Mechanisms of Estrogens’ Dose-Dependent Neuroprotective and Neurodamaging Effects in Experimental Models of Cerebral Ischemia

Abstract
:1. Introduction
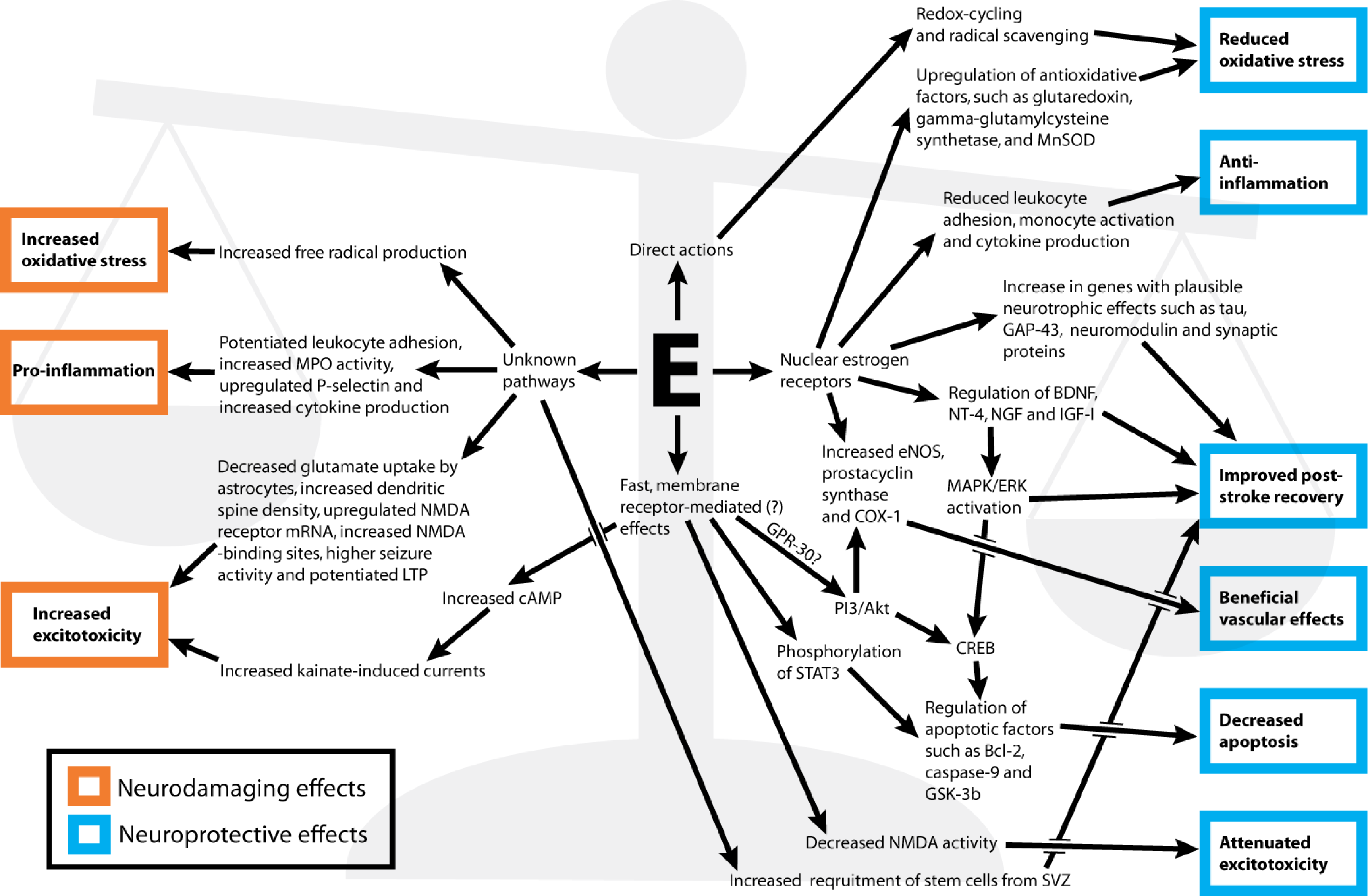
2. Mechanisms for Estrogens’ Neuroprotective and Neurodamaging Effects
2.1. Decreased and Increased Oxidative Stress as Mechanisms of Estrogen Neuroprotection and Neurodamage
2.1.1. Direct Anti-Oxidative Effects
2.1.2. Indirect Anti-Oxidative Effects
2.1.3. Pro-Oxidative Effects
2.2. Anti- and Pro-Inflammatory Actions as Mechanisms of Estrogen Neuroprotection and Neurodamage
2.2.1. Anti-Inflammatory Effects
2.2.2. Pro-Inflammatory Effects
2.3. Increased Excitotoxicity as a Mechanism of Estrogen Neurodamage
2.4. Decreased Apoptosis as a Mechanism of Estrogen Neuroprotection
2.5. Growth Factor Regulation as a Mechanism of Estrogen Neuroprotection
2.6. Vascular Modulation as a Mechanism of Estrogen Neuroprotection
3. Conclusions
3.1. Quality of Mechanism Experiments
- The lowest degree of “evidence” for a certain mechanism comes from the discovery of a biological alteration, which potentially could bring about the biological target effect, in response to the investigated substance. An example is the finding that estrogens increase the concentration of the synaptic protein syntaxin, which hypothetically (without direct experimental evidence) could facilitate recovery after cerebral ischemia [219].
- If the investigated substance has an effect on a presumed mechanism that in itself has been proven to exert the biological target effect the evidence is evidently stronger, even if the relative contribution of the mechanism cannot be quantified. An example is the fact that estrogens upregulate Bcl-2 [164], a protein in itself proven to decrease the damage from cerebral ischemia [156,157].
- A yet higher degree of evidence for a mechanism’s importance is afforded when the presence of a specific blockage inactivates the biological target effect. An example is estrogens’ lack of protective effects in iNOS knocked out mice [111].
3.2. Summary of Mechanism Evaluations
3.3. Difficulties in Studying the Complex Estrogenic Mechanisms
3.4. Final Remarks
List of Abbreviations
| ACh | Acetylcholine |
| AMPA | α-Amino-3-hydroxyl-5-methyl-4-isoxazole-propionate |
| Apaf-1 | Apoptotic Protein-Activating Factor-1 |
| BDNF | Brain-Derived Neurotrophic Factor |
| BH3 | Bcl Homology domain-3 |
| CA1 | Cornu Ammonis area-1 |
| cAMP | Cyclic Adenosine Monophosphate |
| cGMP | Cyclic Guanosine Monophosphate |
| COX | Cyclooxygenase |
| CREB | cAMP Response Element Binding protein |
| eNOS | Extracellular Nitric Oxide Synthase |
| ER | Estrogen Receptor |
| ERE | Estrogen Response Elements |
| ERK | Extracellular signal-Regulated Kinases |
| GABA | Gamma-Aminobutyric Acid |
| GAP-43 | Growth-Associated Protein-43 |
| GPR-30 | G-Protein coupled Receptor-30 |
| GSK-3β | Glycogen synthase kinase 3β |
