Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists

Abstract
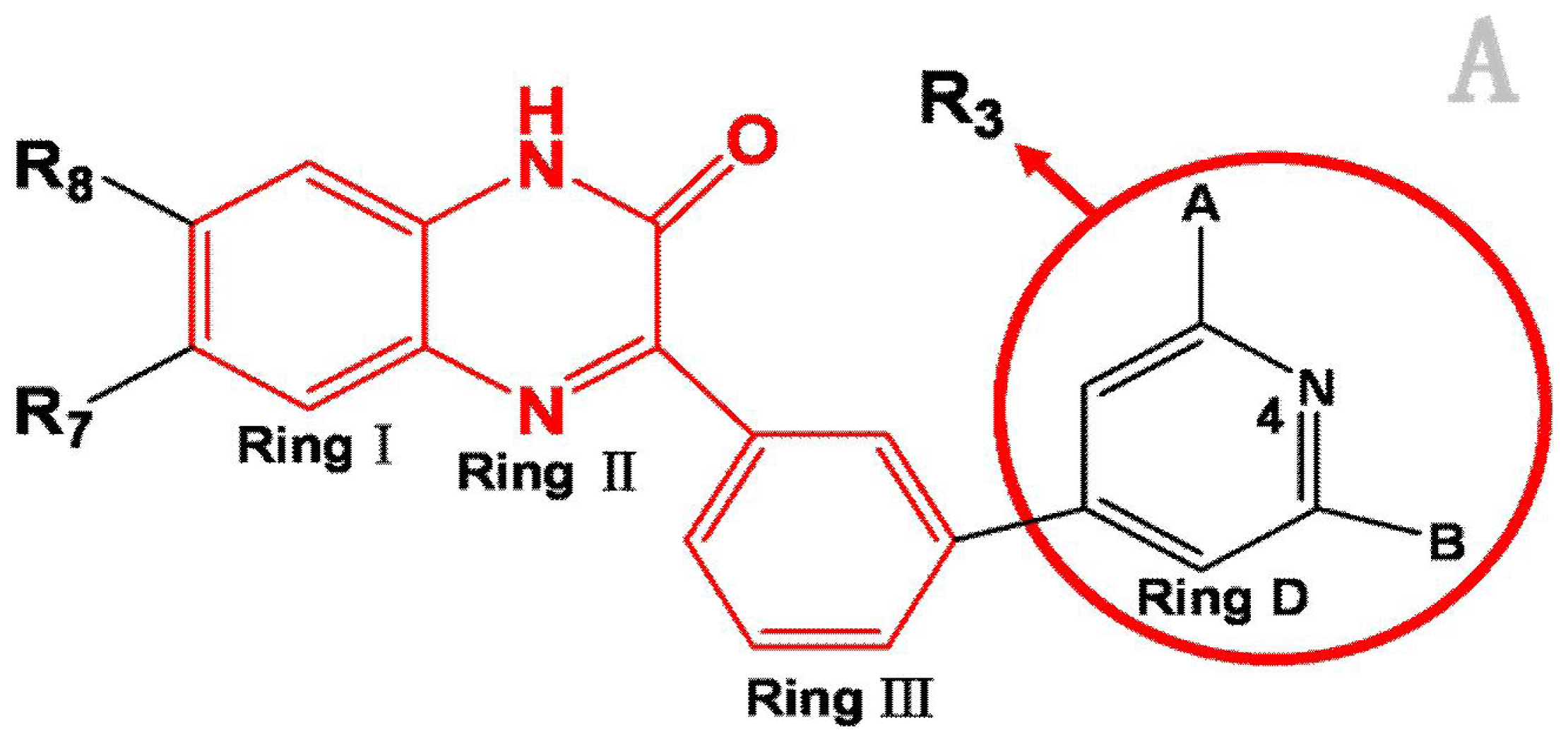
:1. Introduction
2. Results and Discussion
2.1. Results for Activity I
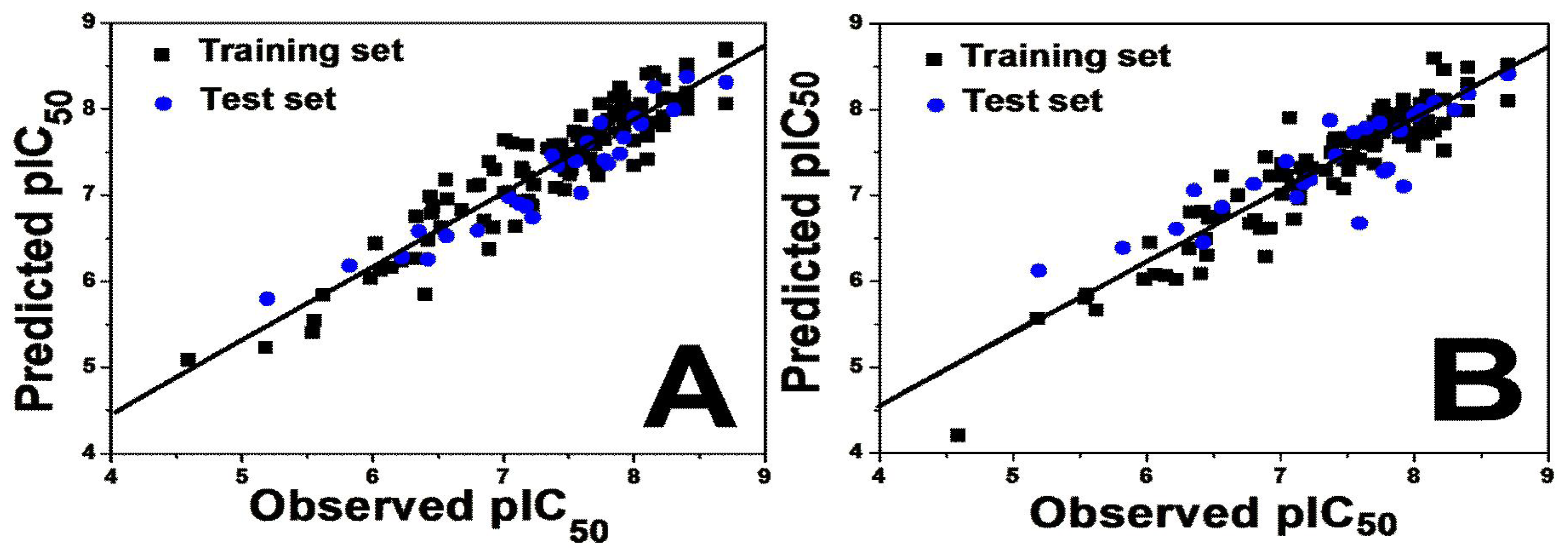
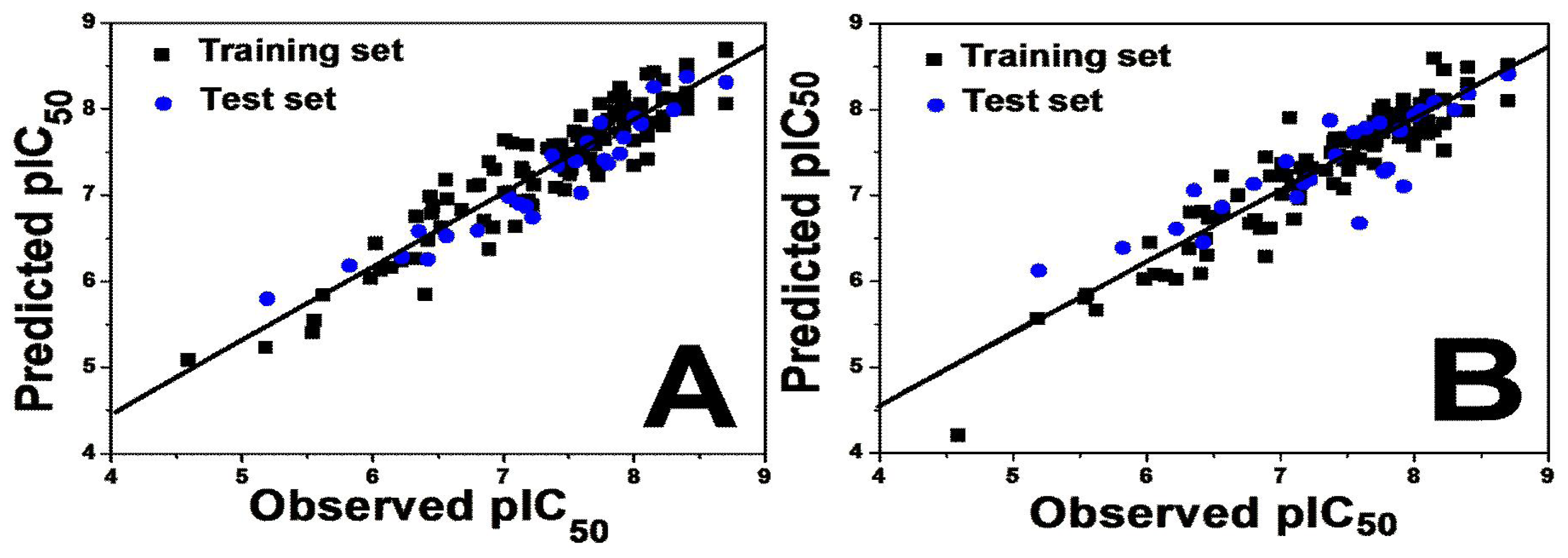
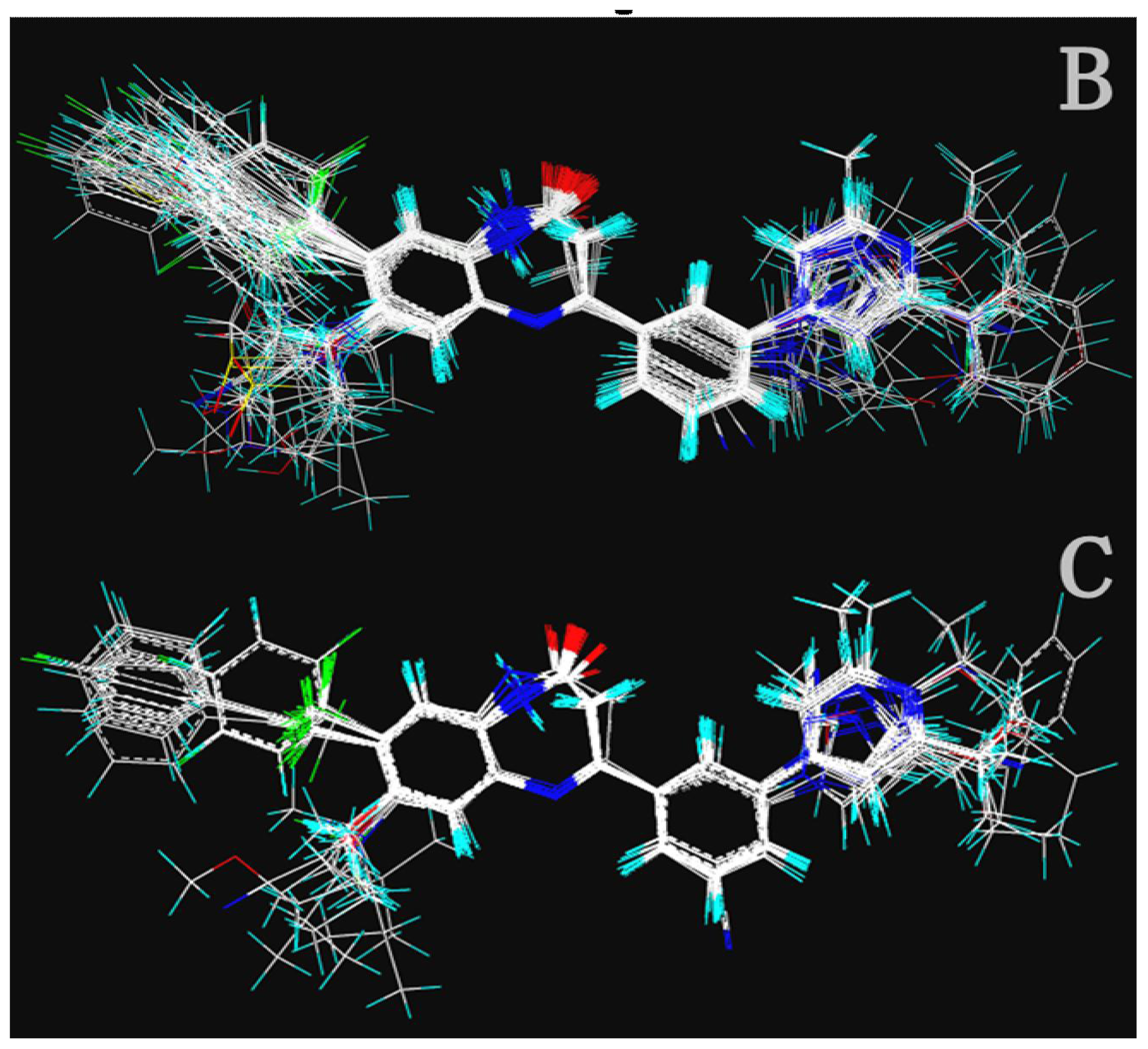
2.1.1. 3D-QSAR Statistical Results
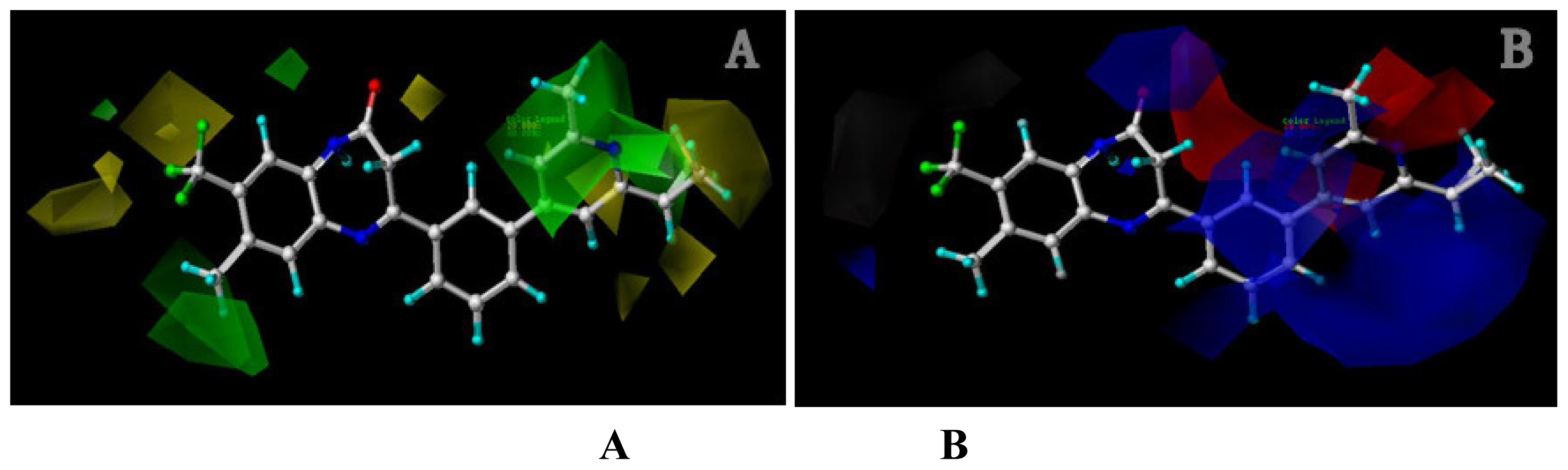
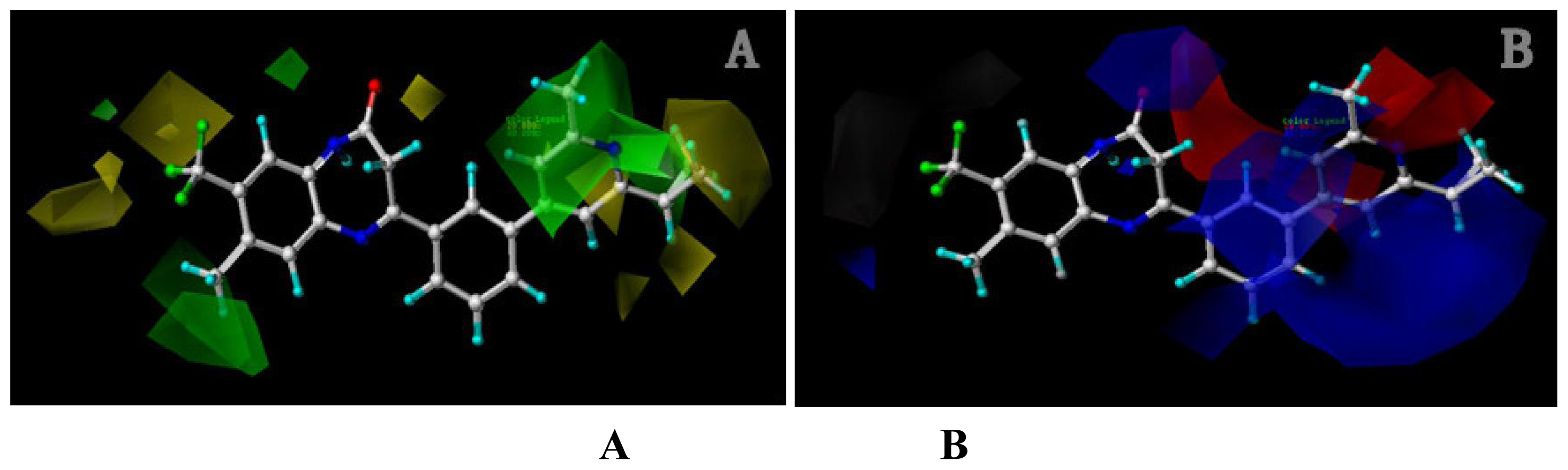
2.1.2. Contour Maps for Activity I
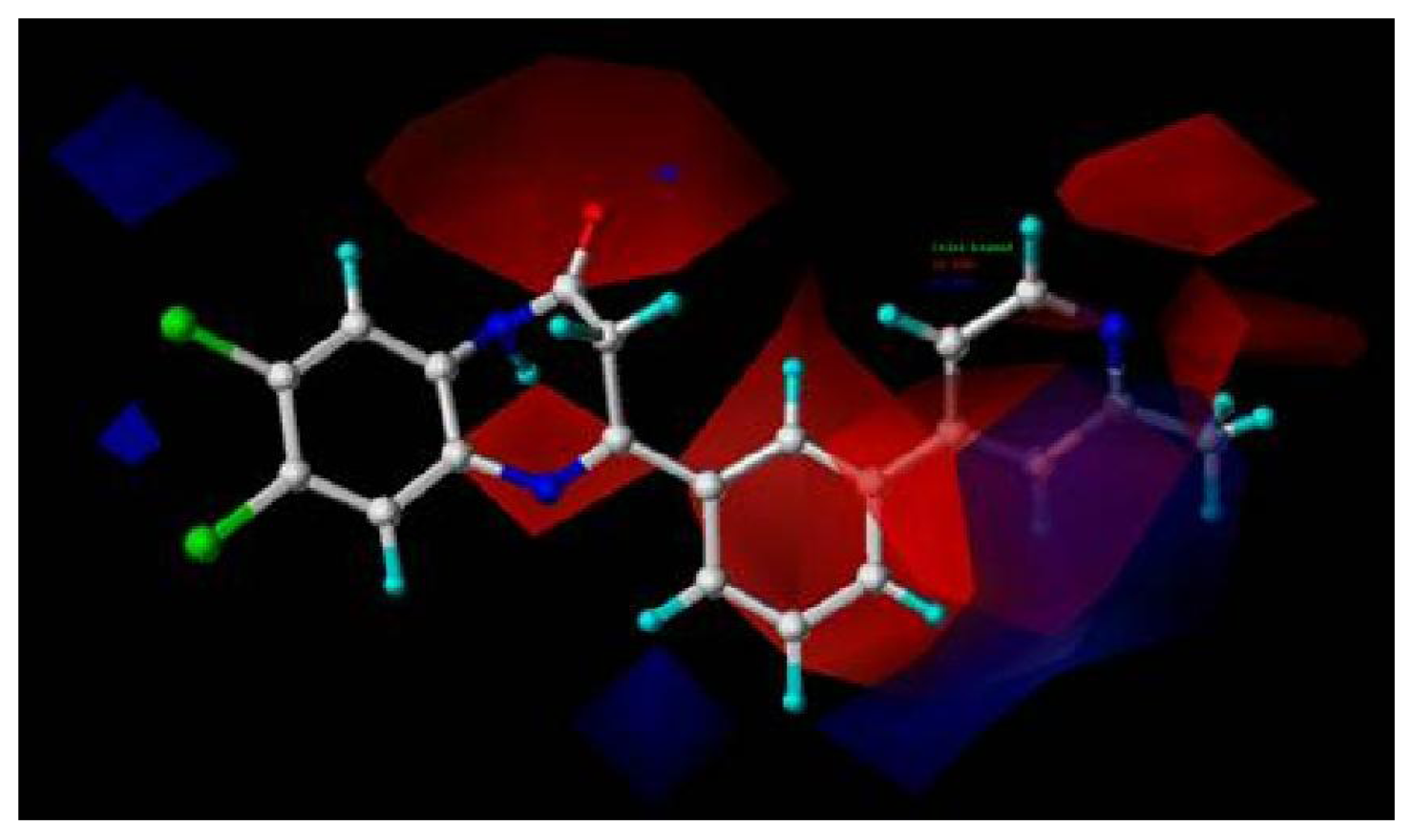
2.1.2.1. CoMFA Contour Maps
2.1.2.2. CoMSIA Contour Maps
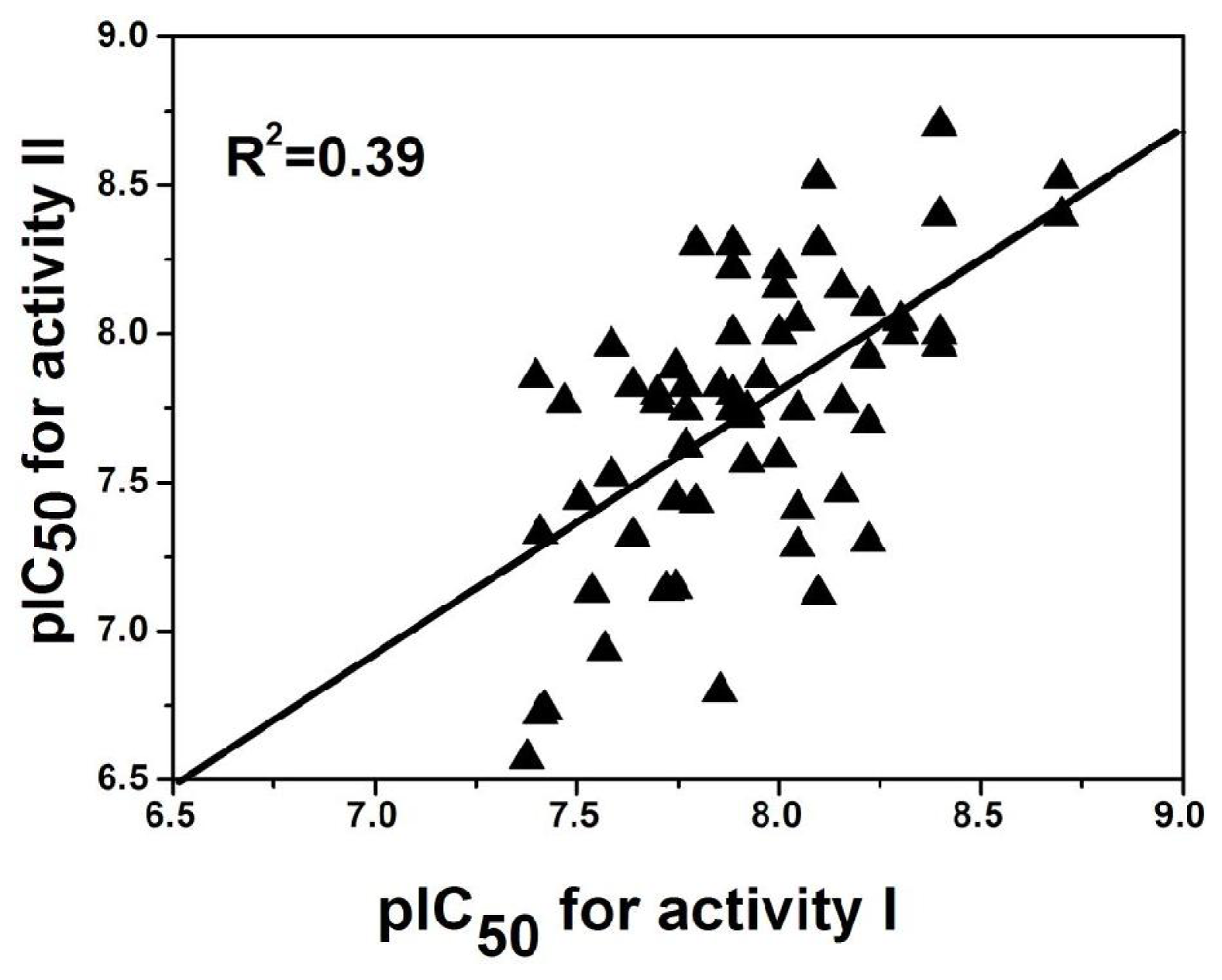
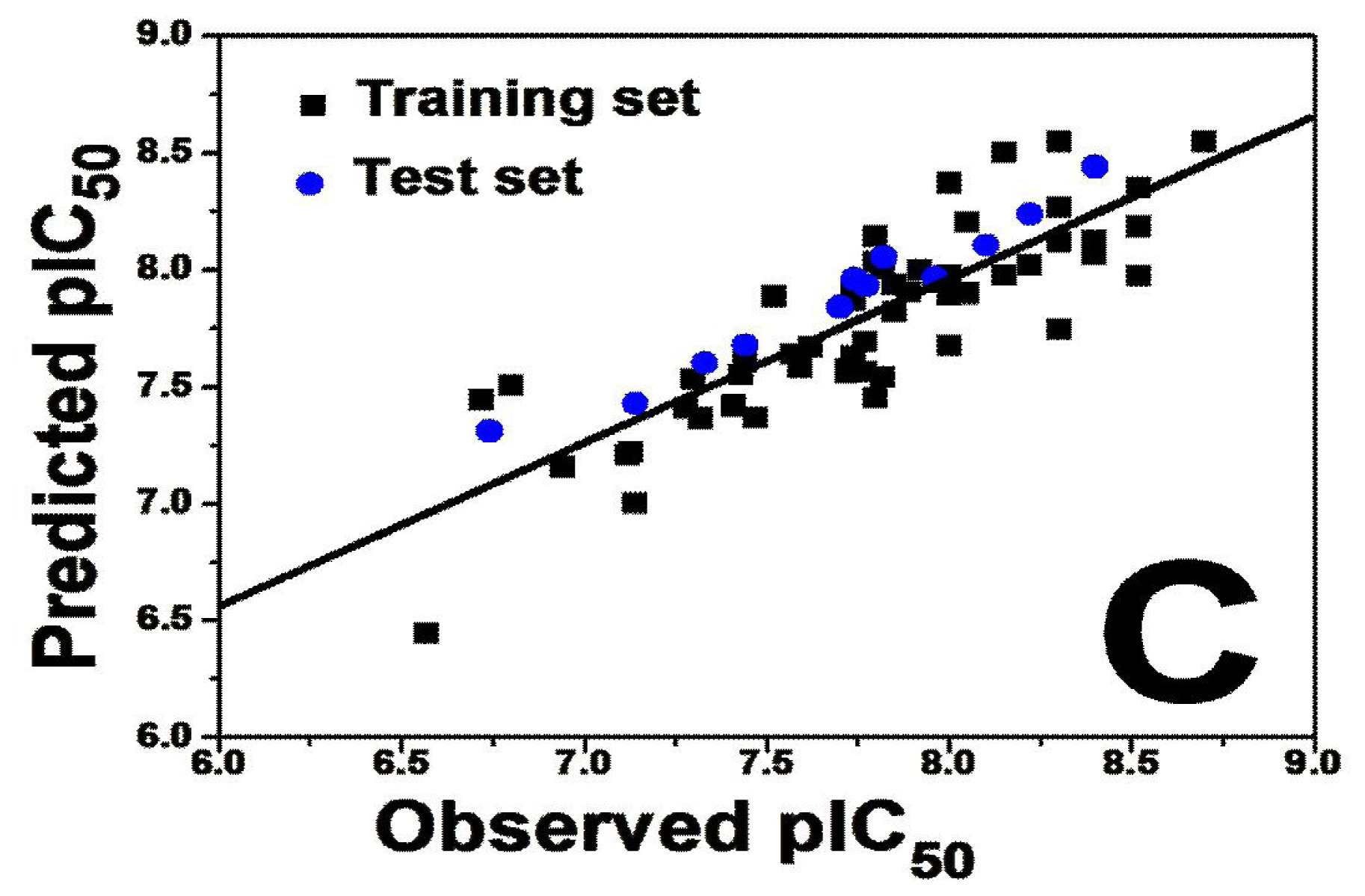
2.2. Results for Activity II
2.2.1. 3D-QSAR Statistical Results
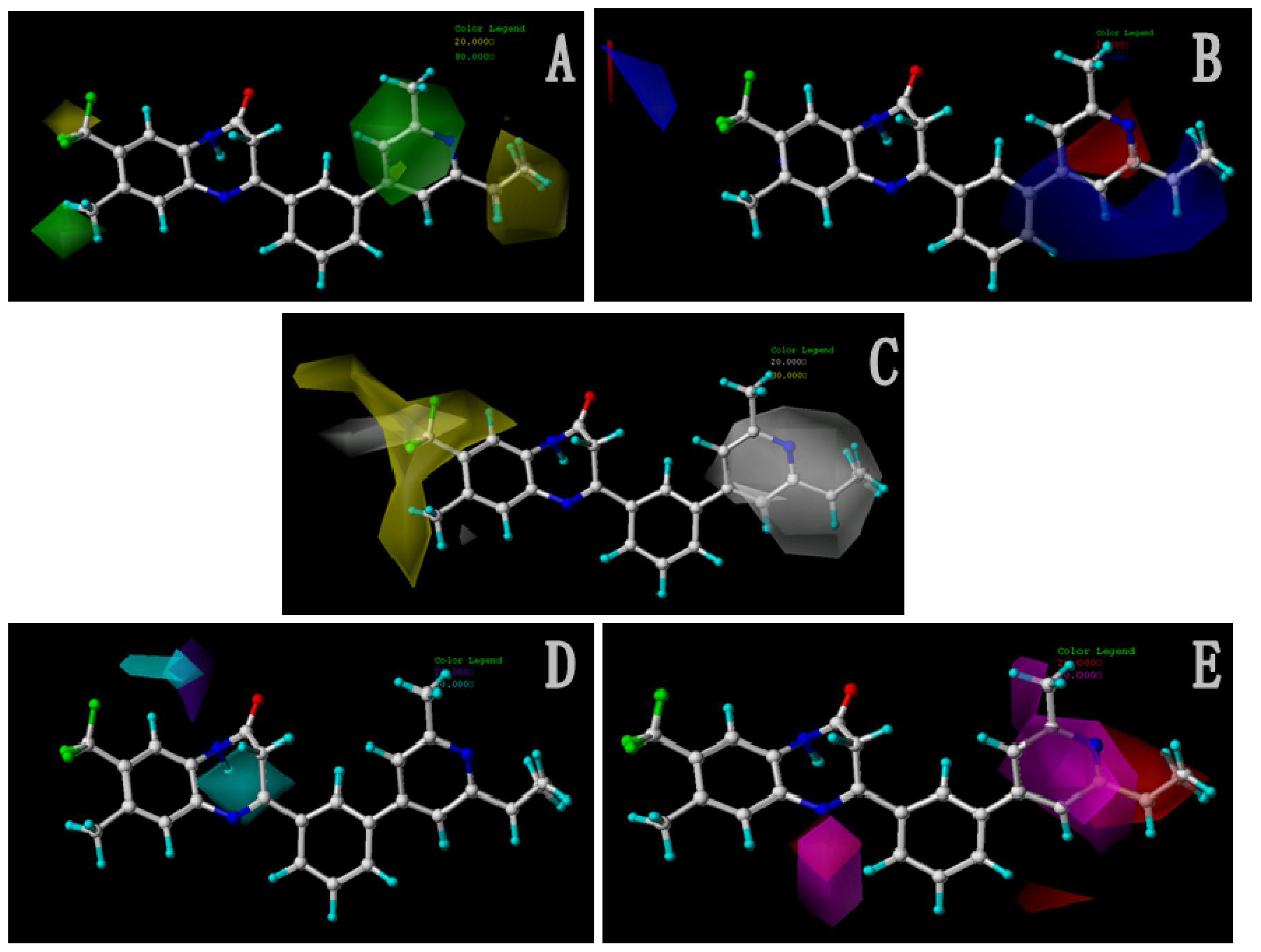
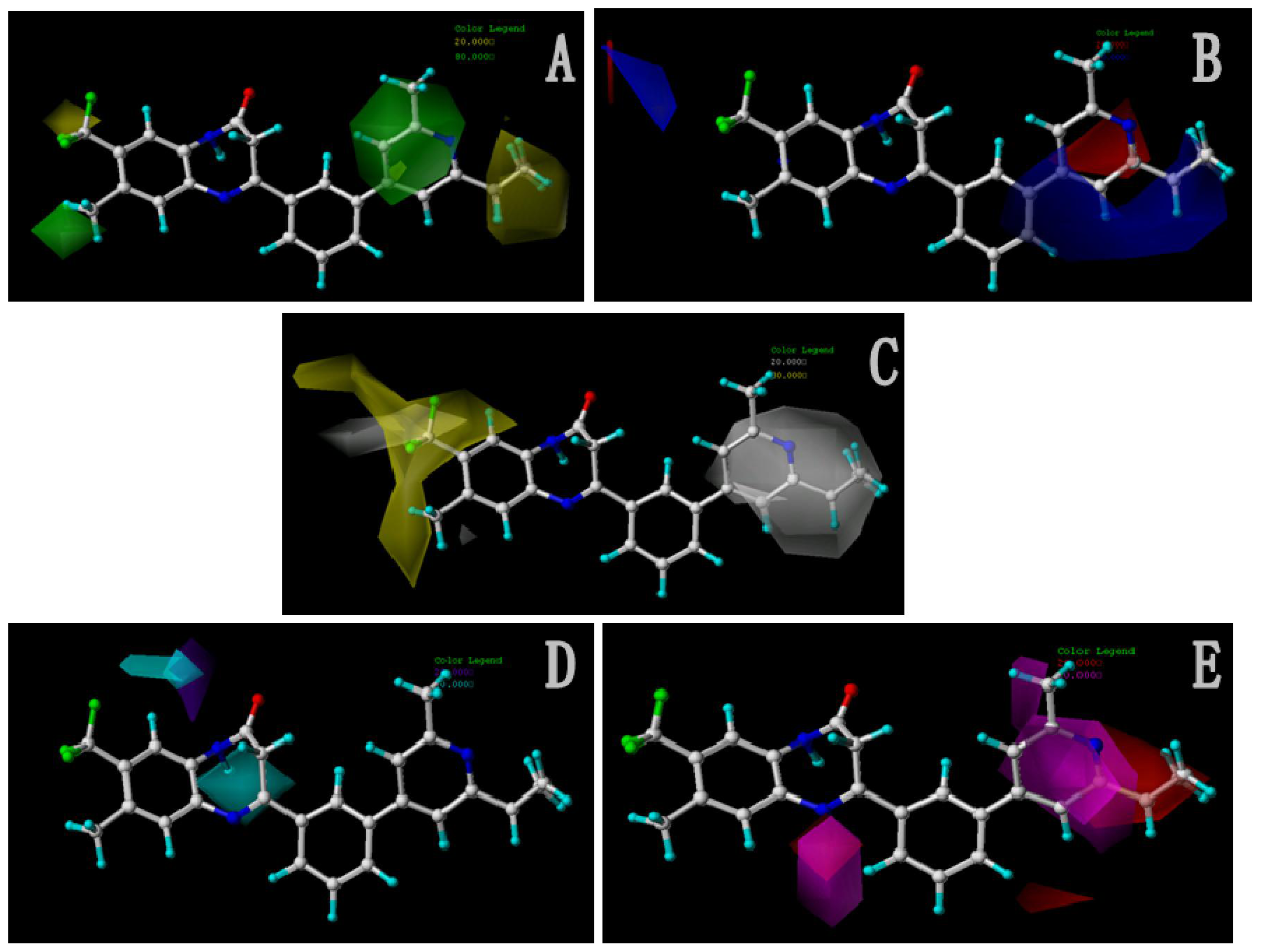
2.2.2. Contour Maps for Activity II
2.3. Homology Modeling Results and Docking Study
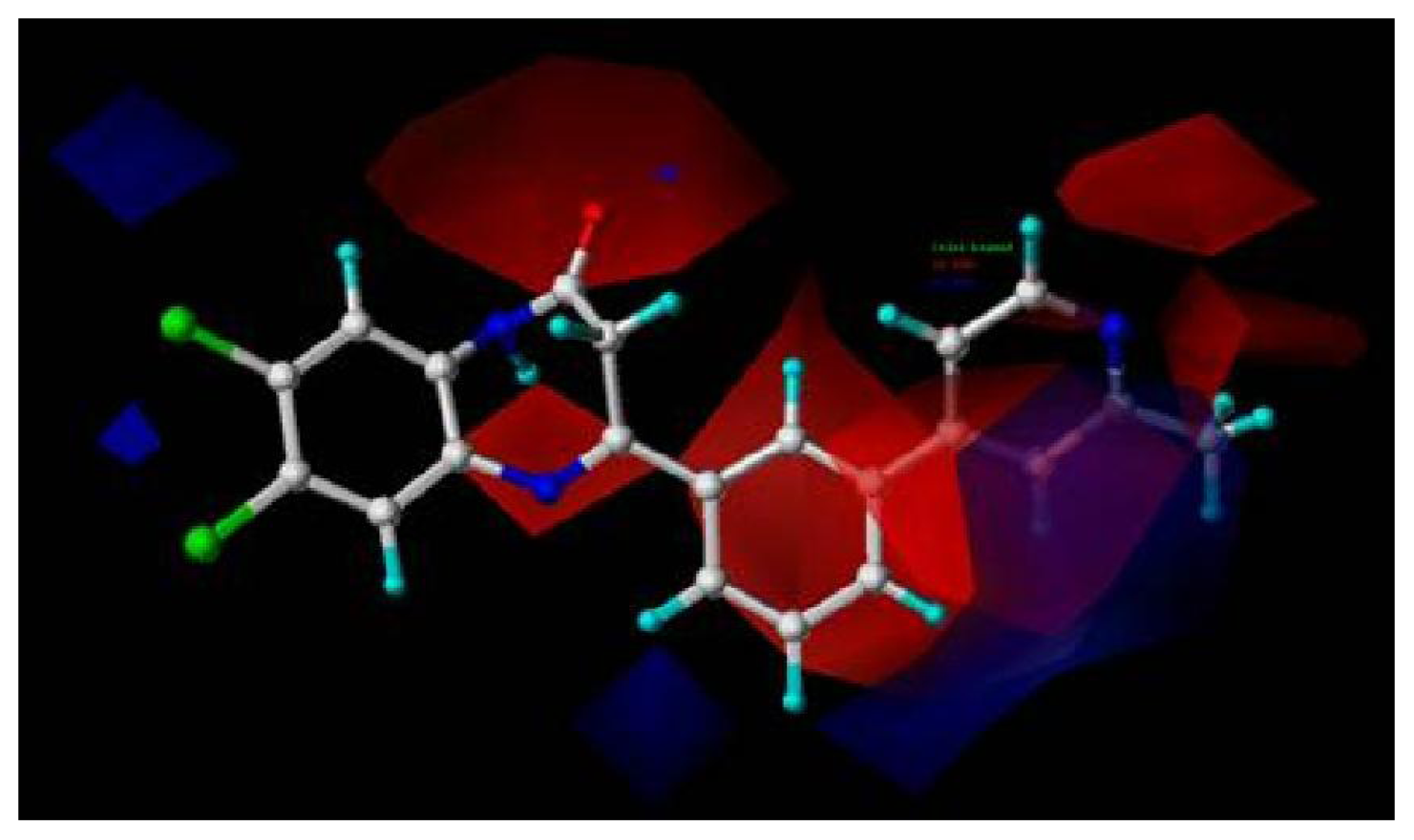
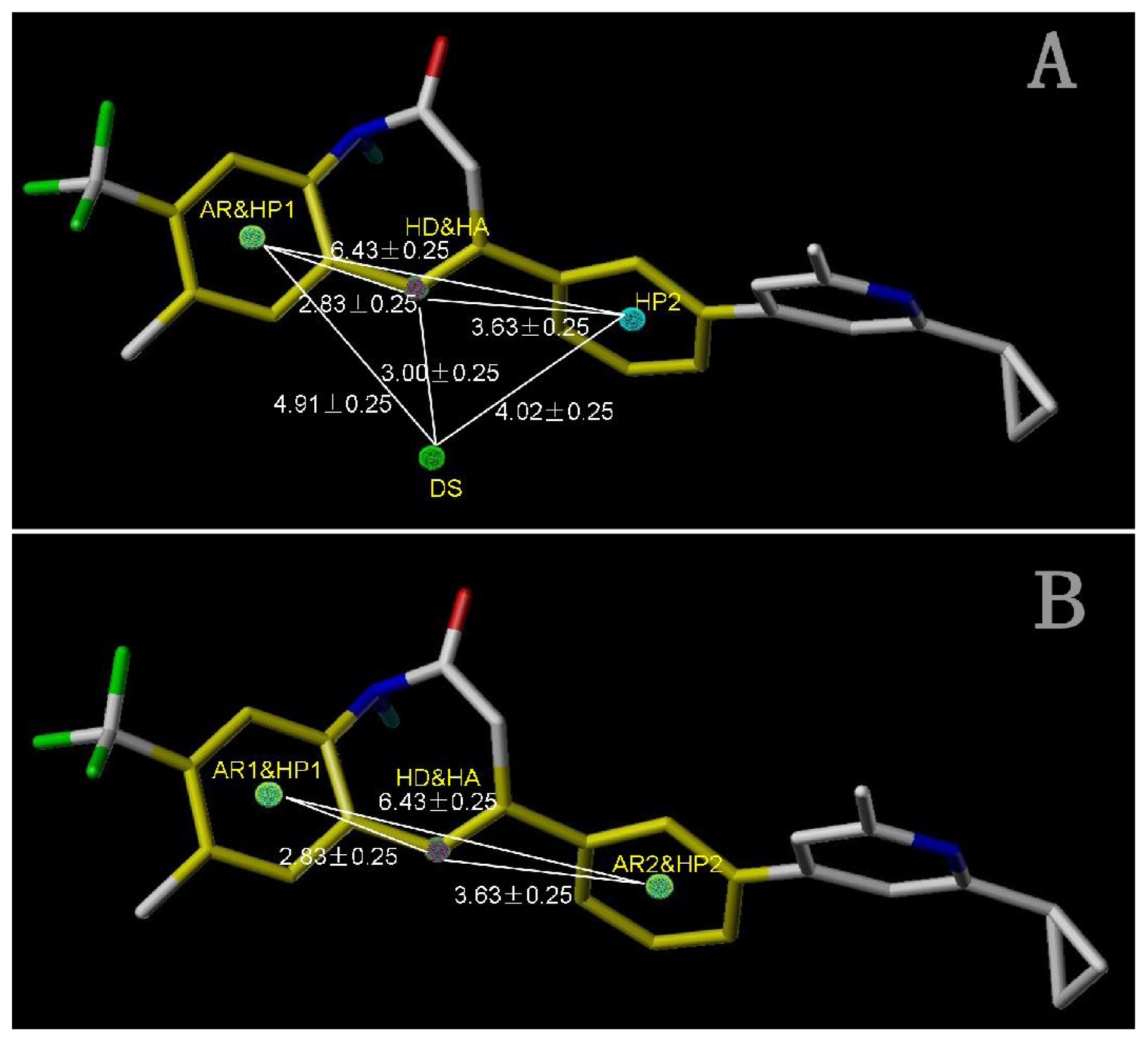
2.4. Pharmacophore Modeling
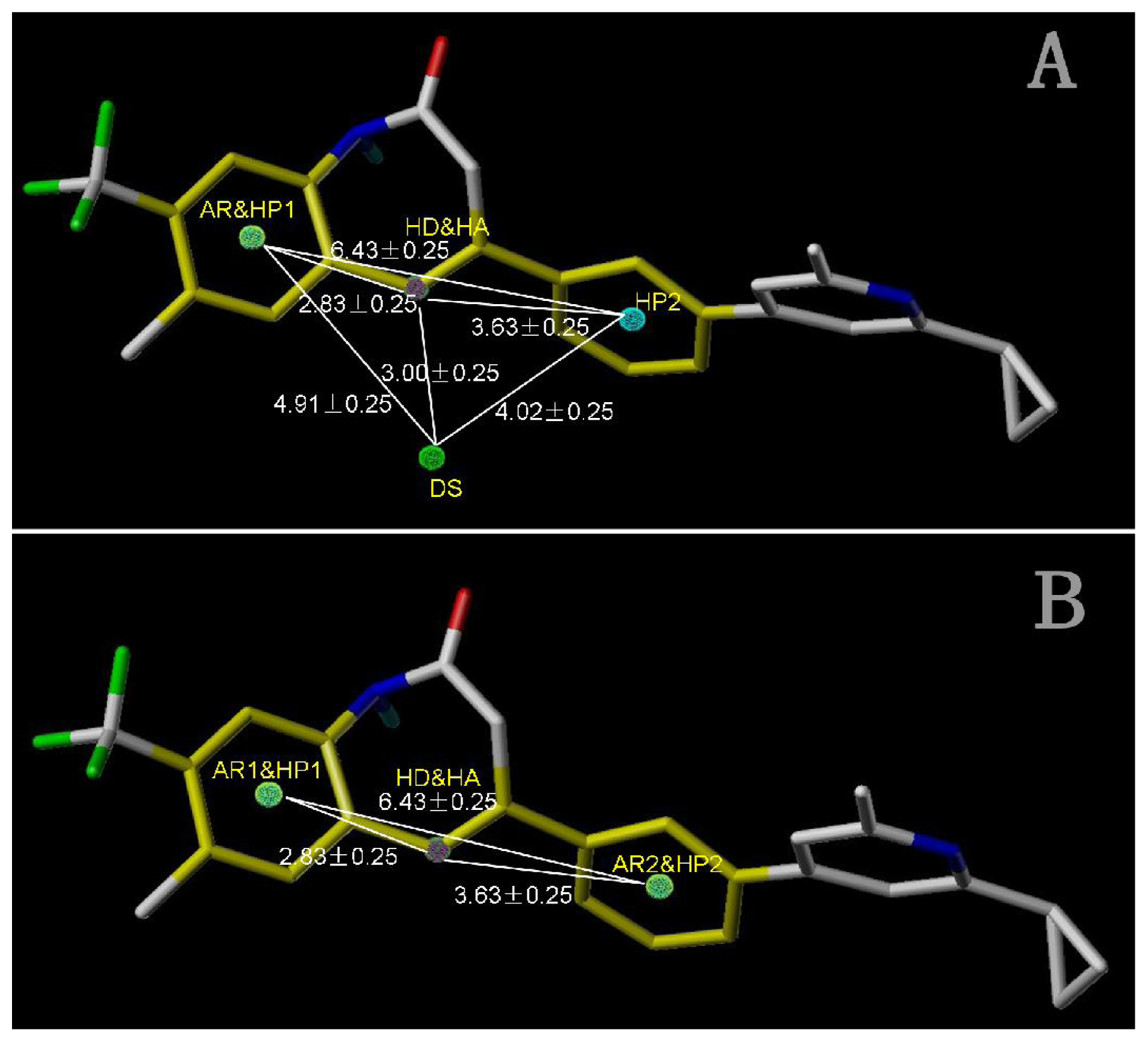
2.4.1. Pharmacophore Model for Activity I
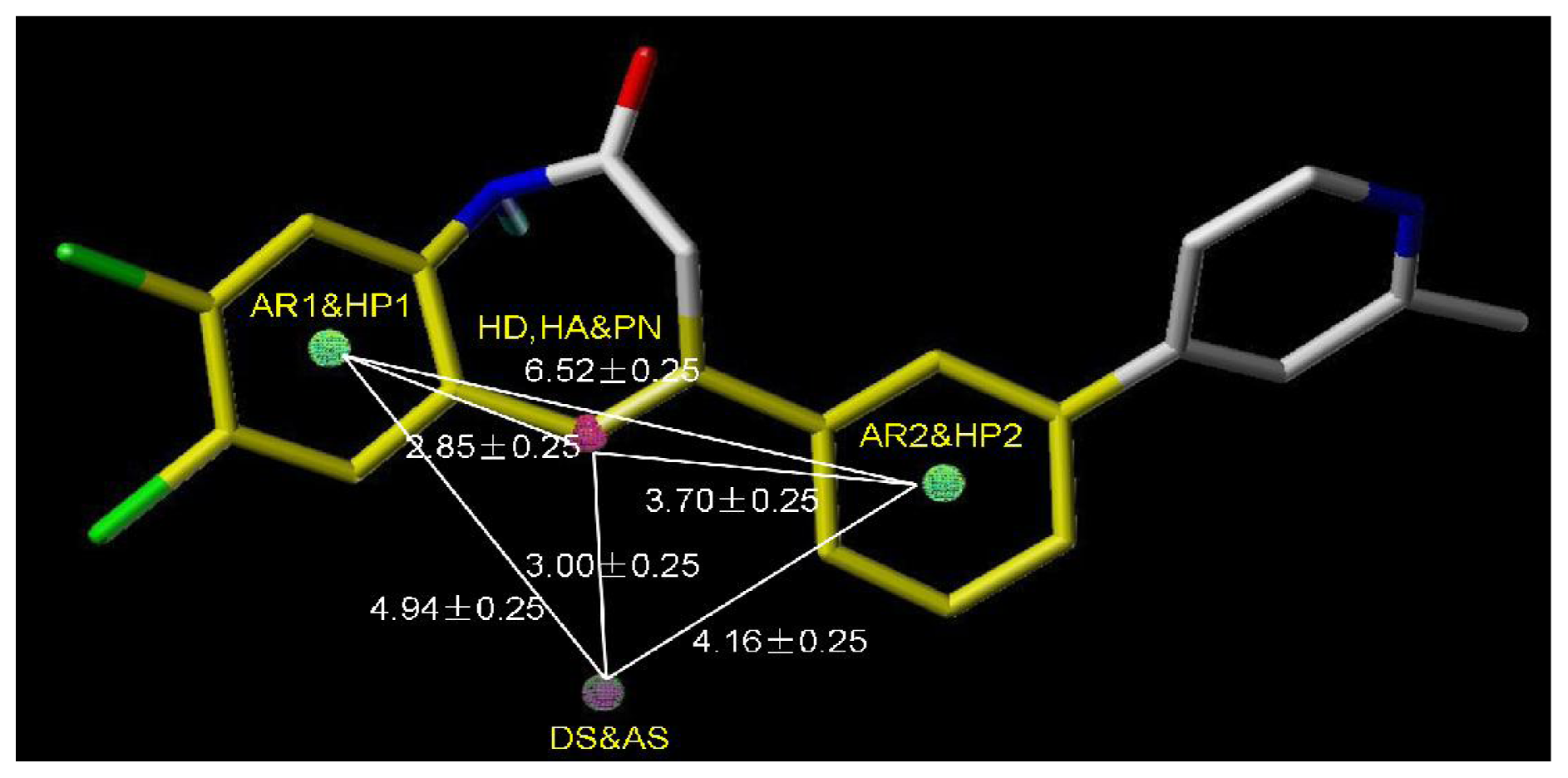
2.4.2. Pharmacophore Model for Activity II
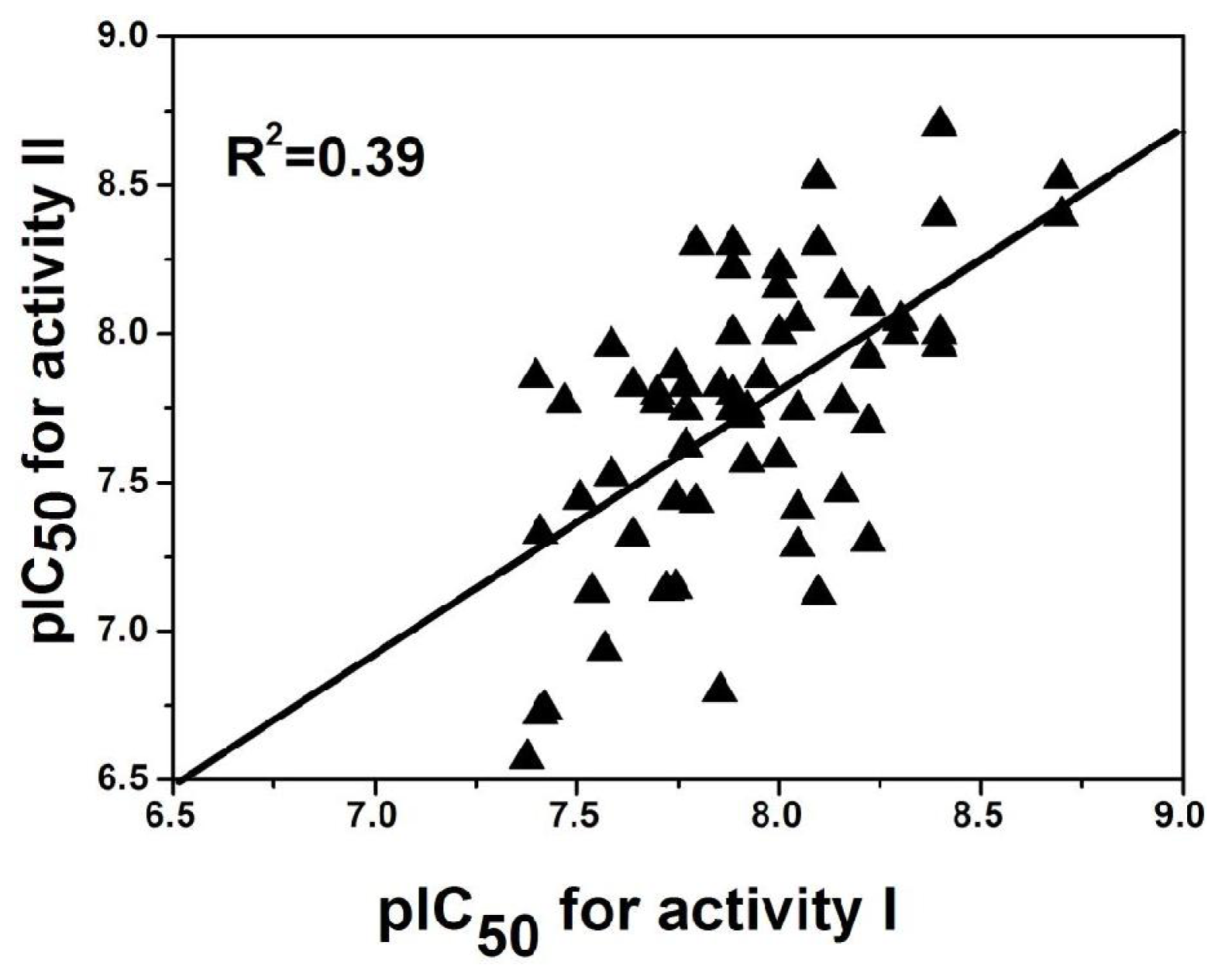
2.5. Comparison between the Two Activities
3. Material and Methods
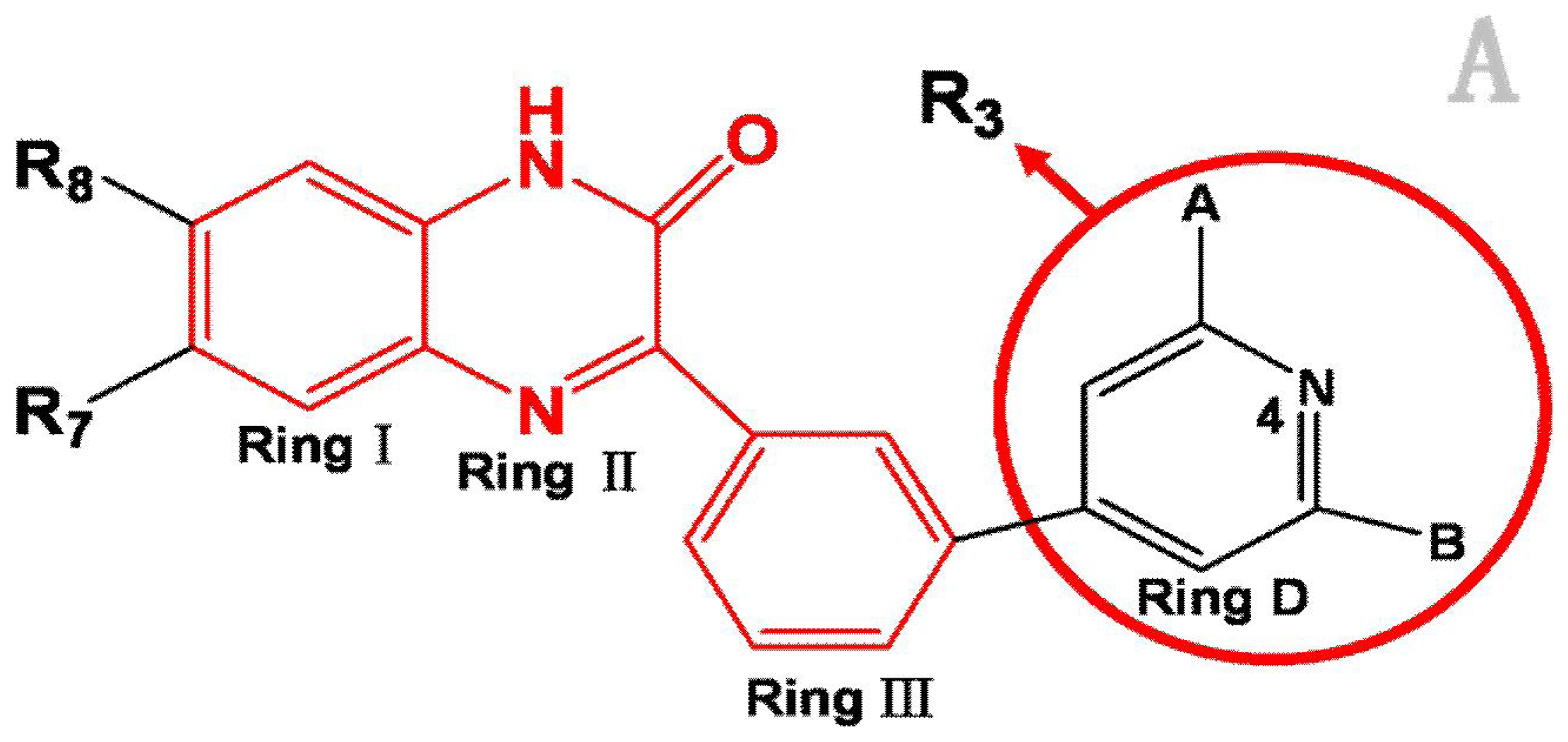
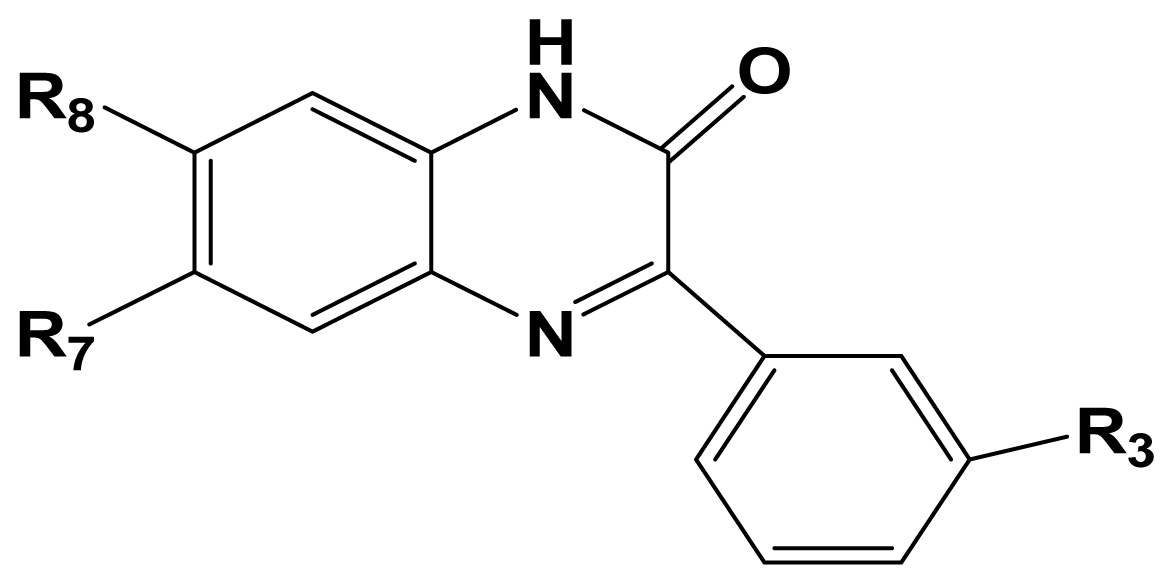
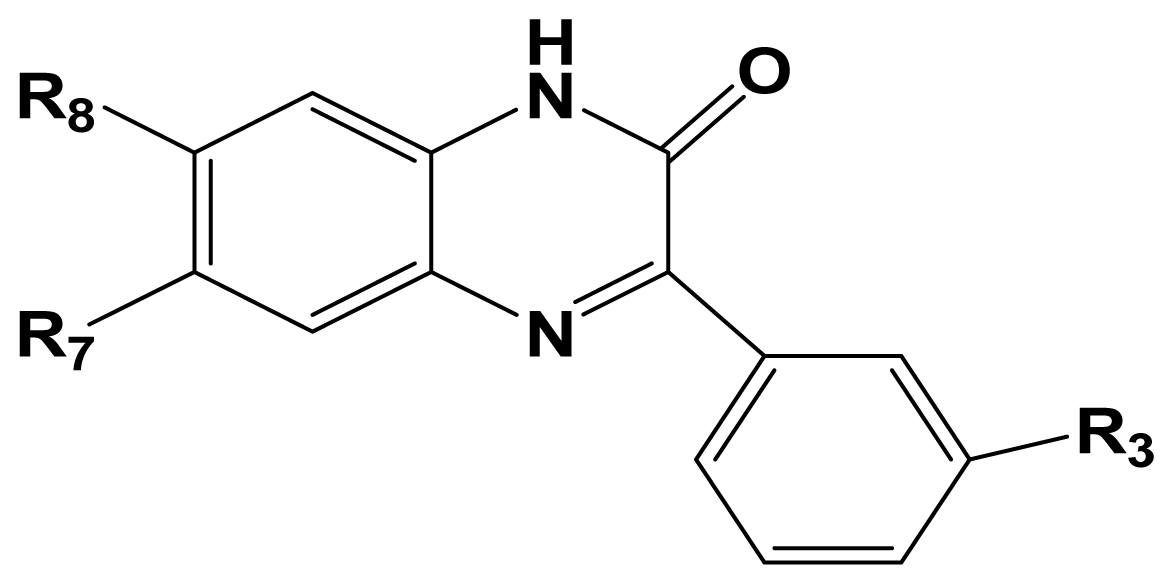
3.1. Dataset and Biological Activity
3.2. Conformational Sampling and Alignment
3.3. CoMFA and CoMSIA Field Calculation
3.4. Partial Least Square Analysis
3.5. DISCOtech Analysis
4. Conclusions
Supplementary Information
ijms-12-05999-s001.pdfAcknowledgements
Reference
- Lipton, SA. Paradigm shift in neuroprotection by nmda receptor blockade: Memantine and beyond. Nat Rev Drug Discov 2006, 5, 160–170. [Google Scholar]
