Intermediates in the Protein Folding Process: A Computational Model



Abstract
:1. Introduction
Materials and Methods
2. Results and Discussion
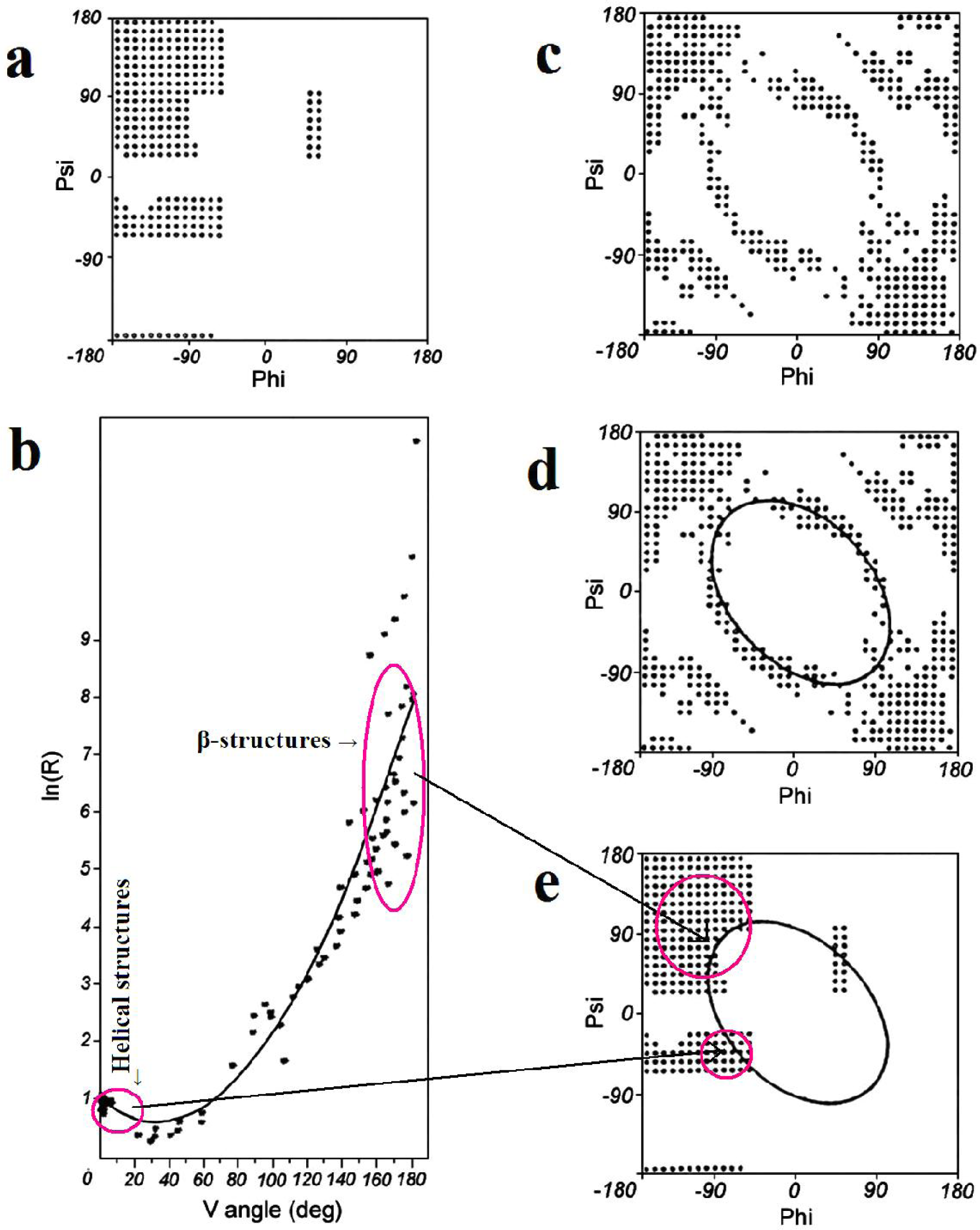
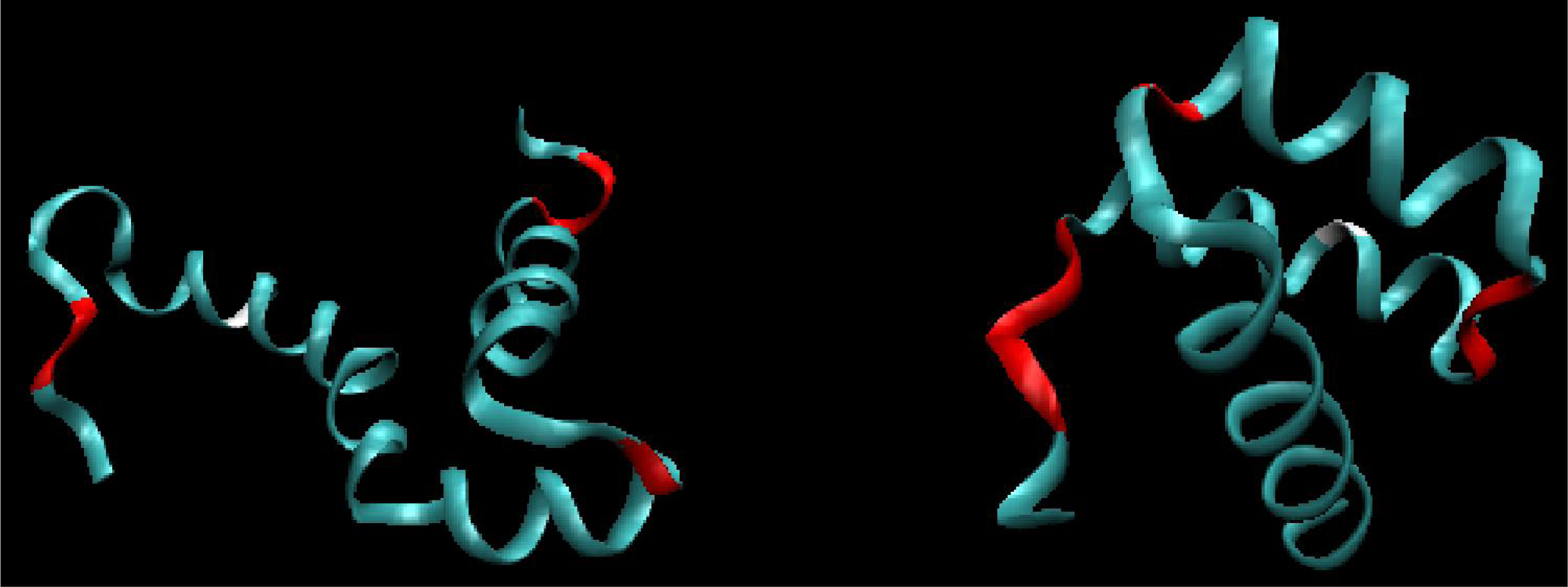
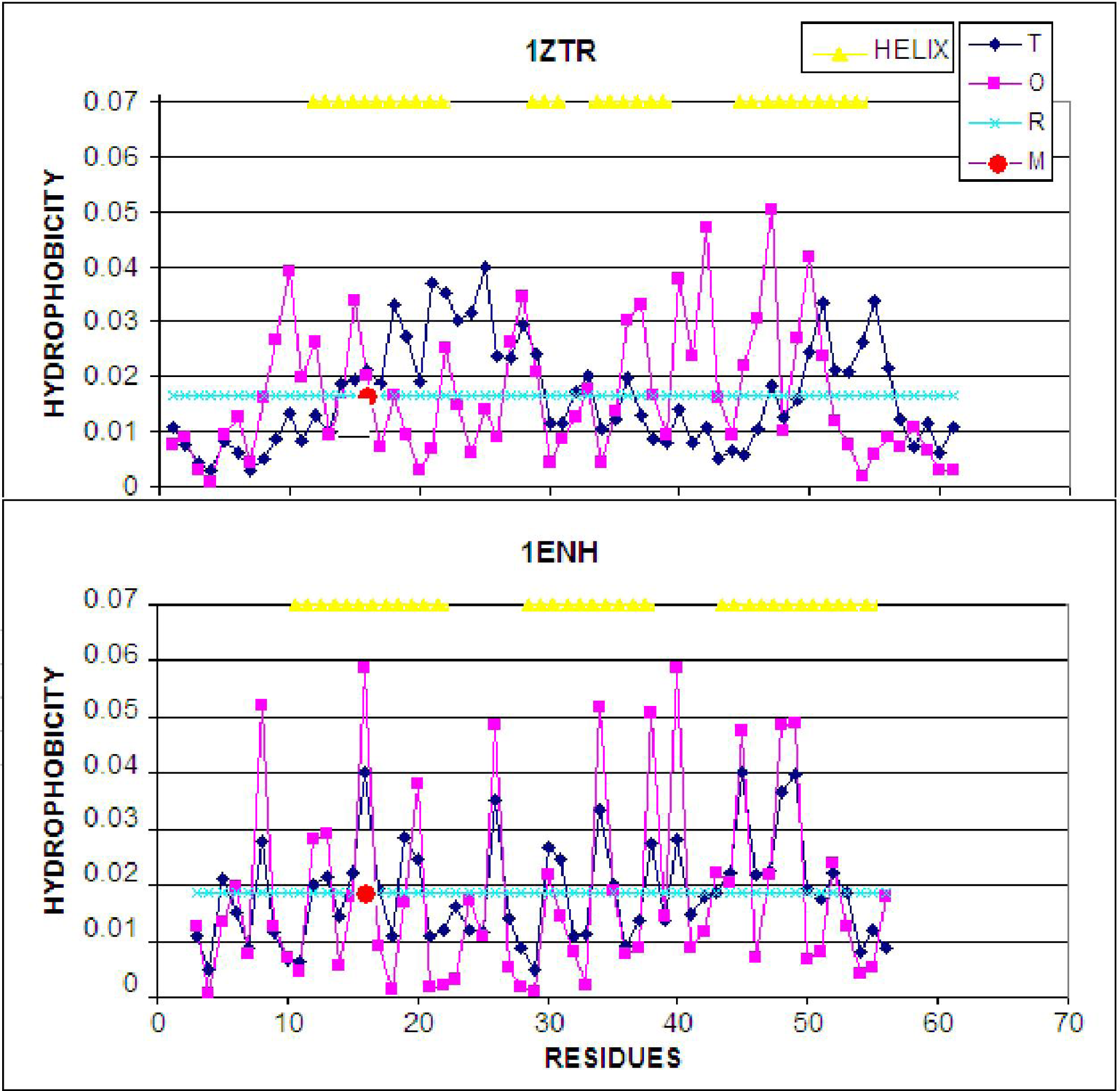
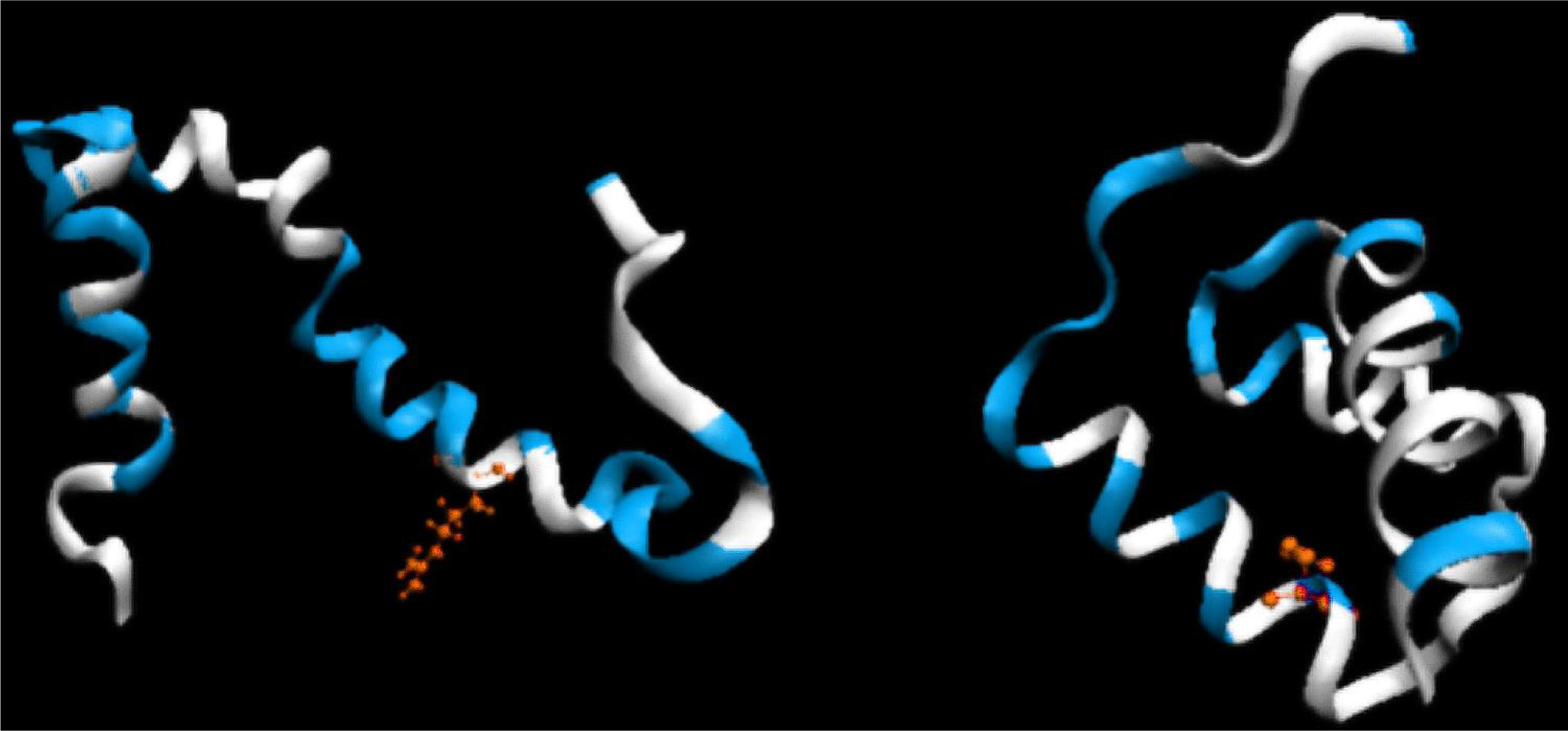
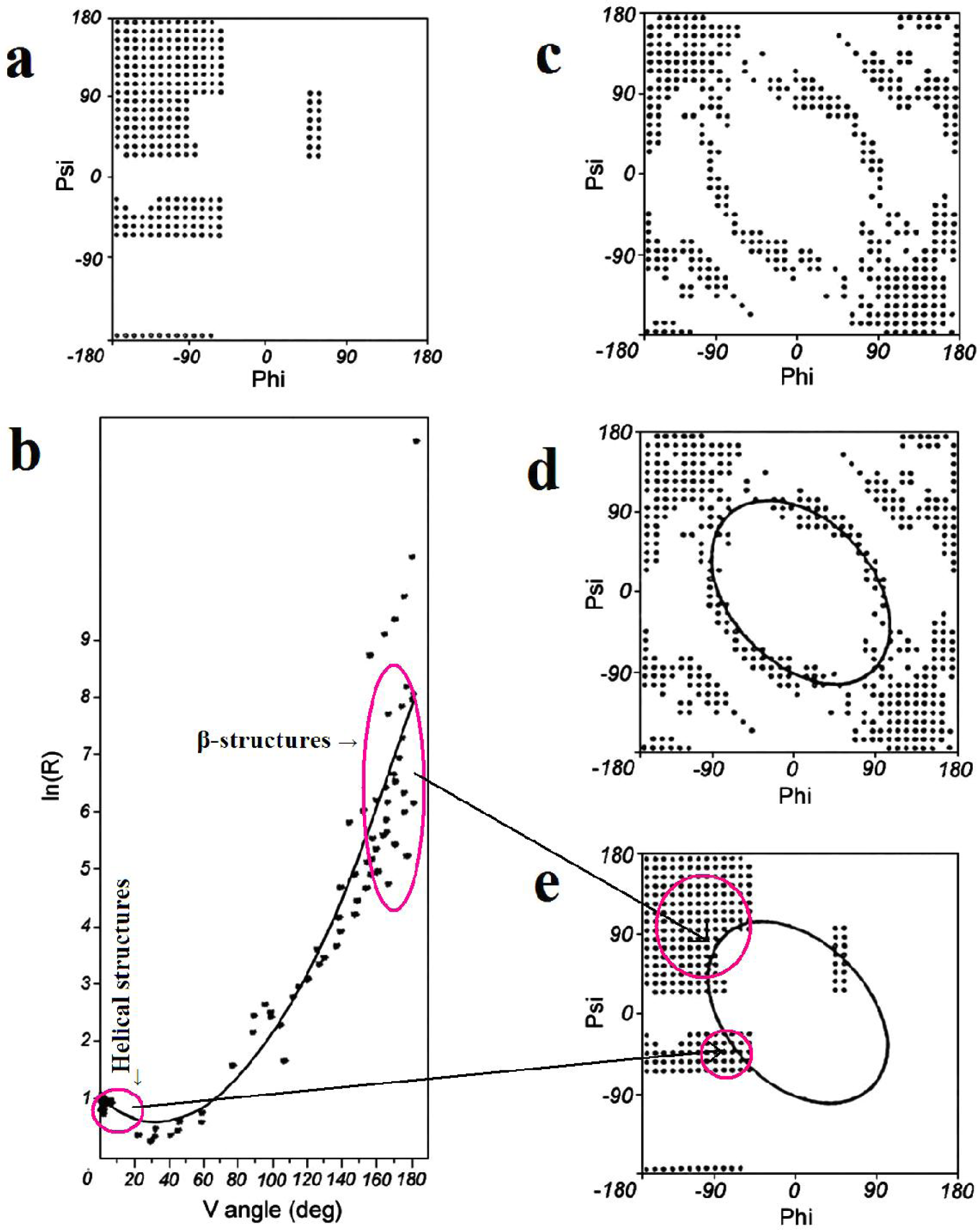
2.1. ES Intermediate
2.2. LS Model
3. Conclusions
- Limiting the conformational subspace for early folding stages seems to be accurate, as proven by the experimentally-verified structure of 1ZTR.
- The LS step may be simulated through the generation of a hydrophobic core (using a 3D Gauss function), which results in the highest concentration of hydrophobicity at the center of the protein body with simultaneous exposure of hydrophilic residues on the protein surface.
- The generation of a hydrophobic core (triggered by an external force field) should be taken as an accompanying procedure in the course of internal energy minimization.
Acknowledgments
References
- Feng, H; Zhou, Z; Bai, Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 5026–5031. [Google Scholar]
- Konieczny, L; Bryliński, M; Roterman, I. Gauss-function-based model of hydrophobicity density in proteins. In Silico Biol 2006, 6, 15–22. [Google Scholar]
- Religa, TL; Marksom, JS; Mayor, U; Freund, SM; Fersht, AR. Solution structure of a protein denatured state and folding intermediate. Nature 2005, 437, 1053–1056. [Google Scholar]
- Clarke, ND; Kissinger, CR; Desjarlais, J; Gilliland, GL; Pabo, CO. Structural studies of the engrailed homeodomain. Protein Sci 1994, 3, 1779–1787. [Google Scholar]
- Roterman, I. Modelling the optimal simulation path in the peptide chain folding-Studies based on geometry of alanine heptapeptide. J. Theor. Biol 1995, 177, 283–288. [Google Scholar]
- Jurkowski, W; Wiśniowski, Z; Konieczny, L; Roterman, I. The conformational sub-space in simulation of early-stage protein folding. Proteins 2004, 55, 115–127. [Google Scholar]
- Brylinski, M; Konieczny, L; Czerwonko, P; Jurkowski, W; Roterman, I. Early-stage folding in proteins (in silico) sequence-to-structure relation. J. Biomed. Biotechnol 2005, 30, 65–79. [Google Scholar]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol 1976, 104, 59–107. [Google Scholar]
- Aboderin, A. An empirical hydrophobicity scale for alfa-amino-acids and some of its applications. Int. J. Biochem 1971, 2, 537–544. [Google Scholar]
- Prymula, K; Jadczyk, T; Roterman, I. Catalytic residues in hydroplases: Analysis of methods designer for ligand binding site prediction. J. Comput. Aided Mol. Des 2011, 25, 117–133. [Google Scholar]
- Nalewajski, RF. Information Theory of Molecular Systems; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Bryliński, M; Konieczny, L; Roterman, I. Fuzzy-oil-drop hydrophobic force field a model to represent late-stage folding (in silico) of lysozyme. J. Biomol. Struct. Dyn 2006, 23, 519–527. [Google Scholar]
- Kumar, R; Prabhu, NP; Bhuyan, AK. Ultrafast events in the folding of ferrocytochrome C. Biochemistry 2005, 44, 9359–9367. [Google Scholar]
- Banach, M; Prymula, K; Jurkowski, W; Konieczny, L; Roterman, I. Fuzzy oil drop model to interpret the structure of antifreeze proteins and their mutants. J Mol Model 2011, in press. [Google Scholar]
- Brylinski, M; Konieczny, L; Roterman, I. Hydrophobic collapse in late-stage folding (in silico) of bovine pancreatic trypsin inhibitor. Biochimie 2006, 88, 1229–1239. [Google Scholar]
- Eliezer, D; Palmer, AG, III. Biophysics: proteins hunt and gather. Nature 2007, 447, 920–921. [Google Scholar]
- Zobnina, V; Roterman, I. Application of the fuzzy-oil-drop model to membrane protein simulation. Proteins 2009, 77, 378–394. [Google Scholar]
- Bryliński, M; Konieczny, L; Roterman, I. Hydrophobic collapse in (in silico) protein folding. Comput. Biol. Chem 2006, 30, 255–267. [Google Scholar]
- Brylinski, M; Konieczny, L; Roterman, I. Is the protein folding an aim-oriented process? Human haemoglobin as example. Int. J. Bioinf. Res. Appl 2007, 3, 234–260. [Google Scholar]
- Bryliński, M; Prymula, K; Jurkowski, W; Kochańczyk, M; Stawowczyk, E; Konieczny, L; Roterman, I. Prediction of functional sites based on the fuzzy oil drop model. PLoS Comput. Biol 2007, 3, E94. [Google Scholar]
- Bryliński, M; Kochanczyk, M; Broniatowska, E; Roterman, I. Localization of ligand binding site in proteins identified in silico. J. Mol. Model 2007, 13, 655–675. [Google Scholar]
- Jurkowski, W; Kułaga, T; Roterman, I. Geometric parameters to define the structure of proteins—relation to their early-stage folding step. J. Biomol. Struct. Dyn 2011, 29, 79–104. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ES MODEL | LS MODEL | ||
|---|---|---|---|
| PROTEIN | Daverage | O/T | O/R |
| 1ZTR (ES structure) | 0.342 | 0.4978 | 0.3638 |
| 1ENH (LS structure) | 0.623 | 0.1286 | 0.2137 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roterman, I.; Konieczny, L.; Banach, M.; Jurkowski, W. Intermediates in the Protein Folding Process: A Computational Model. Int. J. Mol. Sci. 2011, 12, 4850-4860. https://doi.org/10.3390/ijms11084850
Roterman I, Konieczny L, Banach M, Jurkowski W. Intermediates in the Protein Folding Process: A Computational Model. International Journal of Molecular Sciences. 2011; 12(8):4850-4860. https://doi.org/10.3390/ijms11084850
Chicago/Turabian StyleRoterman, Irena, Leszek Konieczny, Mateusz Banach, and Wiktor Jurkowski. 2011. "Intermediates in the Protein Folding Process: A Computational Model" International Journal of Molecular Sciences 12, no. 8: 4850-4860. https://doi.org/10.3390/ijms11084850