| IGF-I | Insulin-like Growth Factor-I |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| IRA | Innovative Research of America |
| LPS | Lipopolysaccharide |
| LTP | Long Term Potentiation |
| MAPK | Mitogen-Activated Protein Kinase |
| MCAo | Middle Cerebral Artery Occlusion |
| MPO | Myeloperoxidase |
| NADPH | Reduced form of Nicotinamide Adenine Dinucleotide Phosphate |
| NFKB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve Growth Factor |
| NMDA | N-Methyl-d-Aspartate |
| nNOS | Neuronal Nitric Oxide Synthase |
| NO | Nitric Oxide |
| NT-4 | Neurotrophin-4 |
| PGE2 | Prostaglandin E2 |
| PI3 | Phosphatidylinositol-3 |
| PUMA | p53-Upregulated Modulator of Apoptosis |
| ROS | Reactive Oxygen Species |
| SOD | Superoxide Dismutase |
| Sp1 | Specificity Protein-1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| SVZ | Subventricular Zone |
| TGF-α | Transforming Growth Factor-α |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor-α |
| VEGF | Vascular Endothelial Growth Factor |
References
- Hall, ED; Pazara, KE; Linseman, KL. Sex differences in postischemic neuronal necrosis in gerbils. J. Cereb. Blood Flow Metab 1991, 11, 292–298. [Google Scholar]
- Simpkins, JW; Rajakumar, G; Zhang, YQ; Simpkins, CE; Greenwald, D; Yu, CJ; Bodor, N; Day, AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J. Neurosurg 1997, 87, 724–730. [Google Scholar]
- Hurn, PD; Littleton-Kearney, MT; Kirsch, JR; Dharmarajan, AM; Traystman, RJ. Postischemic cerebral blood flow recovery in the female: Effect of 17beta-estradiol. J. Cereb. Blood Flow Metab 1995, 15, 666–672. [Google Scholar]
- Lobo, RA. The risk of stroke in postmenopausal women receiving hormonal therapy. Climacteric 2009, 12(Suppl 1), 81–85. [Google Scholar]
- Rossouw, JE; Anderson, GL; Prentice, RL; LaCroix, AZ; Kooperberg, C; Stefanick, ML; Jackson, RD; Beresford, SA; Howard, BV; Johnson, KC; Kotchen, JM; Ockene, J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. Jama 2002, 288, 321–333. [Google Scholar]
- Harukuni, I; Hurn, PD; Crain, BJ. Deleterious effect of beta-estradiol in a rat model of transient forebrain ischemia. Brain Res 2001, 900, 137–142. [Google Scholar]
- Yong, Y; Xie, HJ; Zhang, YF; Yang, QD; Liao, DF; Yang, HL; Yan, PK; Liu, ZJ. 17beta-estradiol potentiates ischemia-reperfusion injury in diabetic ovariectomized female rats. Brain Res 2005, 1054, 192–199. [Google Scholar]
- Theodorsson, A; Theodorsson, E. Estradiol increases brain lesions in the cortex and lateral striatum after transient occlusion of the middle cerebral artery in rats: No effect of ischemia on galanin in the stroke area but decreased levels in the hippocampus. Peptides 2005, 26, 2257–2264. [Google Scholar]
- Gordon, KB; Macrae, IM; Carswell, HV. Effects of 17beta-oestradiol on cerebral ischaemic damage and lipid peroxidation. Brain Res 2005, 1036, 155–162. [Google Scholar]
- Carswell, HV; Bingham, D; Wallace, K; Nilsen, M; Graham, DI; Dominiczak, AF; Macrae, IM. Differential effects of 17beta-estradiol upon stroke damage in stroke prone and normotensive rats. J. Cereb. Blood Flow Metab 2004, 24, 298–304. [Google Scholar]