- Schoepp, DD; Johnson, BG; Wright, RA; Salhoff, CR; Mayne, NG; Wu, S; Cockerham, SL; Paul Burnett, J; Belegaje, R; Bleakman, D. Ly354740 is a potent and highly selective group ii metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology 1997, 36, 1–11. [Google Scholar]
- Gasic, GP; Hollmann, M. Molecular neurobiology of glutamate receptors. Annu Rev Physiol 1992, 54, 507–536. [Google Scholar]
- Conn, PJ; Pin, JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 1997, 37, 205–237. [Google Scholar]
- Gregory, KJ; Dong, EN; Meiler, J; Conn, PJ. Allosteric modulation of metabotropic glutamate receptors: Structural insights and therapeutic potential. Neuropharmacology 2011, 60, 66–81. [Google Scholar]
- Spinelli, S; Ballard, T; Gatti-McArthur, S; Richards, GJ; Kapps, M; Woltering, T; Wichmann, J; Stadler, H; Feldon, J; Pryce, CR. Effects of the mglur2/3 agonist ly354740 on computerized tasks of attention and working memory in marmoset monkeys. Psychopharmacology 2005, 179, 292–302. [Google Scholar]
- Schoepp, DD; Jane, DE; Monn, JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 1999, 38, 1431–1476. [Google Scholar]
- Schoepp, DD; Wright, RA; Levine, LR; Gaydos, B; Potter, WZ. Ly354740, an mglu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 2003, 6, 189–197. [Google Scholar]
- Holden, C. Excited by glutamate. Science 2003, 300, 1866–1868. [Google Scholar]
- Pilc, A. Ly-354740 (eli lilly). IDrugs 2003, 6, 66–71. [Google Scholar]
- Higgins, GA; Ballard, TM; Kew, JNC; Grayson Richards, J; Kemp, JA; Adam, G; Woltering, T; Nakanishi, S; Mutel, V. Pharmacological manipulation of mglu2 receptors influences cognitive performance in the rodent. Neuropharmacology 2004, 46, 907–917. [Google Scholar]
- Woltering, TJ; Wichmann, J; Goetschi, E; Knoflach, F; Ballard, TM; Huwyler, J; Gatti, S. Synthesis and characterization of 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett 2010, 20, 6969–6974. [Google Scholar]
