Comparison between Wild and Hatchery Populations of Korean Pen Shell (Atrina pectinata) Using Microsatellite DNA Markers

Abstract
:1. Introduction
2. Results and Discussion
2.1. Microsatellite Markers Isolation
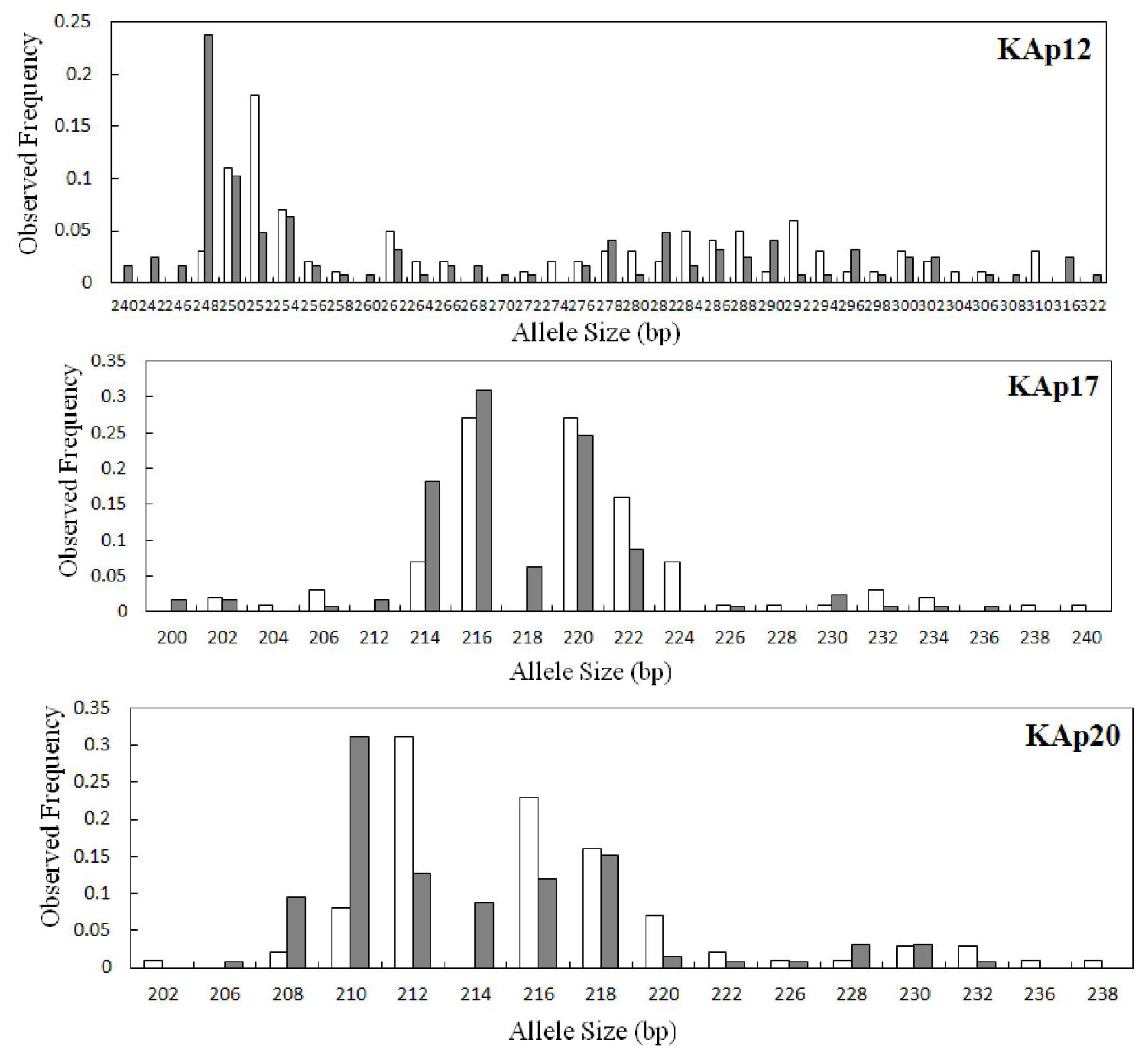
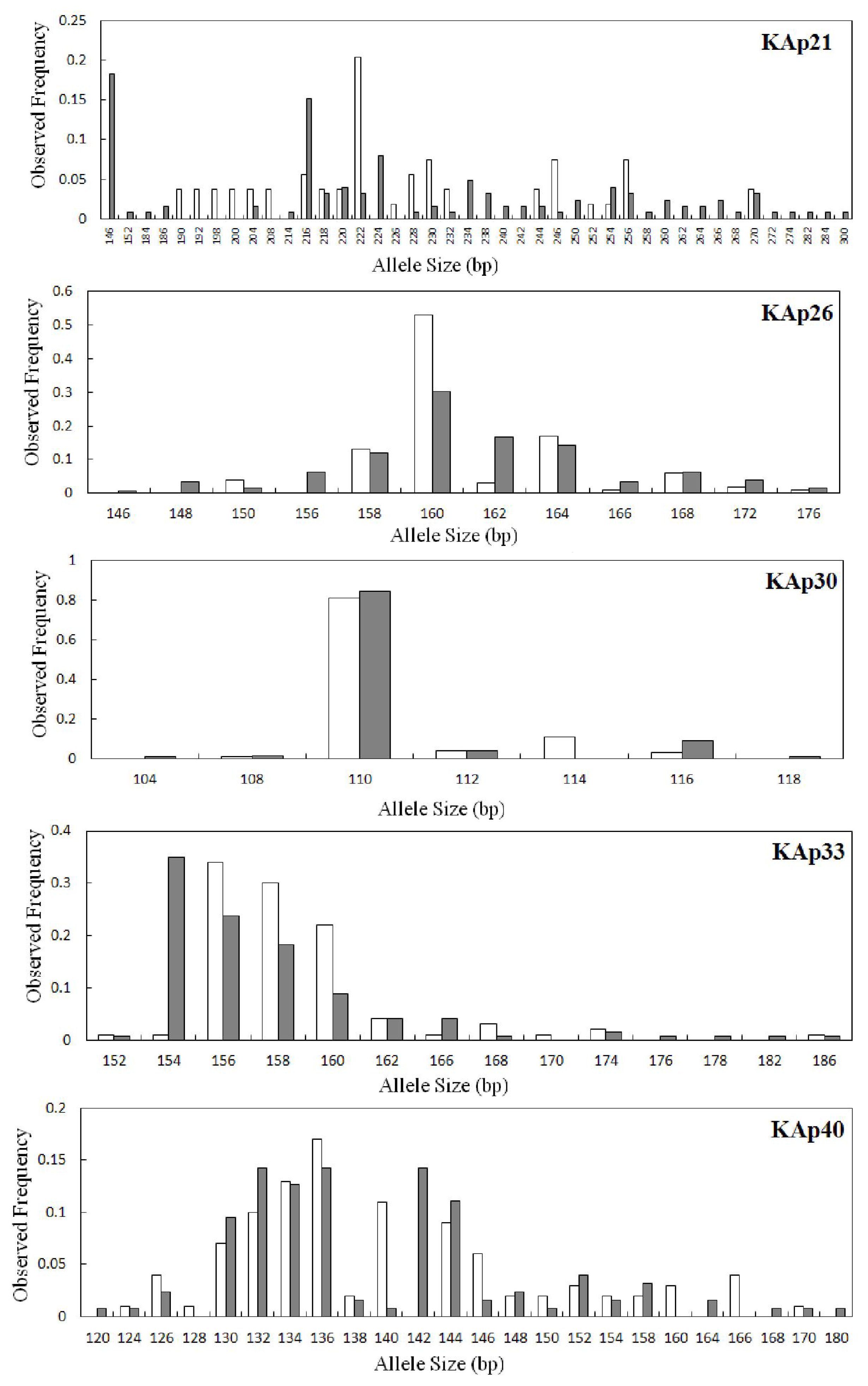
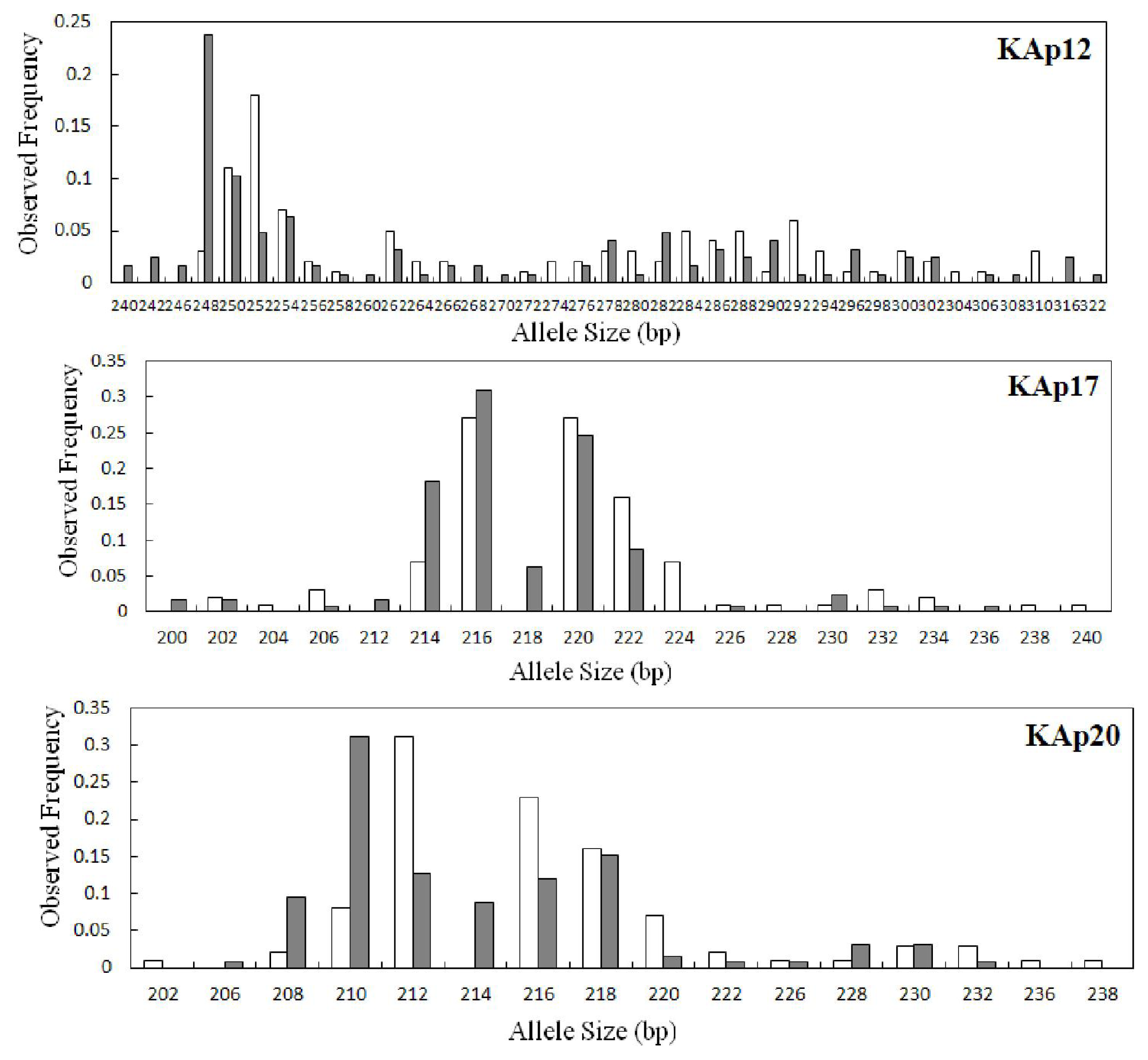
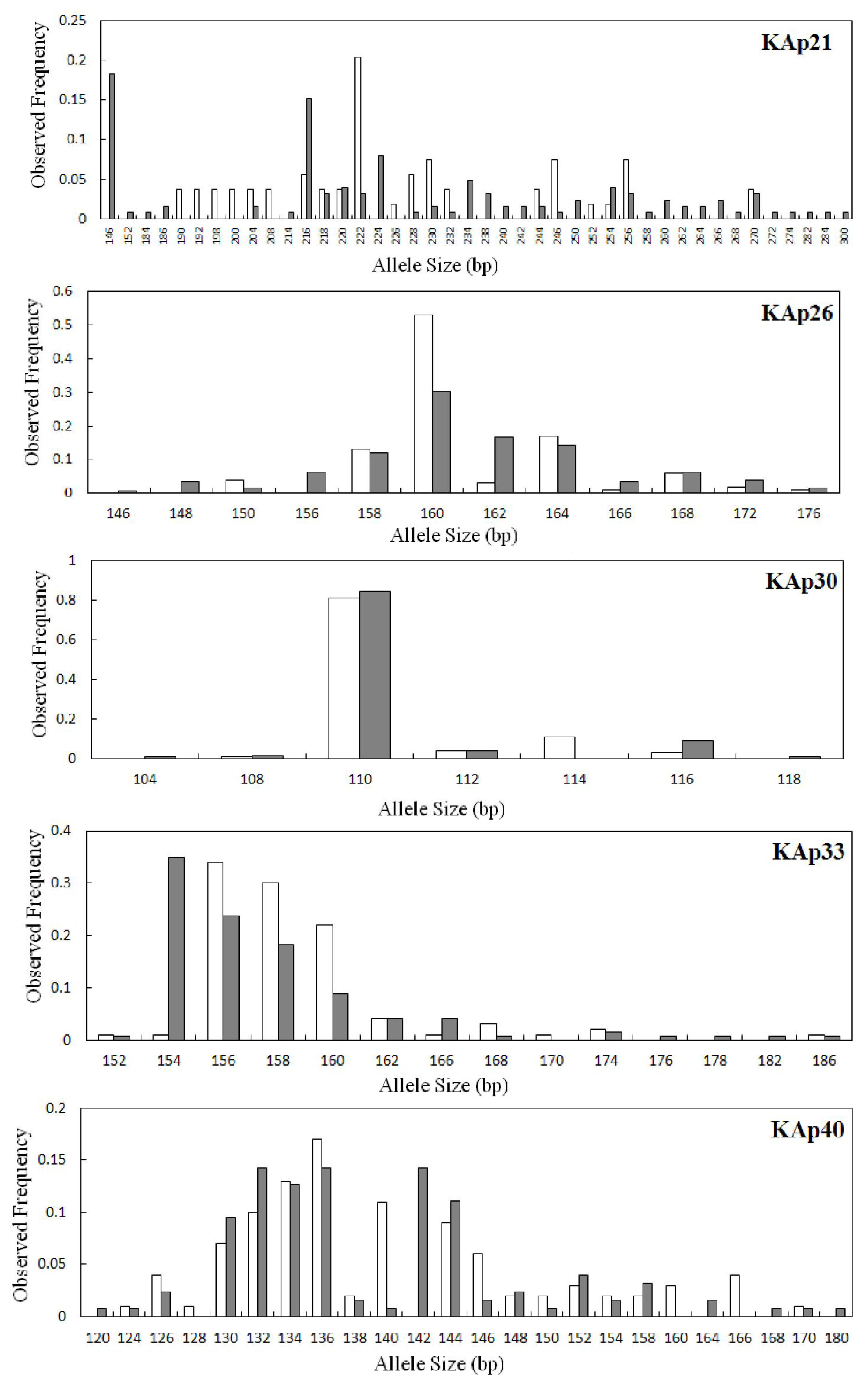
2.2. Genetic Variation within Populations
2.3. Genetic Diversity between the Wild and Hatchery Populations
3. Experimental Section
3.1. Sample Collection and DNA Extraction
3.2. Genomic Library Construction and Microsatellite Sequencing
3.3. Primer Design and Allele Scoring
3.4. Sample Comparisons
4. Conclusions
Acknowledgments
References
- Okutani, T. Basic data on scarcely wild species in Japan–IV. Jpn Fish Resour Conserv Assoc Tokyo 1997, 43–47. [Google Scholar]
- Yurimoto, Y; Watanabe, Y; Nasu, H; Tobase, N; Matsui, S; Yoshioka, N. Relationship between Environmental Food and Glycogen Contents in Pen Shells. Proceedings of 32nd U.S. Japan Symposium on Aquaculture, California, CA, USA, 16–23 November 2003.
- MOMAFK–Ministry of Maritime Affairs and Fisheries of Korea, Annual Statistics of Fisheries Production in 2007; Ministry of Maritime Affairs and Fisheries: Seoul, Korean, 2007. (In Korean)
- Allendorf, FW; Phelps, N. Loss of genetic variation in a hatchery stock of cutthroat trout. Trans Am Fish Soc 1980, 109, 537–543. [Google Scholar]
- Liu, Y; Chen, S; Li, B. Assessing the genetic structure of three Japanese flounder (Paralichthys olivaceus) stocks by microsatellite markers. Aquaculure 2005, 243, 103–111. [Google Scholar]
- Reiss, H; Hoarau, G; Dickey-Collas, M; Wolff, WJ. Genetic population structure of marine fish: mismatch between biological and fisheries management units. Fish Fish 2009, 10, 361–395. [Google Scholar]
- Jerry, DR; Preston, N; Crocos, PJ; Keys, S; Meadows, JRS; Li, Y. Parentage determination of Kuruma prawn shrimp Penaeus (Marsupenaeus) japonicus using microsatellite markers. Aquaculture 2004, 235, 237–247. [Google Scholar]