- Bingham, D; Macrae, IM; Carswell, HV. Detrimental effects of 17beta-oestradiol after permanent middle cerebral artery occlusion. J. Cereb. Blood Flow Metab 2005, 25, 414–420. [Google Scholar]
- Liu, M; Dziennis, S; Hurn, PD; Alkayed, NJ. Mechanisms of gender-linked ischemic brain injury. Restor. Neurol. Neurosci 2009, 27, 163–179. [Google Scholar]
- Brown, CM; Suzuki, S; Jelks, KA; Wise, PM. Estradiol is a potent protective, restorative, and trophic factor after brain injury. Semin. Reprod. Med 2009, 27, 240–249. [Google Scholar]
- Suzuki, S; Brown, CM; Wise, PM. Neuroprotective effects of estrogens following ischemic stroke. Front. Neuroendocrinol 2009, 30, 201–211. [Google Scholar]
- Strom, JO; Theodorsson, A; Theodorsson, E. Dose-related neuroprotective versus neurodamaging effects of estrogens in rat cerebral ischemia: A systematic analysis. J. Cereb. Blood Flow Metab 2009, 29, 1359–1372. [Google Scholar]
- Strom, JO; Theodorsson, E; Holm, L; Theodorsson, A. Different methods for administering 17beta-estradiol to ovariectomized rats result in opposite effects on ischemic brain damage. BMC Neurosci 2010, 11, 39. [Google Scholar]
- Strom, JO; Theodorsson, E; Theodorsson, A. Order of magnitude differences between methods for maintaining physiological 17beta-oestradiol concentrations in ovariectomized rats. Scand. J. Clin. Lab. Invest 2008, 68, 814–822. [Google Scholar]
- Calabrese, EJ; Baldwin, LA. Hormesis: The dose-response revolution. Annu. Rev. Pharmacol. Toxicol 2003, 43, 175–197. [Google Scholar]
- Dziennis, S; Jia, T; Ronnekleiv, OK; Hurn, PD; Alkayed, NJ. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J. Neurosci 2007, 27, 7268–7274. [Google Scholar]
- Schreihofer, DA; Do, KD; Schreihofer, AM. High-soy diet decreases infarct size after permanent middle cerebral artery occlusion in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 2005, 289, R103–108. [Google Scholar]
- Rau, SW; Dubal, DB; Bottner, M; Gerhold, LM; Wise, PM. Estradiol attenuates programmed cell death after stroke-like injury. J. Neurosci 2003, 23, 11420–11426. [Google Scholar]
- He, Z; He, YJ; Day, AL; Simpkins, JW. Proestrus levels of estradiol during transient global cerebral ischemia improves the histological outcome of the hippocampal CA1 region: Perfusion-dependent and-independent mechanisms. J. Neurol. Sci 2002, 193, 79–87. [Google Scholar]
- Sandstrom, NJ; Rowan, MH. Acute pretreatment with estradiol protects against CA1 cell loss and spatial learning impairments resulting from transient global ischemia. Horm. Behav 2007, 51, 335–345. [Google Scholar]
- Pelligrino, DA; Santizo, R; Baughman, VL; Wang, Q. Cerebral vasodilating capacity during forebrain ischemia: Effects of chronic estrogen depletion and repletion and the role of neuronal nitric oxide synthase. Neuroreport 1998, 9, 3285–3291. [Google Scholar]
- Saleh, TM; Connell, BJ; Legge, C; Cribb, AE. Estrogen attenuates neuronal excitability in the insular cortex following middle cerebral artery occlusion. Brain Res 2004, 1018, 119–129. [Google Scholar]
- Plamondon, H; Morin, A; Charron, C. Chronic 17beta-estradiol pretreatment and ischemia-induced hippocampal degeneration and memory impairments: A 6-month survival study. Horm. Behav 2006, 50, 361–369. [Google Scholar]
- Choi, YC; Lee, JH; Hong, KW; Lee, KS. 17Beta-estradiol prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam. Clin. Pharmacol 2004, 18, 547–557. [Google Scholar]
- Yu, SJ; Kim, JR; Lee, CK; Kim, JH; Kam, KY; Hong, JH; Kang, SG. Involvement of purα gene in neuroprotection effects of estrogen in rat ischemic brain model. Korean J. Genet 2006, 28, 403–412. [Google Scholar]
- Gulinello, M; Lebesgue, D; Jover-Mengual, T; Zukin, RS; Etgen, AM. Acute and chronic estradiol treatments reduce memory deficits induced by transient global ischemia in female rats. Horm. Behav 2006, 49, 246–260. [Google Scholar]
- Kii, N; Adachi, N; Liu, K; Arai, T. Acute effects of 17beta-estradiol on oxidative stress in ischemic rat striatum. J. Neurosurg. Anesthesiol 2005, 17, 27–32. [Google Scholar]
- Littleton-Kearney, MT; Klaus, JA; Hurn, PD. Effects of combined oral conjugated estrogens and medroxyprogesterone acetate on brain infarction size after experimental stroke in rat. J. Cereb. Blood Flow Metab 2005, 25, 421–426. [Google Scholar]
- Strom, JO; Theodorsson, A; Theodorsson, E. Substantial discrepancies in 17beta-oestradiol concentrations obtained with three different commercial direct radioimmunoassay kits in rat sera. Scand. J. Clin. Lab. Invest 2008, 68, 806–813. [Google Scholar]
- Suzuki, S; Gerhold, LM; Bottner, M; Rau, SW; Dela Cruz, C; Yang, E; Zhu, H; Yu, J; Cashion, AB; Kindy, MS; Merchenthaler, I; Gage, FH; Wise, PM. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors alpha and beta. J. Comp. Neurol 2007, 500, 1064–1075. [Google Scholar]
- Simpkins, JW; Yi, KD; Yang, SH. Role of protein phosphatases and mitochondria in the neuroprotective effects of estrogens. Front. Neuroendocrinol 2009, 30, 93–105. [Google Scholar]
- Singer, CA; Rogers, KL; Strickland, TM; Dorsa, DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci. Lett 1996, 212, 13–16. [Google Scholar]
- Weaver, CE, Jr; Park-Chung, M; Gibbs, TT; Farb, DH. 17Beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res 1997, 761, 338–341. [Google Scholar]
- Carswell, HV; Macrae, IM; Farr, TD. Complexities of oestrogen in stroke. Clin. Sci. (London) 2010, 118, 375–389. [Google Scholar]
- Macrae, IM; Carswell, HV. Oestrogen and stroke: The potential for harm as well as benefit. Biochem. Soc. Trans 2006, 34, 1362–1365. [Google Scholar]
- Duckles, SP; Krause, DN. Cerebrovascular effects of oestrogen: Multiplicity of action. Clin. Exp. Pharmacol. Physiol 2007, 34, 801–808. [Google Scholar]
- Simpkins, JW; Singh, M. More than a decade of estrogen neuroprotection. Alzheimers Dement 2008, 4, S131–136. [Google Scholar]
- Lebesgue, D; Chevaleyre, V; Zukin, RS; Etgen, AM. Estradiol rescues neurons from global ischemia-induced cell death: Multiple cellular pathways of neuroprotection. Steroids 2009, 74, 555–561. [Google Scholar]
- Arnold, S; Beyer, C. Neuroprotection by estrogen in the brain: The mitochondrial compartment as presumed therapeutic target. J. Neurochem 2009, 110, 1–11. [Google Scholar]
- Sherwin, BB. Estrogen therapy: Is time of initiation critical for neuroprotection? Nat. Rev. Endocrinol 2009, 5, 620–627. [Google Scholar]
- Fujimura, M; Tominaga, T; Chan, PH. Neuroprotective effect of an antioxidant in ischemic brain injury: Involvement of neuronal apoptosis. Neurocrit. Care 2005, 2, 59–66. [Google Scholar]
- Behl, C; Skutella, T; Lezoualc’h, F; Post, A; Widmann, M; Newton, CJ; Holsboer, F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol. Pharmacol 1997, 51, 535–541. [Google Scholar]
- Culmsee, C; Vedder, H; Ravati, A; Junker, V; Otto, D; Ahlemeyer, B; Krieg, JC; Krieglstein, J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: Evidence for a receptor-independent antioxidative mechanism. J. Cereb. Blood Flow Metab 1999, 19, 1263–1269. [Google Scholar]
- Vedder, H; Anthes, N; Stumm, G; Wurz, C; Behl, C; Krieg, JC. Estrogen hormones reduce lipid peroxidation in cells and tissues of the central nervous system. J. Neurochem 1999, 72, 2531–2538. [Google Scholar]
- Ayres, S; Abplanalp, W; Liu, JH; Subbiah, MT. Mechanisms involved in the protective effect of estradiol-17beta on lipid peroxidation and DNA damage. Am. J. Physiol 1998, 274, E1002–1008. [Google Scholar]
- Rattanajarasroj, S; Unchern, S. Comparable attenuation of Abeta(25–35)-induced neurotoxicity by quercitrin and 17beta-estradiol in cultured rat hippocampal neurons. Neurochem. Res 2010, 35, 1196–1205. [Google Scholar]
- Wang, X; Dykens, JA; Perez, E; Liu, R; Yang, S; Covey, DF; Simpkins, JW. Neuroprotective effects of 17beta-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol. Pharmacol 2006, 70, 395–404. [Google Scholar]
- Keller, JN; Germeyer, A; Begley, JG; Mattson, MP. 17Beta-estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J. Neurosci. Res 1997, 50, 522–530. [Google Scholar]
- Prokai, L; Simpkins, JW. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol. Ther 2007, 114, 1–12. [Google Scholar]
- Behl, C; Widmann, M; Trapp, T; Holsboer, F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem. Biophys. Res. Commun 1995, 216, 473–482. [Google Scholar]
- Bonnefont, AB; Munoz, FJ; Inestrosa, NC. Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase-amyloid complexes. FEBS Lett 1998, 441, 220–224. [Google Scholar]
- Prokai, L; Prokai-Tatrai, K; Perjesi, P; Zharikova, AD; Perez, EJ; Liu, R; Simpkins, JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc. Natl. Acad. Sci. USA 2003, 100, 11741–11746. [Google Scholar]
- Prokai-Tatrai, K; Perjesi, P; Rivera-Portalatin, NM; Simpkins, JW; Prokai, L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids 2008, 73, 280–288. [Google Scholar]
- Picazo, O; Azcoitia, I; Garcia-Segura, LM. Neuroprotective and neurotoxic effects of estrogens. Brain Res 2003, 990, 20–27. [Google Scholar]
- Chen, C; Hu, Q; Yan, J; Lei, J; Qin, L; Shi, X; Luan, L; Yang, L; Wang, K; Han, J; Nanda, A; Zhou, C. Multiple effects of 2ME2 and D609 on the cortical expression of HIF-1alpha and apoptotic genes in a middle cerebral artery occlusion-induced focal ischemia rat model. J. Neurochem 2007, 102, 1831–1841. [Google Scholar]
- Bruce-Keller, AJ; Keeling, JL; Keller, JN; Huang, FF; Camondola, S; Mattson, MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar]
- Diwakar, L; Kenchappa, RS; Annepu, J; Ravindranath, V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: Implications in excitotoxicity. Neurochem. Int 2007, 51, 37–46. [Google Scholar]
- Schmidt, AJ; Krieg, JC; Vedder, H. Differential effects of glucocorticoids and gonadal steroids on glutathione levels in neuronal and glial cell systems. J. Neurosci. Res 2002, 67, 544–550. [Google Scholar]
- Diwakar, L; Kenchappa, RS; Annepu, J; Saeed, U; Sujanitha, R; Ravindranath, V. Down-regulation of glutaredoxin by estrogen receptor antagonist renders female mice susceptible to excitatory amino acid mediated complex I inhibition in CNS. Brain Res 2006, 1125, 176–184. [Google Scholar]
- Urata, Y; Ihara, Y; Murata, H; Goto, S; Koji, T; Yodoi, J; Inoue, S; Kondo, T. 17Beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J. Biol. Chem 2006, 281, 13092–13102. [Google Scholar]
- Ozacmak, VH; Sayan, H. The effects of 17beta estradiol, 17alpha estradiol and progesterone on oxidative stress biomarkers in ovariectomized female rat brain subjected to global cerebral ischemia. Physiol. Res 2009, 58, 909–912. [Google Scholar]
- Gottipati, S; Cammarata, PR. Mitochondrial superoxide dismutase activation with 17 beta-estradiol-treated human lens epithelial cells. Mol. Vis 2008, 14, 898–905. [Google Scholar]
- Pedram, A; Razandi, M; Wallace, DC; Levin, ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol. Biol. Cell 2006, 17, 2125–2137. [Google Scholar]
- Tripanichkul, W; Sripanichkulchai, K; Duce, JA; Finkelstein, DI. 17Beta-estradiol reduces nitrotyrosine immunoreactivity and increases SOD1 and SOD2 immunoreactivity in nigral neurons in male mice following MPTP insult. Brain Res 2007, 1164, 24–31. [Google Scholar]
- Strehlow, K; Rotter, S; Wassmann, S; Adam, O; Grohe, C; Laufs, K; Bohm, M; Nickenig, G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res 2003, 93, 170–177. [Google Scholar]
- Nilsen, J; Irwin, RW; Gallaher, TK; Brinton, RD. Estradiol in vivo regulation of brain mitochondrial proteome. J. Neurosci 2007, 27, 14069–14077. [Google Scholar]
- Irwin, RW; Yao, J; Hamilton, RT; Cadenas, E; Brinton, RD; Nilsen, J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology 2008, 149, 3167–3175. [Google Scholar]
- Miller, AA; Drummond, GR; