Förster Resonance Energy Transfer (FRET) as a Tool for Dissecting the Molecular Mechanisms for Maturation of the Shigella Type III Secretion Needle Tip Complex

Abstract
:1. Introduction
2. Use of Fluorescence in Exploring the Lives of Bacteria and Bacterial Pathogens
2.1. Limitations of Traditional Fluorescence Measurements and Alternative Techniques
3. Type III Secretion and Secretion Systems
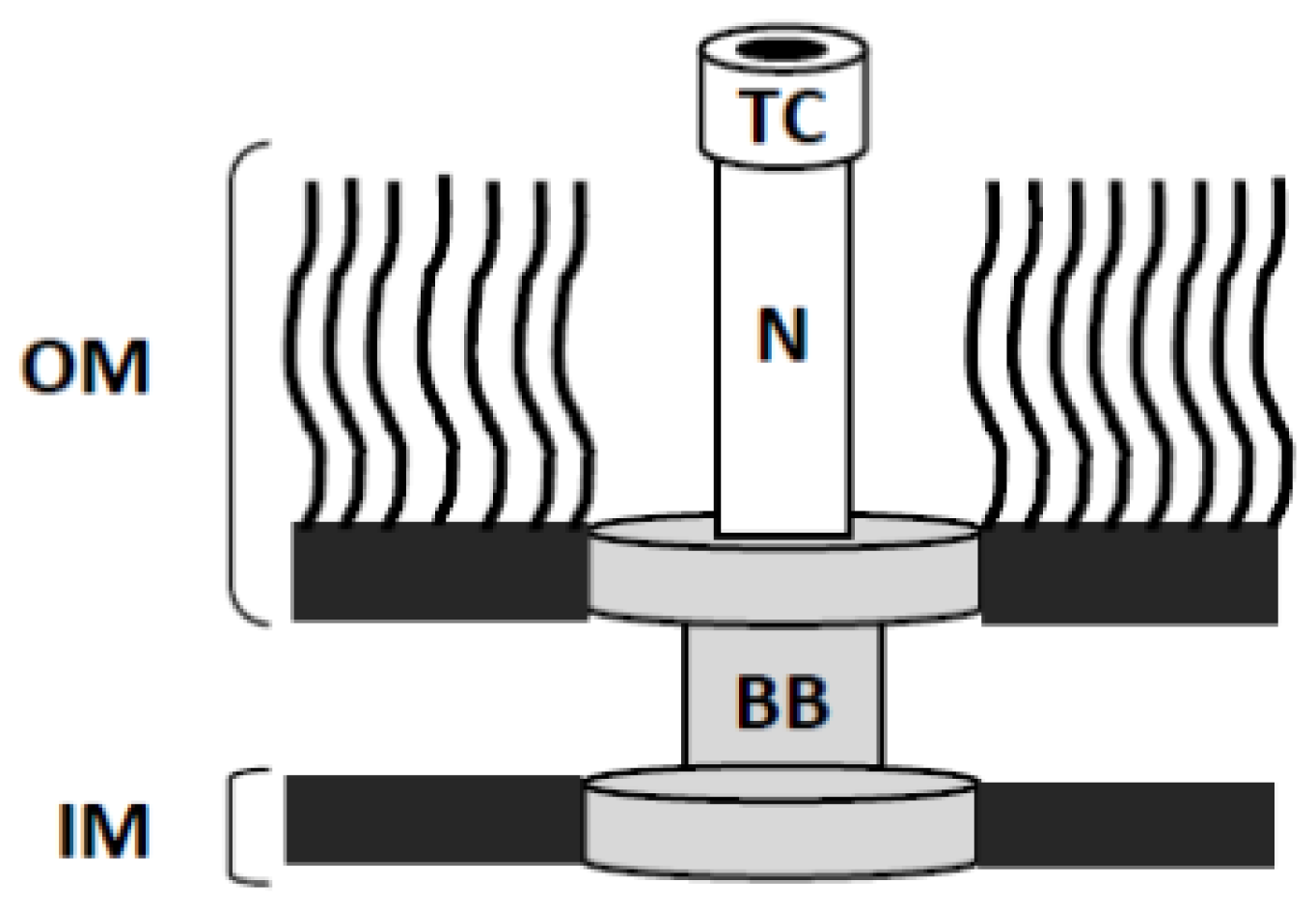
3.1. Shigella as a Model for Dissecting the Structure and Function of the T3SA Needle Tip Complex
3.2. High-Resolution Structures of the External Portions of the T3SA
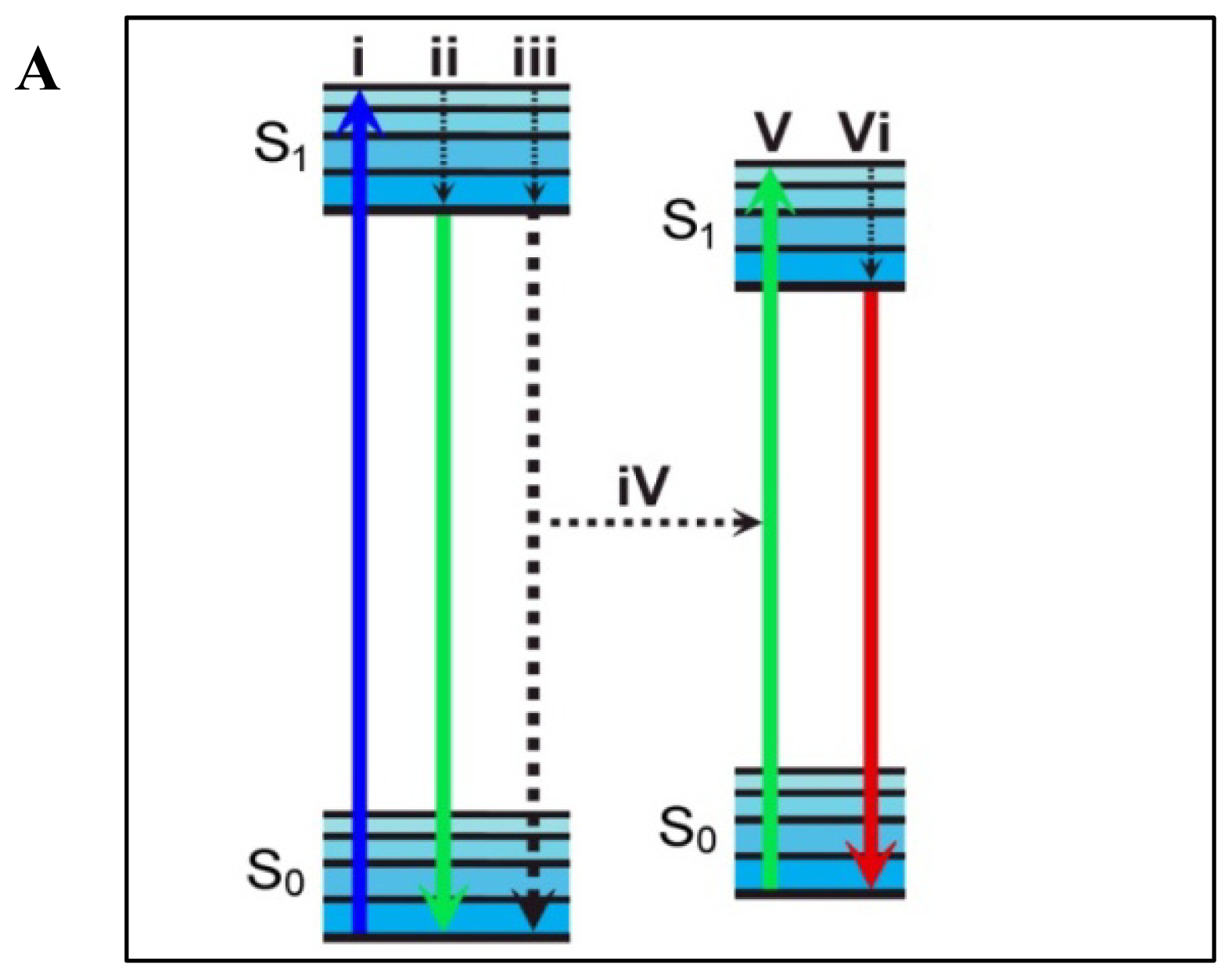
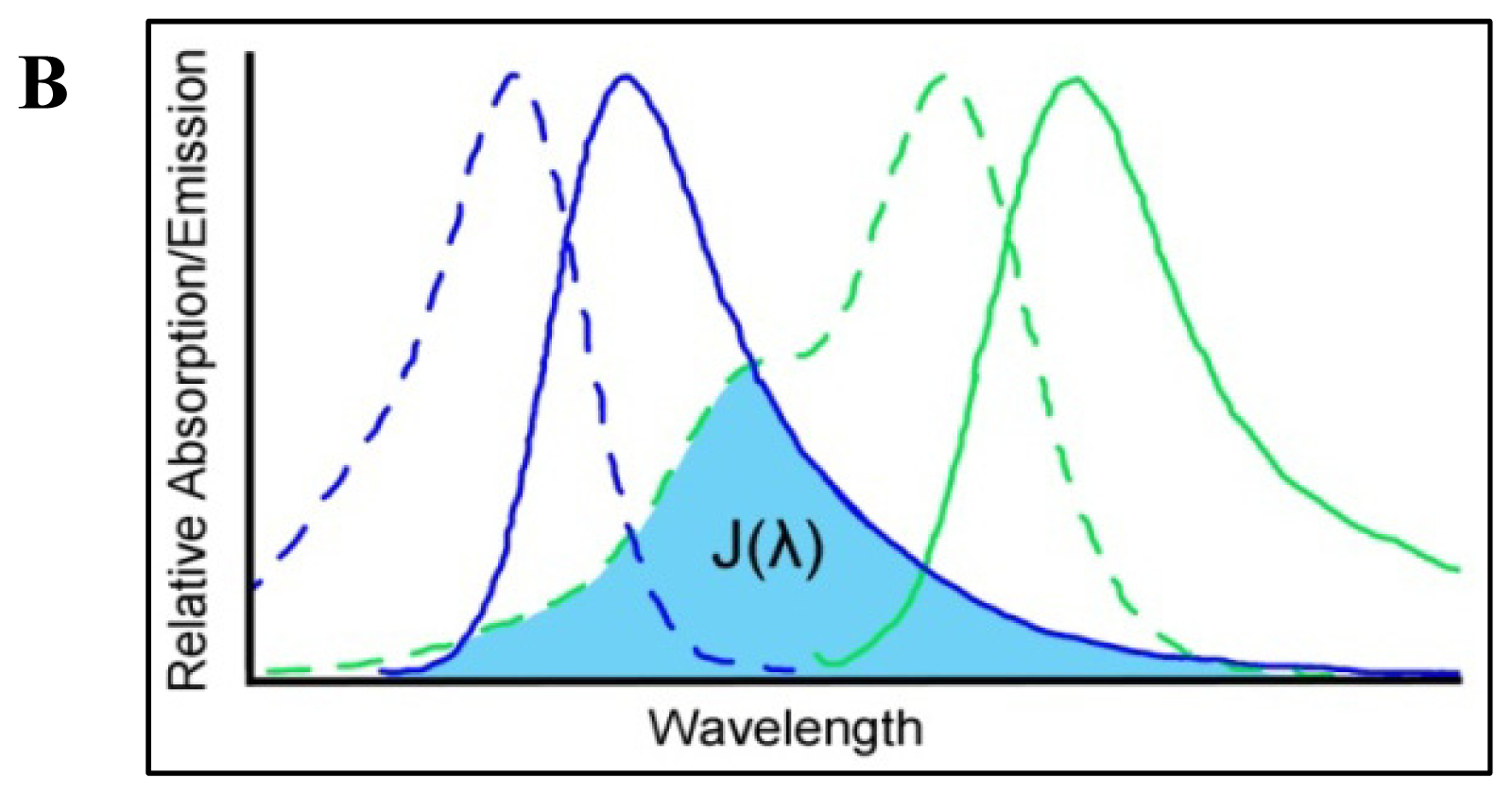
4. Förster Resonance Energy Transfer
4.1. Measuring FRET
4.2. Consideration of FRET Limitations
4.2.1. Donor and Acceptor Labeling
4.2.2. Technical Considerations
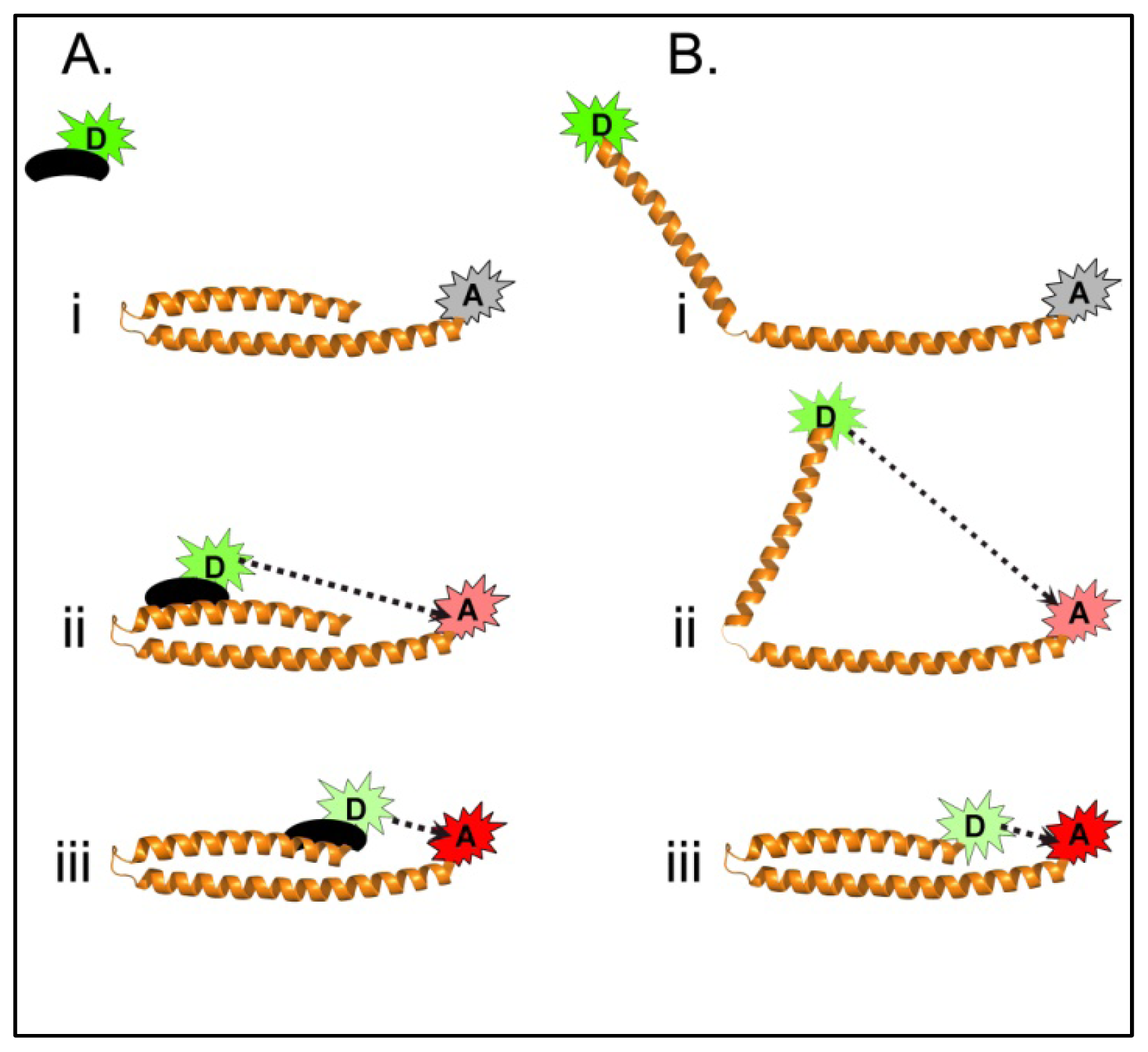
4.3. Using FRET to Dissect Shigella T3SA Maturation
4.3.1. Using FRET to Identify Intermolecular Interactions That Occur During Maturation of the Shigella T3SA Needle Tip Complex
4.3.2. Identifying Ligand Binding Effects on Protein Structure—the Interesting Case of IpaD
4.3.3. Monitoring the Structural States of the Shigella Translocator IpaB with an Intrinsic FRET Donor
4.3.4. Ongoing Studies Involving FRET Analysis of the Shigella T3SA Needle Tip
4.4. The Future of FRET in Bacterial Pathogenesis
5. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Ashida, H.; Ogawa, M.; Mimuro, H.; Kobayashi, T.; Sanada, T.; Sasakawa, C. Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr. Opin. Immunol 2011, 23, 448–455. [Google Scholar]
- Picking, W.L.; Picking, W.D. Early Steps in Inducing Type III Secretion in Shigella. Microbes Infect 2009, 4, 554–559. [Google Scholar]