- Zarraonaindia, I; Pardo, MA; Iriondo, M; Manzano, C; Estonba, A. Microsatellite variability in European anchovy (Engraulis encrasicolus) calls for further investigation of its genetic structure and biogeography. ICES J Mar Sci 2009, 66, 2176–2182. [Google Scholar]
- Hansen, MM; Meier, K; Mensberg, K-LD. Identifying footprints of selection in stocked brown trout populations: a spatio-temporal approach. Mol Ecol 2010, 19, 1787–1800. [Google Scholar]
- An, HS; Lee, JW; Kim, HC; Myeong, J-I. Genetic characterization of five hatchery populations of the Pacific abalone (Haliotis discus hannai) using microsatellite markers. Int J Mol Sci 2011. In Press. [Google Scholar]
- Fu, H; Wu, Y; Gong, Y; Jiang, S; Xiong, Y; Qiao, H; Cheng, L; Tong, J; Li, M. Isolation and characterization of 15 polymorphic microsatellite markers for comb pen shell (Atrina pectinata). Aquac Res 2010, 41, e703–e705. [Google Scholar]
- Liu, J; Li, Q; Kong, L-F. Isolation and characterization of 13 microsatellite loci in the pen shell Atrina pectinata (Bivalvia: Pinnidae). Conserv Genet 2009, 10, 1369–1371. [Google Scholar]
- Hamilton, MB; Pincus, EL; DiFiore, A; Fleischer, RC. Universal linker and ligation procedures for construction of genomic DNA libraries enriched for microsatellites. BioTechniques 1999, 27, 500–507. [Google Scholar]
- Gardner, MG; Cooper, SJB; Bull, CM; Grant, WN. Isolation of microsatellite loci from a social lizard, Egernia stokesii, using a modified enrichment procedure. J Hered 1999, 90, 301–304. [Google Scholar]
- Carleton, KL; Streelman, JT; Lee, BY; Garnhart, N; Kidd, M; Kocher, TD. Rapid isolation of CA microsatellites from the tilapia genome. Anim Genet 2002, 33, 140–144. [Google Scholar]
- Evans, B; Knauer, J; Taylor, J; Jerry, D. Development and characterization of six new microsatellite markers for the silver- or gold-lipped pearl oyster, Pinctada maxima (Pteriidae). Mol Ecol Notes 2006, 6, 835–837. [Google Scholar]
