Scholar]
- Takano, T.; Tsukiyama-Kohara, K.; Hayashi, M.; Hirata, Y.; Satoh, M.; Tokunaga, Y.; Tateno, C.; Hayashi, Y.; Hishima, T.; Funata, N.; et al. Augmentation of DHCR24 expression by hepatitis C virus infection facilitates viral replication in hepatocytes. J. Hepatol 2011, 55, 512–521. [Google Scholar]
- Satoh, M.; Saito, M.; Takano, T.; Kasama, Y.; Nishimura, T.; Nishito, Y.; Hirata, Y.; Arai, M.; Sudoh, M.; Kai, C.; et al. Monoclonal antibody 2–152a suppresses hepatitis C virus infection through betaine/GABA transporter-1. J. Infect. Dis 2011, 204, 1172–1180. [Google Scholar]
- Takenaka, M.; Preston, A.S.; Kwon, H.M.; Handler, J.S. The tonicity-sensitive element that mediates increased transcription of the betaine transporter gene in response to hypertonic stress. J. Biol. Chem 1994, 269, 29379–29381. [Google Scholar]
- Offensperger, W.B.; Offensperger, S.; Stoll, B.; Gerok, W.; Haussinger, D. Effects of anisotonic exposure on duck hepatitis B virus replication. Hepatology 1994, 20, 1–7. [Google Scholar]
- Lever, M.; George, P.M.; Atkinson, W.; Molyneux, S.L.; Elmslie, J.L.; Slow, S.; Richards, A.M.; Chambers, S.T. Plasma lipids and betaine are related in an acute coronary syndrome cohort. PLoS One 2011, 6, e21666. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsukiyama-Kohara, K. Role of Oxidative Stress in Hepatocarcinogenesis Induced by Hepatitis C Virus. Int. J. Mol. Sci. 2012, 13, 15271-15278. https://doi.org/10.3390/ijms131115271
Tsukiyama-Kohara K. Role of Oxidative Stress in Hepatocarcinogenesis Induced by Hepatitis C Virus. International Journal of Molecular Sciences. 2012; 13(11):15271-15278. https://doi.org/10.3390/ijms131115271
Chicago/Turabian StyleTsukiyama-Kohara, Kyoko. 2012. "Role of Oxidative Stress in Hepatocarcinogenesis Induced by Hepatitis C Virus" International Journal of Molecular Sciences 13, no. 11: 15271-15278. https://doi.org/10.3390/ijms131115271