- Stensrud, K.F.; Adam, P.R.; La Mar, C.D.; Olive, A.J.; Lushington, G.H.; Sudharsan, R.; Shelton, N.L.; Givens, R.S.; Picking, W.L.; Picking, W.D. Deoxycholate interacts with IpaD of Shigella flexneri in inducing the recruitment of IpaB to the type III secretion apparatus needle tip. J. Biol. Chem 2008, 283, 18646–18654. [Google Scholar]
- Dickenson, N.E.; Zhang, L.; Epler, C.R.; Adam, P.R.; Picking, W.L.; Picking, W.D. Conformational changes in IpaD from Shigella flexneri upon binding bile salts provide insight into the second step of type III secretion. Biochemistry 2011, 50, 172–180. [Google Scholar]
- Olive, A.J.; Kenjale, R.; Espina, M.; Moore, D.S.; Picking, W.L.; Picking, W.D. Bile salts stimulate recruitment of IpaB to the Shigella flexneri surface, where it colocalizes with IpaD at the tip of the type III secretion needle. Infect. Immun 2007, 75, 2626–2629. [Google Scholar]
- Adam, P.R.; Patil, M.K.; Dickenson, N.E.; Choudhari, S.; Barta, M.; Geisbrecht, B.V.; Picking, W.L.; Picking, W.D. Binding affects the tertiary and quaternary structures of the Shigella translocator protein IpaB and its chaperone IpgC. Biochemistry 2012, 51, 4062–4071. [Google Scholar]
- Van Teeffelen, S.; Shaevitz, J.W.; Gitai, Z. Image analysis in fluorescence microscopy: Bacterial dynamics as a case study. Bioessays 2012, 34, 427–436. [Google Scholar]
- Margolin, W. Green fluorescent protein as a reporter for macromolecular localization in bacterial cells. Methods 2000, 20, 62–72. [Google Scholar]
- Skoble, J.; Portnoy, D.A.; Welch, M.D. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J. Cell Biol 2000, 150, 527–538. [Google Scholar]
- Zijnge, V.; Ammann, T.; Thurnheer, T.; Gmur, R. Subgingival biofilm structure. Front. Oral. Biol 2012, 15, 1–16. [Google Scholar]
- Kentner, D.; Sourjik, V. Use of fluorescence microscopy to study intracellular signaling in bacteria. Annu. Rev. Microbiol 2010, 64, 373–390. [Google Scholar]
- Van den Wildenberg, S.M.; Bollen, Y.J.; Peterman, E.J. How to quantify protein diffusion in the bacterial membrane. Biopolymers 2011, 95, 312–321. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Kukulski, W.; Schorb, M.; Kaksonen, M.; Briggs, J.A. Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell 2012, 150, 508–520. [Google Scholar]
- Chong, K.; Deng, Y. The three dimensionality of cell membranes: Lamellar to cubic membrane transition as investigated by electron microscopy. Methods Cell Biol 2012, 108, 319–343. [Google Scholar]
- Epler, C.R.; Dickenson, N.E.; Bullitt, E.; Picking, W.L. Ultrastructural analysis of IpaD at the tip of the nascent MxiH type III secretion apparatus of Shigella flexneri. J. Mol. Biol 2012, 420, 29–39. [Google Scholar]
- Muller, S.A.; Aebi, U.; Engel, A. What transmission electron microscopes can visualize now and in the future. J. Struct. Biol 2008, 163, 235–245. [Google Scholar]
- Dedonder, S.E.; Cheng, C.; Willard, L.H.; Boyle, D.L.; Ganta, R.R. Transmission electron microscopy reveals distinct macrophage- and tick cell-specific morphological stages of Ehrlichia chaffeensis. PLoS One 2012, 7, e36749. [Google Scholar]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem 2009, 78, 993–1016. [Google Scholar]
- Leung, B.O.; Chou, K.C. Review of super-resolution fluorescence microscopy for biology. Appl. Spectrosc 2011, 65, 967–980. [Google Scholar]
- Knox, R.S. Forster’s resonance excitation transfer theory: Not just a formula. J. Biomed. Opt 2012, 17, 011003. [Google Scholar]
- Hayward, R.D.; Goguen, J.D.; Leong, J.M. No better time to FRET: Shedding light on host pathogen interactions. J. Biol 2010, 9, 12. [Google Scholar]
- Förster, T. Intermolecular energy migration and fluorescence. Ann. Phys 1948, 2, 55–75. [Google Scholar]
- Hueck, C.J. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev 1998, 62, 379–433. [Google Scholar]