- Slabbert, R; Ruivo, NR; Van den Berg, NC; Lizamore, DL; Roodt-Wilding, R. Isolation and characterization of 63 microsatellite loci for the abalone, Haliotis midae. J World Aquac Soc 2008, 39, 429–435. [Google Scholar]
- Wang, Y; Wang, A; Guo, X. Development and characterization of polymorphic microsatellite markers for the northern quahog Mercenaria mercenaria (Linnaeus, 1758). J Shell Res 2010, 29, 77–82. [Google Scholar]
- Cruz, F; Pérez, M; Presa, P. Distribution and abundance of microsatellites in the genome of bivalves. Gene 2005, 241–247. [Google Scholar]
- Li, G; Hubert, S; Bucklin, K; Ribes, V; Hedgecock, D. Characterization of 79 microsatellite DNA markers in the Pacific oysters Crassostrea gigas. Mol Ecol Notes 2003, 3, 228–232. [Google Scholar]
- An, HS; Park, JY. Ten new highly polymorphic microsatellite loci in the blood clam Scapharca broughtonii. Mol Ecol Notes 2005, 5, 896–898. [Google Scholar]
- Kenchington, EL; Patwary, MU; Zouros, E; Bird, CJ. Genetic differentiation in relation to marine landscape in a broadcast-spawning bivalve mollusc (Placopecten magellanicus). Mol Ecol 2006, 15, 1781–1796. [Google Scholar]
- Launey, S; Hedgecock, D. High genetic load in the Pacific oyster Crassostrea gigas. Genetics 2001, 159, 255–265. [Google Scholar]
- Rice, WR. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar]
- Xu, ZK; Primavera, JH; dela Pena, LD; Belak, J; Alcivar-Warren, A. Genetic diversity of wild and cultured Black tiger Shrimp (Penaeus monodon) in the Philippines using microsatellites. Aquaculture 2001, 199, 13–40. [Google Scholar]
- Evans, B; Bartlett, J; Sweijd, N; Cook, P; Elliott, NG. Loss of genetic variation at microsatellite loci in hatchery produced abalone in Australia (Haliotis rubra) and South Africa (Haliotis midae). Aquaculture 2004, 233, 109–127. [Google Scholar]
- Zhang, Q; Allen, SK, Jr; Reece, KS. Genetic variation in wild and hatchery stocks of Suminoe oyster (Crassostrea ariakensis) assessed by PCR-RFLP and microsatellite markers. Mar Biotechnol 2005, 7, 588–599. [Google Scholar]