- Addex makes plans to move mGluR2 antagonist into clinical trials for Alzheimer’s disease. Available online: http://www.Bioportfolio.Com/news/article/120812/addex-makes-plans-tomove-mglur2-antagonist-into-clinical-trials-for-alzheimer.Html accessed on 15 September 2011.
- Wang, F; Ma, Z; Li, Y; Zhu, S; Xiao, Z; Zhang, H; Wang, Y. Development of in silico models for pyrazoles and pyrimidine derivatives as cyclin-dependent kinase 2 inhibitors. J Mol Graph Model 2011, 30, 67–81. [Google Scholar]
- Liu, J; Zhang, H; Xiao, Z; Wang, F; Wang, X; Wang, Y. Combined 3d-qsar, molecular docking and molecular dynamics study on derivatives of peptide epoxyketone and tyropeptinboronic acid as inhibitors against the beta5 subunit of human 20s proteasome. Int J Mol Sci 2011, 12, 1807–1835. [Google Scholar]
- Wang, G; Li, Y; Liu, X; Wang, Y. Understanding the aquatic toxicity of pesticide: Structureactivity relationship and molecular descriptors to distinguish the ratings of toxicity. QSAR Comb Sci 2009, 28, 11–12. [Google Scholar]
- Da Cunha, EFF; Sippl, W; de Castro Ramalho, T; Ceva Antunes, OA; de Alencastro, RB; Albuquerque, MG. 3d-qsar comfa/comsia models based on theoretical active conformers of hoe/bay-793 analogs derived from hiv-1 protease inhibitor complexes. Eur J Med Chem 2009, 44, 4344–4352. [Google Scholar]
- Cramer, RD; Patterson, DE; Bunce, JD. Comparative molecular field analysis (comfa). 1. Effect of shape on binding of steroids to carrier proteins. Eur J Med Chem 1988, 110, 5959–5967. [Google Scholar]
- Zhou, H-Y; Chen, S-R; Chen, H; Pan, H-L. Functional plasticity of group ii metabotropic glutamate receptors in regulating spinal excitatory and inhibitory synaptic input in neuropathic pain. J Pharmacol Exp Ther 2011, 336, 254–264. [Google Scholar]