- Pugsley, A.P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev 1993, 57, 50–108. [Google Scholar]
- Desvaux, M.; Parham, N.J.; Henderson, I.R. The autotransporter secretion system. Res. Microbiol 2004, 155, 53–60. [Google Scholar]
- Erhardt, M.; Namba, K.; Hughes, K.T. Bacterial nanomachines: The flagellum and type III injectisome. Cold Spring Harb. Perspect. Biol 2010, 2, a000299. [Google Scholar]
- Marlovits, T.C.; Stebbins, C.E. Type III secretion systems shape up as they ship out. Curr. Opin. Microbiol 2010, 13, 47–52. [Google Scholar]
- Ibuki, T.; Imada, K.; Minamino, T.; Kato, T.; Miyata, T.; Namba, K. Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat. Struct. Mol. biol 2011, 18, 277–282. [Google Scholar]
- Mueller, C.A.; Broz, P.; Cornelis, G.R. The type III secretion system tip complex and translocon. Mol. Microbiol 2008, 68, 1085–1095. [Google Scholar]
- Espina, M.; Olive, A.J.; Kenjale, R.; Moore, D.S.; Ausar, S.F.; Kaminski, R.W.; Oaks, E.V.; Middaugh, C.R.; Picking, W.D.; Picking, W.L. IpaD Localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect. Immun 2006, 74, 4391–4400. [Google Scholar]
- Mueller, C.A.; Broz, P.; Muller, S.A.; Ringler, P.; Erne-Brand, F.; Sorg, I.; Kuhn, M.; Engel, A.; Cornelis, G.R. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 2005, 310, 674–676. [Google Scholar]
- Epler, C.R.; Dickenson, N.E.; Olive, A.J.; Picking, W.L.; Picking, W.D. Liposomes recruit IpaC to the Shigella flexneri type III secretion apparatus needle as a final step in secretion induction. Infect. Immun 2009, 77, 2754–2761. [Google Scholar]
- Blocker, A.; Holden, D.; Cornelis, G. Type III secretion systems: What is the translocator and what is translocated? Cell. Microbiol 2000, 2, 387–390. [Google Scholar]