- Li, Q; Yu, H; Yu, RH. Genetic variability assessed by microsatellites in cultured populations of the Pacific oyster (Crassostrea gigas) in China. Aquaculture 2006, 259, 95–102. [Google Scholar]
- Callen, DF; Thompson, AD; Shen, Y; Phillips, HA; Mulley, JC; Sutherland, GR. Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am J Hum Genet 1993, 52, 922–927. [Google Scholar]
- Zhan, AB; Bao, ZM; Hu, XL; Hui, M; Wang, ML; Peng, W; Zhao, HB; Hu, JJ. Isolation and characterization of 150 novel microsatellite markers for Zhikong scallop (Chlamys farreri). Mol Ecol Notes 2007, 7, 1015–1022. [Google Scholar]
- Hansen, MM. Estimating the long-term effects of stocking domesticated trout into wild brown trout (Salmo trutta) populations: An approach using microsatellite DNA analysis of historical and contemporary samples. Mol Ecol 2002, 11, 1003–1015. [Google Scholar]
- Li, Q; Park, C; Endo, T; Kijima, A. Loss of genetic variation at microsatellite loci in hatchery strains of the Pacific abalone (Haliotis discus hannai). Aquaculture 2004, 235, 207–222. [Google Scholar]
- Hara, M; Sekino, M. Genetic differences between hatchery stocks and natural populations in Pacific Abalone (Haliotis discus) estimated using microsatellite DNA markers. Mar Biotechnol 2007, 9, 74–81. [Google Scholar]
- Asahida, T; Kobayashi, T; Saitoh, K; Nakayama, I. Tissue preservation and total DNA extraction from fish stored at ambient temperature using buffers containing high concentrations of urea. Fish Sci Tokyo 1996, 62, 727–730. [Google Scholar]
- Li, Q; Park, C; Kijima, A. Isolation and characterization of microsatellite loci in the Pacific abalone, Haliotis discus hannai. J Shell Res 2002, 212, 811–815. [Google Scholar]
- van Herwerden, L; Choat, JH; Dudgeon, CL; Carlos, G; Newman, SJ; Frisch, A; van Oppen, M. Contrasting patterns of genetic structure in two species of the coral trout Pletropomus (Serranidae) from east and west Australia: introgressive hybridization or ancestral polymorphisms. Mol Phylogenet Evol 2006, 41, 420–435. [Google Scholar]
- Weir, BS; Cockerham, CC. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- El Mousadik, A; Petit, RJ. High level of genetic differentiation for allelic richness among populations of the argan tree (Argania spinosa (L.) Skeels) endemic to Morocco. Theor Appl Genet 1996, 92, 832–839. [Google Scholar]
- Excoffier, L; Smouse, PE; Quattro, JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Excoffier, L; Laval, G; Schneider, S. ARLEQUIN version 3.0: An integrated software package for population genetics data analysis. Evol Bioinf 2005, 1, 47–50. [Google Scholar]
- Slatkin, M; Excoffier, L. Testing for linkage disequilibrium in genotypic data using the EM algorithm. Heredity 1996, 76, 377–383. [Google Scholar]
- Cornuet, JM; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]

{kind=link}
{kind=link}
| Locus | Repeat Motif | Primer Sequence (5′→3′) | Ta (°C) | Genebank Accession No. |
|---|---|---|---|---|
| KAp2 | (GT)12 | F: CAATGTTGATGATGGATGTTA ned R: GCTTCATGTGGGTTTGG | 60 | EU026353 |
| KAp6 | (GT)12 | F: CAGCTACCAAAACCATAAATC hex R: CCAGTCAGCTTGAGTTACAGA | 60 | EU026354 |
| KAp9 | (GT)9 | F: AATGGTACAGTTGTACAGCAC ned R: CAAGCATATTTGTCATTTGAT | 55 | EU026355 |
| KAp11 | (CA)12 | F: GCTGTTGGAAATACGGACTAC 6-fam R: CCACCAAAATTCGGTAAAA | 55 | EU026356 |
| KAp12 | (GT)13 | F: CGATCCTATCCAAGGGTTAT 6-fam R: CTCTATCCGTTCTCCATTTCT | 55 | EU026357 |
| KAp17 | (CA)11 | F: GCAAGGCAAAATGTATTACC 6-fam R: CGAGTACTTGCCGTAGTGAC | 55 | EU026358 |
| KAp18 | (CA)12 | F: TTGGAAATACGGACTACTCA hex R: CCTTGACGTGACCACTTA | 57 | EU026359 |
| KAp19 | (GT)15 | F: CAAAATCCAGGAGTATCTCA hex R: TTCCCTAGAGTGCATAAACT | 55 | EU026360 |
| KAp20 | (GT)11 | F: ACCGTAGTGACACTGAAGGA hex R: GAGGCAAGGCAAAATGTAT | 55 | EU026361 |
| KAp21 | (GT)15 | F: GGAATCATTCTCGCAATA 6-fam R: GAAGCACGTATCATCACTAA | 55 | EU026362 |
| KAp23 | (CA)11 | F: ATCAAGTCATTGCCACAC ned R: AGAAGCACTTGCCGTAG | 57 | EU026363 |
| KAp24 | (GT)13 | F: CCGTGCTGTGGTAATGTA hex R: TTGGCATAAATAGAAAGGTT | 52 | EU026364 |
| KAp25 | (CA)22 | F: CGCGTTCGACTCTTGA ned R: CAAAATTTGGCCTATGCT | 52 | EU026365 |
| KAp26 | (GT)10 | F: GGCGTGTCTATACTTGAACT 6-fam R: TGTCACTTGCGTCACTTTA | 55 | EU026366 |