- Yanamala, N; Tirupula, K; Klein-Seetharaman, J. Preferential binding of allosteric modulators to active and inactive conformational states of metabotropic glutamate receptors. BMC Bioinformatics 2008, 9, S16. [Google Scholar]
- Bruno, A; Guadix, AE; Costantino, G. Molecular dynamics simulation of the heterodimeric mglur2/5ht2a complex. An atomistic resolution study of a potential new target in psychiatric conditions. J Chem Inf Mod 2009, 49, 1602–1616. [Google Scholar]
- Liu, J; Li, Y; Zhang, S; Xiao, Z; Ai, C. Studies of new fused benzazepine as selective dopamine d3 receptor antagonists using 3d-qsar, molecular docking and molecular dynamics. Int J Mol Sci 2011, 12, 1196–1221. [Google Scholar]
- Costantino, G; Macchiarulo, A; Pellicciari, R. Pharmacophore models of group i and group ii metabotropic glutamate receptor agonists. Analysis of conformational, steric, and topological parameters affecting potency and selectivity. J Med Chem 1999, 42, 2816–2827. [Google Scholar]
- Harley, EA; Middlemiss, DN; Ragan, CI. Relationship between inhibition of cyclic amp production in chinese hamster ovary cells expressing the rat d2(444) receptor and antagonist/agonist binding ratios. Br J Pharmacol 1995, 115, 1307–1313. [Google Scholar]
- Taylor, SS; Kim, C; Cheng, CY; Brown, SHJ; Wu, J; Kannan, N. Signaling through camp and camp-dependent protein kinase: Diverse strategies for drug design. Biochim Biophys Acta 2008, 1784, 16–26. [Google Scholar]
- De Jong, LAA; Uges, DRA; Franke, JP; Bischoff, R. Receptor-ligand binding assays: Technologies and applications. J Chromatogr B 2005, 829, 1–25. [Google Scholar]
- Gasteiger, J; Marsili, M. Iterative partial equalization of orbital electronegativity--a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- Clark, M; Cramer, RD; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J Comput Chem 1989, 10, 982–1012. [Google Scholar]