- Blocker, A.; Gounon, P.; Larquet, E.; Niebuhr, K.; Cabiaux, V.; Parsot, C.; Sansonetti, P. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol 1999, 147, 683–693. [Google Scholar]
- Tran Van Nhieu, G.; Bourdet-Sicard, R.; Dumenil, G.; Blocker, A.; Sansonetti, P.J. Bacterial signals and cell responses during Shigella entry into epithelial cells. Cell. Microbiol 2000, 2, 187–193. [Google Scholar]
- Fujii, T.; Cheung, M.; Blanco, A.; Kato, T.; Blocker, A.J.; Namba, K. Structure of a type III secretion needle at 7-A resolution provides insights into its assembly and signaling mechanisms. Proc. Natl. Acad. Sci. USA 2012, 109, 4461–4466. [Google Scholar]
- Kenjale, R.; Wilson, J.; Zenk, S.F.; Saurya, S.; Picking, W.L.; Picking, W.D.; Blocker, A. The needle component of the type III secreton of Shigella regulates the activity of the secretion apparatus. J. Biol. Chem 2005, 280, 42929–42937. [Google Scholar]
- Blocker, A.; Komoriya, K.; Aizawa, S. Type III secretion systems and bacterial flagella: Insights into their function from structural similarities. Proc. Natl. Acad. Sci. USA 2003, 100, 3027–3030. [Google Scholar]
- Barrett, B.S.; Markham, A.P.; Esfandiary, R.; Picking, W.L.; Picking, W.D.; Joshi, S.B.; Middaugh, C.R. Formulation and immunogenicity studies of type III secretion system needle antigens as vaccine candidates. J Pharm. Sci 2010, 99, 4488–4496. [Google Scholar]
- Deane, J.E.; Roversi, P.; Cordes, F.S.; Johnson, S.; Kenjale, R.; Daniell, S.; Booy, F.; Picking, W.D.; Picking, W.L.; Blocker, A.J.; et al. Molecular model of a type III secretion system needle: Implications for host-cell sensing. Proc. Natl. Acad. Sci. USA 2006, 103, 12529–12533. [Google Scholar]
- Zhang, L.; Wang, Y.; Picking, W.L.; Picking, W.D.; de Guzman, R.N. Solution structure of monomeric BsaL, the type III secretion needle protein of Burkholderia pseudomallei. J. Mol. Biol 2006, 359, 322–330. [Google Scholar]
- Wang, Y.; Ouellette, A.N.; Egan, C.W.; Rathinavelan, T.; Im, W.; de Guzman, R.N. Differences in the electrostatic surfaces of the type III secretion needle proteins PrgI, BsaL, and MxiH. J. Mol. Biol 2007, 371, 1304–1314. [Google Scholar]
- Cordes, F.S.; Komoriya, K.; Larquet, E.; Yang, S.; Egelman, E.H.; Blocker, A.; Lea, S.M. Helical structure of the needle of the type III secretion system of Shigella flexneri. J. Biol. Chem 2003, 278, 17103–17107. [Google Scholar]
- Torruellas, J.; Jackson, M.W.; Pennock, J.W.; Plano, G.V. The Yersinia pestis type III secretion needle plays a role in the regulation of Yop secretion. Mol. Microbiol 2005, 57, 1719–1733. [Google Scholar]
- Barta, M.L.; Guragain, M.; Adam, P.; Dickenson, N.E.; Patil, M.; Geisbrecht, B.V.; Picking, W.L.; Picking, W.D. Identification of the bile salt binding site on IpaD from Shigella flexneri and the influence of ligand binding on IpaD structure. Proteins 2012, 80, 935–945. [Google Scholar]
- Montagner, C.; Arquint, C.; Cornelis, G.R. Translocators YopB and YopD from Yersinia enterocolitica form a multimeric integral membrane complex in eukaryotic cell membranes. J. Bacteriol 2011, 193, 6923–6928. [Google Scholar]
- Sun, Y.; Wallrabe, H.; Seo, S.A.; Periasamy, A. FRET microscopy in 2010: The legacy of Theodor Forster on the 100th anniversary of his birth. Chemphyschem 2011, 12, 462–474. [Google Scholar]
- Kopp, K.; Buntru, A.; Pils, S.; Zimmermann, T.; Frank, R.; Zumbusch, A.; Hauck, C.R. GRB14 is a negative regulator of ceacam3-mediated phagocytosis of pathogenic bacteria. J. Biol. Chem 2012, 287, 39158–39170. [Google Scholar]
- Wong, K.W.; Isberg, R.R. Yersinia pseudotuberculosis spatially controls activation and misregulation of host cell Rac1. PLoS Pathog 2005, 1, e16. [Google Scholar]
- Griffin, B.A.; Adams, S.R.; Tsien, R.Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. [Google Scholar]
- Menard, R.; Sansonetti, P.; Parsot, C.; Vasselon, T. Extracellular association and cytoplasmic partitioning of the IpaB and IpaC invasins of S. flexneri. Cell 1994, 79, 515–525. [Google Scholar]
- Birket, S.E.; Harrington, A.T.; Espina, M.; Smith, N.D.; Terry, C.M.; Darboe, N.; Markham, A.P.; Middaugh, C.R.; Picking, W.L.; Picking, W.D. Preparation and characterization of translocator/chaperone complexes and their component proteins from Shigella flexneri. Biochemistry 2007, 46, 8128–8137. [Google Scholar]
- Barta, M.L.; Dickenson, N.E.; Patil, M.; Keightley, A.; Wyckoff, G.J.; Picking, W.D.; Picking, W.L.; Geisbrecht, B.V. The structures of coiled-coil domains from type III secretion system translocators reveal homology to pore-forming toxins. J. Mol. Biol 2012, 417, 395–405. [Google Scholar]
- Lokareddy, R.K.; Lunelli, M.; Eilers, B.; Wolter, V.; Kolbe, M. Combination of two separate binding domains defines stoichiometry between type III secretion system chaperone IpgC and translocator protein IpaB. J. Biol. Chem 2010, 285, 39965–39975. [Google Scholar]
- Betts-Hampikian, H.J.; Fields, K.A. Disulfide bonding within components of the Chlamydia type III secretion apparatus correlates with development. J. Bacteriol 2011, 193, 6950–6959. [Google Scholar]
- Markham, A.P.; Jaafar, Z.A.; Kemege, K.E.; Middaugh, C.R.; Hefty, P.S. Biophysical characterization of Chlamydia trachomatis CT584 supports its potential role as a type III secretion needle tip protein. Biochemistry 2009, 48, 10353–10361. [Google Scholar]
- Ulrich, R.L.; DeShazer, D. Type III secretion: A virulence factor delivery system essential for the pathogenicity of Burkholderia mallei. Infect. Immun 2004, 72, 1150–1154. [Google Scholar]
- Journet, L.; Hughes, K.T.; Cornelis, G.R. Type III secretion: A secretory pathway serving both motility and virulence (review). Mol. Membr. Biol 2005, 22, 41–50. [Google Scholar]
- Nothelfer, K.; Dias Rodrigues, C.; Bobard, A.; Phalipon, A.; Enninga, J. Monitoring Shigella flexneri vacuolar escape by flow cytometry. Virulence 2011, 2, 54–57. [Google Scholar]
- Simeone, R.; Bobard, A.; Lippmann, J.; Bitter, W.; Majlessi, L.; Brosch, R.; Enninga, J. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 2012, 8, e1002507. [Google Scholar]
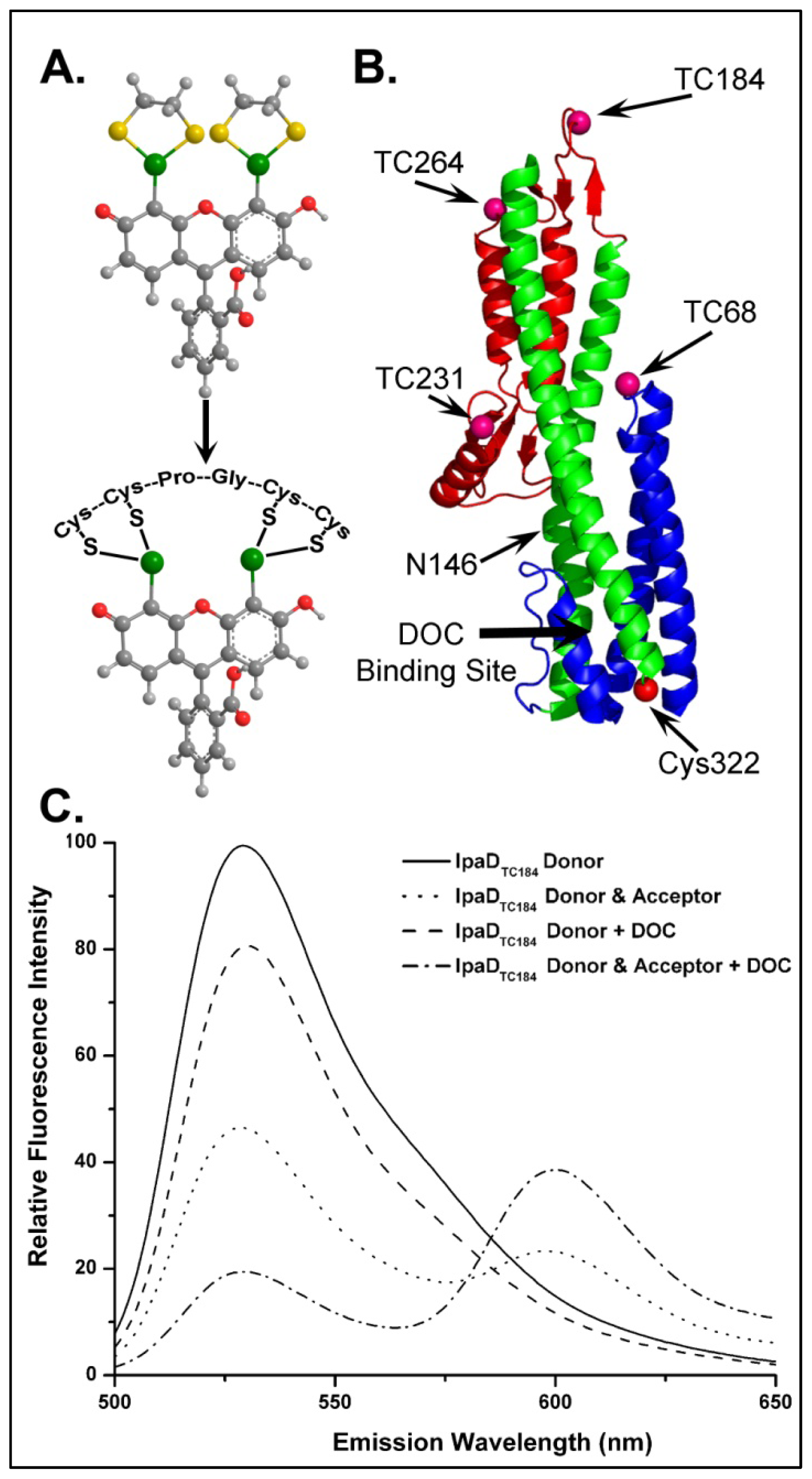
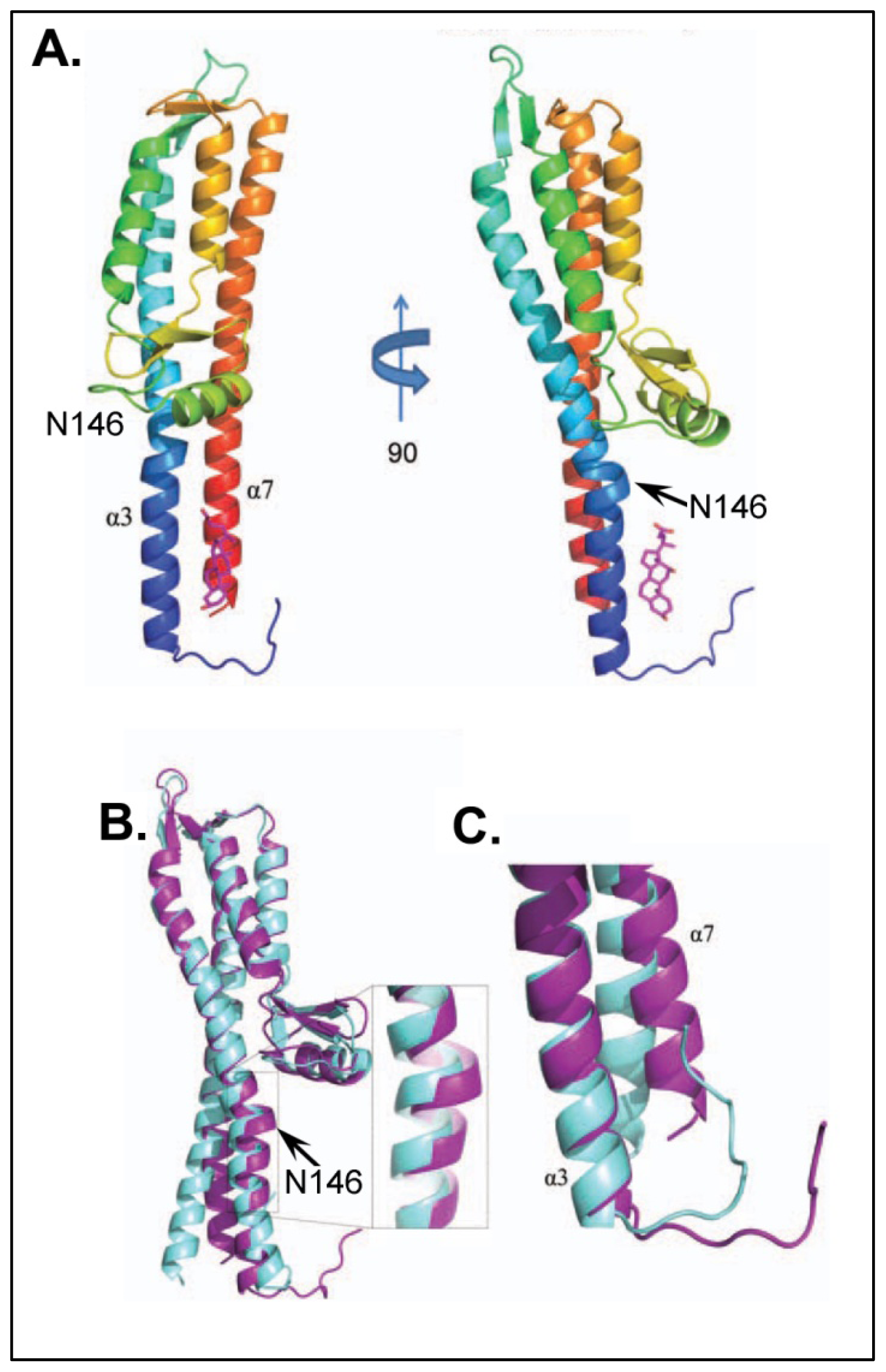


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
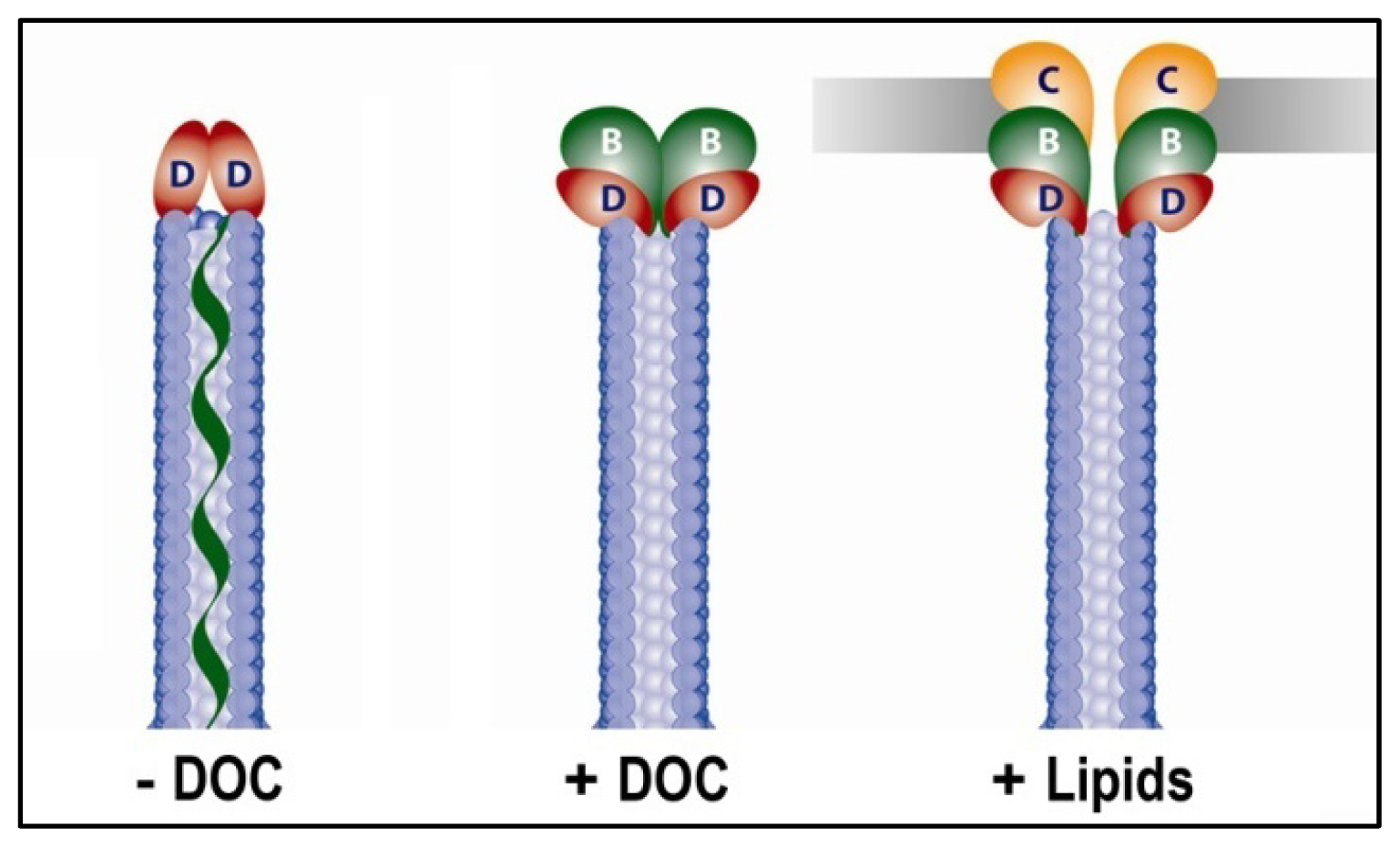{kind=link}
| Protein | No IpgC | 1 ìM IpgC | Är (Å) | ||
|---|---|---|---|---|---|
| FRET a (%) | r (Å) | FRET (%) | r (Å) | ||
| IpaBN28.226 | 47.1 ± 3.2 | 21.4 | 37.9 ± 14.8 | 22.8 | 1.4 |
| IpaBC28.226 | 74.5 ± 5.9 | 17.6 | 46.1 ± 6.8 | 21.6 | 4.0 |
| IpaBN1.226 | 37.6 ± 3.3 | 22.8 | 6.5 ± 3.6 | 32.8 | 10.0 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dickenson, N.E.; Picking, W.D. Förster Resonance Energy Transfer (FRET) as a Tool for Dissecting the Molecular Mechanisms for Maturation of the Shigella Type III Secretion Needle Tip Complex. Int. J. Mol. Sci. 2012, 13, 15137-15161. https://doi.org/10.3390/ijms131115137
Dickenson NE, Picking WD. Förster Resonance Energy Transfer (FRET) as a Tool for Dissecting the Molecular Mechanisms for Maturation of the Shigella Type III Secretion Needle Tip Complex. International Journal of Molecular Sciences. 2012; 13(11):15137-15161. https://doi.org/10.3390/ijms131115137
Chicago/Turabian StyleDickenson, Nicholas E., and William D. Picking. 2012. "Förster Resonance Energy Transfer (FRET) as a Tool for Dissecting the Molecular Mechanisms for Maturation of the Shigella Type III Secretion Needle Tip Complex" International Journal of Molecular Sciences 13, no. 11: 15137-15161. https://doi.org/10.3390/ijms131115137