| KAp30 | (GT)11 | F: TTGAACAAAGACTTGTCA ned R: TCACGTTGAGACTTCATA | 52 | EU026367 |
| KAp32 | (CA)13 | F: TCATCATGTGGCTGTATA ned R: CTGCAGTTGCTTGAAG | 55 | EU026368 |
| KAp33 | (GT)14AA(GT)4 | F: CCACCTGACTGTCTCTGA hex R: ACAGAATCTCGCCTAAAG | 52 | EU026369 |
| KAp38 | (GT)CT(GT)13 | F: CGCCTACATTTAGTCAGT ned R: CTTGGCTTGTACCATATC | 50 | EU026370 |
| KAp39 | (GT)21T(GT) | F: CCGACGTATTATTAGTGC 6-fam R: GCGTAACCCATGTATTAA | 50 | EU026371 |
| KAp40 | (GT)10TT(GT)4 | F: CCATTCACCAAGAGGTTG ned R: CCGTGCGTTGTCGTAC | 50 | EU026372 |
| KAp42 | (GA)3(GT)8 | F: GATAGGCTTCCGTTGTTCTA hex R: AGCCTATGTTCCACGAGAC | 55 | EU026373 |
| KAp43 | (TG)8 | F: AGCTGTTTCACTCTCATTT 6-fam R: AGAATTTTAACCACAACCTT | 52 | EU026374 |
| Microsatellite loci | Fst | Population (No)
| |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Jangheung Wild (63) | Jangheung Hatchery (50) | ||||||||||||||||||
| NA | AR | S | F | U | He | Ho | Fis | P | NA | AR | S | F | U | He | Ho | Fis | P | ||
| KAp2 | −0.0003 | 18 | 16.41 | 132–172 | 0.175 | 3 | 0.908 | 0.889 | 0.021 | 0.626 | 19 | 19.00 | 130–180 | 0.139 | 4 | 0.921 | 0.889 | 0.036 | 0.688 |
| KAp6 | 0.0044 | 13 | 12.45 | 150–186 | 0.286 | 2 | 0.818 | 0.730 | 0.108 | 0.496 | 15 | 15.00 | 146–178 | 0.330 | 4 | 0.823 | 0.820 | 0.003 | 0.658 |
| KAp9 | −0.0045 | 17 | 15.5 | 100–150 | 0.278 | 7 | 0.849 | 0.794 | 0.066 | 0.702 | 14 | 14.00 | 100–138 | 0.330 | 4 | 0.823 | 0.820 | 0.004 | 0.067 |
| KAp11 | −0.0015 | 21 | 20.32 | 218–262 | 0.119 | 2 | 0.942 | 0.937 | 0.005 | 0.012 | 23 | 23.00 | 216–266 | 0.100 | 4 | 0.949 | 0.960 | −0.011 | 0.500 |
| KAp12 | 0.0294 | 34 | 31.19 | 240–322 | 0.238 | 9 | 0.920 | 0.873 | 0.051 | 0.564 | 28 | 28.00 | 248–310 | 0.180 | 3 | 0.938 | 0.840 | 0.106 | 0.000 |
| KAp17 | 0.0102 | 14 | 12.84 | 200–236 | 0.310 | 4 | 0.803 | 0.714 | 0.112 | 0.214 | 15 | 15.00 | 202–240 | 0.270 | 5 | 0.824 | 0.880 | −0.069 | 0.289 |
| KAp18 | 0.0038 | 22 | 21.28 | 138–182 | 0.111 | 1 | 0.945 | 0.937 | 0.009 | 0.000 | 23 | 23.00 | 140–184 | 0.120 | 2 | 0.947 | 0.940 | 0.007 | 0.427 |
| KAp19 | 0.0037 | 29 | 27.33 | 208–266 | 0.079 | 5 | 0.955 | 0.937 | 0.020 | 0.398 | 23 | 23.00 | 208–264 | 0.130 | 0 | 0.935 | 0.920 | 0.016 | 0.011 |
| KAp20 | 0.0576 | 13 | 12.13 | 206–232 | 0.310 | 2 | 0.839 | 0.746 | 0.111 | 0.173 | 14 | 14.00 | 202–238 | 0.310 | 3 | 0.819 | 0.680 | 0.171 | 0.175 |
| KAp21 | 0.0422 | 35 | 32.11 | 146–300 | 0.183 | 22 | 0.930 | 0.476 | 0.490 | 0.000 | 20 | 20.00 | 190–270 | 0.204 | 7 | 0.937 | 0.111 | 0.883 | 0.000 |
| KAp23 | 0.0131 | 12 | 11.29 | 116–160 | 0.254 | 3 | 0.817 | 0.349 | 0.574 | 0.000 | 11 | 11.00 | 116–140 | 0.380 | 2 | 0.778 | 0.560 | 0.283 | 0.001 |
| KAp26 | 0.0404 | 12 | 11.71 | 146–176 | 0.302 | 3 | 0.841 | 0.524 | 0.379 | 0.000 | 9 | 9.00 | 150–176 | 0.530 | 0 | 0.673 | 0.460 | 0.319 | 0.010 |
| KAp30 | 0.0177 | 6 | 5.55 | 104–118 | 0.841 | 2 | 0.285 | 0.222 | 0.222 | 0.083 | 5 | 5.00 | 108–116 | 0.810 | 1 | 0.333 | 0.380 | −0.144 | 1.000 |
| KAp32 | 0.0049 | 23 | 21.43 | 126–208 | 0.206 | 5 | 0.917 | 0.825 | 0.101 | 0.625 | 26 | 26.00 | 126–196 | 0.190 | 8 | 0.924 | 0.740 | 0.201 | 0.000 |
| KAp33 | 0.0861 | 13 | 11.72 | 152–186 | 0.349 | 3 | 0.783 | 0.571 | 0.272 | 0.000 | 11 | 11.00 | 152–186 | 0.340 | 1 | 0.750 | 0.440 | 0.416 | 0.000 |
| KAp38 | 0.0077 | 32 | 29.28 | 80–146 | 0.246 | 10 | 0.915 | 0.873 | 0.047 | 0.373 | 24 | 24.00 | 78–144 | 0.200 | 2 | 0.906 | 0.840 | 0.074 | 0.072 |
| KAp39 | 0.0041 | 19 | 17.24 | 186–248 | 0.444 | 5 | 0.760 | 0.778 | −0.023 | 0.762 | 20 | 20.00 | 184–244 | 0.350 | 6 | 0.811 | 0.700 | 0.138 | 0.024 |
| KAp40 | 0.0130 | 21 | 19.37 | 120–180 | 0.143 | 5 | 0.903 | 0.730 | 0.193 | 0.000 | 19 | 19.00 | 124–170 | 0.170 | 3 | 0.917 | 0.700 | 0.239 | 0.000 |
| KAp42 | −0.0040 | 5 | 4.59 | 184–200 | 0.659 | 1 | 0.497 | 0.397 | 0.203 | 0.049 | 6 | 6.00 | 172–202 | 0.610 | 2 | 0.534 | 0.500 | 0.065 | 0.594 |
| KAp43 | 0.0027 | 15 | 14.46 | 224–274 | 0.254 | 2 | 0.875 | 0.476 | 0.458 | 0.000 | 16 | 16.00 | 226–268 | 0.210 | 3 | 0.900 | 0.500 | 0.447 | 0.000 |
| Mean | 0.0162 | 18.7 | 17.41 | 0.289 | 4.8 | 0.825 | 0.689 | 17.05 | 17.05 | 0.295 | 3.2 | 0.822 | 0.684 | ||||||
| Locus | P-value | Locus | P-value |
|---|---|---|---|
| KAp2 | 0.383 | KAp23 | 0.012 |
| KAp6 | 0.083 | KAp26 | 0.000* |
| KAp9 | 0.695 | KAp30 | 0.001* |
| KAp11 | 0.575 | KAp32 | 0.040 |
| KAp12 | 0.001* | KAp33 | 0.000* |
| KAp17 | 0.000* | KAp38 | 0.292 |
| KAp18 | 0.191 | KAp39 | 0.486 |
| KAp19 | 0.259 | KAp40 | 0.000* |
| KAp20 | 0.000* | KAp41 | 0.797 |
| KAp21 | 0.000* | KAp42 | 0.602 |
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation (%) | P-Value |
|---|---|---|---|---|
| Among Populations | 11.881 | 0.524 | 1.059 | 0.006 |
| Among individuals with population | 663 | 0 | 5.668 | 0.000 |
| Within individuals | 602.000 | 5.383 | 93.323 | 0.000 |
| Total | 1277.273 | 5.761 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suck An, H.; Kim, B.H.; Lee, J.W.; Dong, C.M.; Kim, S.K.; Kim, Y.C. Comparison between Wild and Hatchery Populations of Korean Pen Shell (Atrina pectinata) Using Microsatellite DNA Markers. Int. J. Mol. Sci. 2011, 12, 6024-6039. https://doi.org/10.3390/ijms12096024
Suck An H, Kim BH, Lee JW, Dong CM, Kim SK, Kim YC. Comparison between Wild and Hatchery Populations of Korean Pen Shell (Atrina pectinata) Using Microsatellite DNA Markers. International Journal of Molecular Sciences. 2011; 12(9):6024-6039. https://doi.org/10.3390/ijms12096024
Chicago/Turabian StyleSuck An, Hye, Byeong Hak Kim, Jang Wook Lee, Chun Mae Dong, Shin Kwon Kim, and Yi Cheong Kim. 2011. "Comparison between Wild and Hatchery Populations of Korean Pen Shell (Atrina pectinata) Using Microsatellite DNA Markers" International Journal of Molecular Sciences 12, no. 9: 6024-6039. https://doi.org/10.3390/ijms12096024
APA StyleSuck An, H., Kim, B. H., Lee, J. W., Dong, C. M., Kim, S. K., & Kim, Y. C. (2011). Comparison between Wild and Hatchery Populations of Korean Pen Shell (Atrina pectinata) Using Microsatellite DNA Markers. International Journal of Molecular Sciences, 12(9), 6024-6039. https://doi.org/10.3390/ijms12096024