- AbdulHameed, MDM; Hamza, A; Liu, J; Zhan, C-G. Combined 3d-qsar modeling and molecular docking study on indolinone derivatives as inhibitors of 3-phosphoinositide-dependent protein kinase-1. J Chem Inf Mod 2008, 48, 1760–1772. [Google Scholar]
- Liu, J; Wang, F; Ma, Z; Wang, X; Wang, Y. Structural determination of three different series of compounds as hsp90 inhibitors using 3d-qsar modeling, molecular docking and molecular dynamics methods. Int J Mol Sci 2011, 12, 946–970. [Google Scholar]
- Li, Y; Wang, Y-H; Yang, L; Zhang, S-W; Liu, C-H; Yang, S-L. Comparison of steroid substrates and inhibitors of p-glycoprotein by 3d-qsar analysis. J Mol Struct 2005, 733, 111–118. [Google Scholar]
- Klebe, G; Abraham, U; Mietzner, T. Molecular similarity indices in a comparative analysis (comsia) of drug molecules to correlate and predict their biological activity. J Med Chem 1994, 37, 4130–4146. [Google Scholar]
- Xu, M; Zhang, A. Studies of 3d-quantitative structure-activity relationships on a set of nitroaromatic compounds: Comfa, advanced comfa and comsia. Chemosphere 2002, 48, 707–715. [Google Scholar]
- Wold, S; Geladi, P; Esbensen, K; Öhman, J. Multi-way principal components-and pls-analysis. J Chemometr 1987, 1, 41–56. [Google Scholar]
- Geladi, P. Notes on the history and nature of partial least squares (pls) modelling. J Chemometr 1988, 2, 231–246. [Google Scholar]
- Li, Y; Wang, Y-H; Yang, L; Zhang, S-W; Liu, C-H; Yang, S-L. Comparison of steroid substrates and inhibitors of p-glycoprotein by 3d-qsar analysis. J Mol Struct 2005, 733, 111–118. [Google Scholar]
- Wang, F; Li, Y; Ma, Z; Wang, X; Wang, Y. Structural determinants of benzodiazepinedione/peptidebased p53-HDM(2) inhibitors using 3d-qsar, docking and molecular dynamics. J Mol Model 2011. [Google Scholar] [CrossRef]
- Spadoni, G; Balsamini, C; Diamantini, G; Di Giacomo, B; Tarzia, G; Mor, M; Plazzi, PV; Rivara, S; Lucini, V; Nonno, R; et al. Conformationally restrained melatonin analogues: Synthesis, binding affinity for the melatonin receptor, evaluation of the biological activity, and molecular modeling study. J Med Chem 1997, 40, 1990–2002. [Google Scholar]
- Marriott, DP; Dougall, IG; Meghani, P; Liu, Y-J; Flower, DR. Lead generation using pharmacophore mapping and three-dimensional database searching: Application to muscarinic m3 receptor antagonists. J Med Chem 1999, 42, 3210–3216. [Google Scholar]
- Martin, YC; Bures, MG; Danaher, EA; DeLazzer, J; Lico, I; Pavlik, PA. A fast new approach to pharmacophore mapping and its application to dopaminergic and benzodiazepine agonists. J Comput Aid Mol Des 1993, 7, 83–102. [Google Scholar]
- Clark, RD. Optisim: An extended dissimilarity selection method for finding diverse representative subsets. J Chem Inf Comput Sci 1997, 37, 1181–1188. [Google Scholar]
- Jun, Z; We-quan, DAI. 3d-qsar study of a series of non-competitive 1,3-dihydrobenzo [b][1,4]diazepine-2-one mglur2/3 antagonists. J Tongji Univ 2008, 29, 5. [Google Scholar]
- Flor, PJ; Lindauer, K; Puttner, I; Ruegg, D; Lukic, S; Knopfel, T; Kuhn, R. Molecular cloning, functional expression and pharmacological characterization of the human metabotropic glutamate receptor type 2. Eur J Neurosci 1995, 7, 622–629. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistics | Activity I | Activity II | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| Q2 | 0.513 | 0.450 | 0.503 | 0.367 |
| R2 ncv | 0.868 | 0.899 | 0.715 | 0.657 |
| SEE | 0.296 | 0.273 | 0.265 | 0.285 |
| F | 96.022 | 101.353 | 20.907 | 24.927 |
| R2 pre | 0.876 | 0.735 | 0.723 | 0.667 |
| SEP | 0.288 | 0.420 | 0.241 | 0.265 |
| OPN | 7 | 8 | 6 | 4 |
| Contribution | ||||
| Steric | 0.488 | 0.112 | ||
| Electrostatic | 0.461 | 0.277 | 0.917 | 0.455 |
| Hydrophobic | 0.184 | 0.409 | ||
| H-donor | 0.135 | |||
| H-acceptor | 0.256 | |||
| Clogp | 0.051 | 0.035 | 0.083 | 0.136 |
| MODEL | SIZE a | HITS b | SCORE c | TOLERANCE d | DMEAN e |
|---|---|---|---|---|---|
| MODEL_002 | 6 | 50 | 2.3577 | 0.25 | 3.4348 |
| MODEL_001 | 6 | 50 | 1.8562 | 0.25 | 3.4163 |
| MODEL_005 | 7 | 50 | 1.7220 | 0.25 | 3.0344 |
| MODEL_009 | 7 | 50 | 1.3646 | 0.25 | 3.4012 |
| MODEL_003 | 7 | 50 | 1.3629 | 0.25 | 3.3955 |
| MODEL_006 | 7 | 50 | 1.3607 | 0.25 | 3.3880 |
| MODEL_008 | 6 | 50 | 0.6783 | 0.25 | 2.0754 |
| MODEL_004 | 6 | 50 | 0.3735 | 0.25 | 2.8621 |
| MODEL_007 | 6 | 50 | 0.3733 | 0.25 | 2.8616 |
| AR1 | HP1 | DS | HP2 | |
|---|---|---|---|---|
| HA | 2.83 | 2.83 | 3.00 | 3.63 |
| HD | 2.83 | 2.83 | 3.00 | 3.63 |
| DS | 4.91 | 4.91 | 4.02 | |
| HP2 | 6.43 | 6.43 | 4.02 |
| MODEL | SIZE a | HITS b | SCORE c | TOLERANCE d | DMEAN e |
|---|---|---|---|---|---|
| MODEL_001 | 9 | 50 | 1.8286 | 0.25 | 3.3280 |
| AR1 | HP1 | DS | HP2 | |
|---|---|---|---|---|
| HA | 2.83 | 2.83 | 3.00 | 3.63 |
| HD | 2.83 | 2.83 | 3.00 | 3.63 |
| DS | 4.91 | 4.91 | 4.02 | |
| HP2 | 6.43 | 6.43 | 4.02 |
 | |||||
|---|---|---|---|---|---|
| NO. | R3 | R8 | R7 | Activity I | Activity II |
| 1 | H | Me | H | 5.1938 | |
| 2 | CN | Ph-C≡C- | H | 7.4685 | 7.7696 |
| 14u | CN | 2-Thiazolyl-C≡C- | H | 6.5229 | |
| 14v | CN | 2-Pyridyl-C≡C- | H | 6.0605 | |
| 14x | CN | H2C=C(Me)-C≡C- | H | 6.3979 | |
| 14aa | CN | Ph-C≡C | 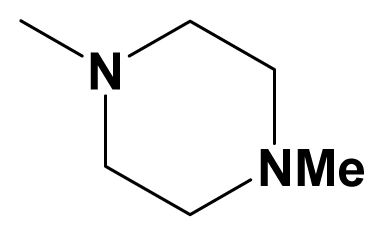 | 6.5560 | |
| 15c | 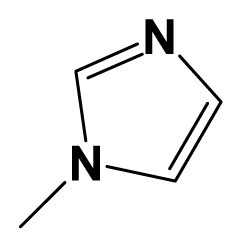 | 2-F-C6H4-C≡C- | H | 7.6990 | 7.7959 |
| 15m | 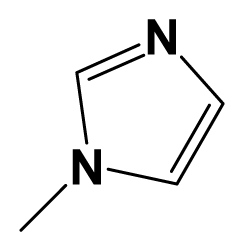 | Ph-C≡C- | -OCH2CN | 7.7447 | 7.8861 |
| 15q |  | 4-F-C6H4-C≡C- | -OH | 7.6990 | 7.7696 |
| 7g |  | Cyclo-propyl | H | 6.6778 | |
| 7o | 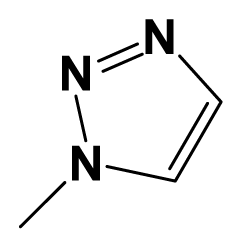 | 4-F-C6H4- | H | 7.5086 | 7.4437 |
| 7z | 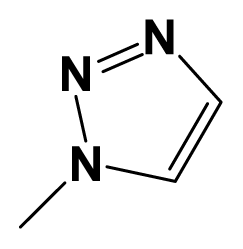 | F3C- | Iso-butylN(Me) | 7.9586 | 7.8539 |
| 7ac |  | F3C- | MeO | 7.5376 | 7.1308 |
| 8a | 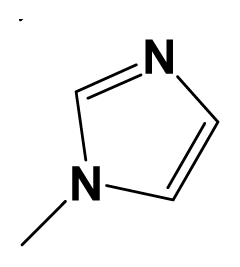 | F3C- | Me | 7.9208 | 7.7212 |
| 8h | 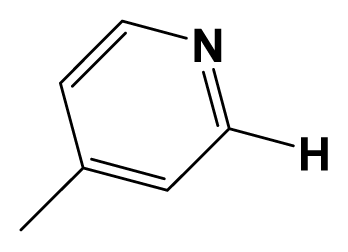 | F3C- | Me | 8.3979 | 7.9586 |
| 8y |  | F3C- | Me | 7.5850 | 7.5229 |
| 8aa | 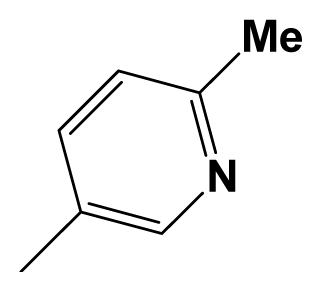 | F3C- | Me | 7.8861 | 8.3010 |
| 8ae |  | F3C- | Me | 8.3010 | 8.0000 |
| 8aj |  | F3C- | Me | 8.2218 | 7.3010 |
| 8ao | 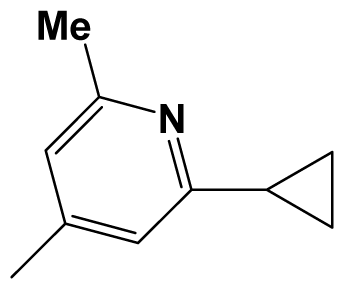 | F3C- | Me | 8.6990 | 8.3979 |
| 8av | 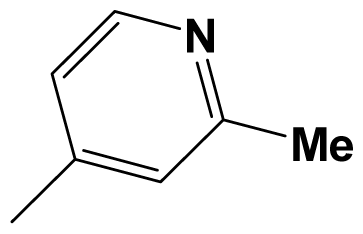 | Cl | Cl | 8.3979 | 8.6990 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, M.-Q.; Zhang, X.-L.; Li, Y.; Fan, W.-J.; Wang, Y.-H.; Hao, M.; Zhang, S.-W.; Ai, C.-Z. Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists. Int. J. Mol. Sci. 2011, 12, 5999-6023. https://doi.org/10.3390/ijms12095999
Zhang M-Q, Zhang X-L, Li Y, Fan W-J, Wang Y-H, Hao M, Zhang S-W, Ai C-Z. Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists. International Journal of Molecular Sciences. 2011; 12(9):5999-6023. https://doi.org/10.3390/ijms12095999
Chicago/Turabian StyleZhang, Meng-Qi, Xiao-Le Zhang, Yan Li, Wen-Jia Fan, Yong-Hua Wang, Ming Hao, Shu-Wei Zhang, and Chun-Zhi Ai. 2011. "Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists" International Journal of Molecular Sciences 12, no. 9: 5999-6023. https://doi.org/10.3390/ijms12095999
APA StyleZhang, M.-Q., Zhang, X.-L., Li, Y., Fan, W.-J., Wang, Y.-H., Hao, M., Zhang, S.-W., & Ai, C.-Z. (2011). Investigation on Quantitative Structure Activity Relationships and Pharmacophore Modeling of a Series of mGluR2 Antagonists. International Journal of Molecular Sciences, 12(9), 5999-6023. https://doi.org/10.3390/ijms12095999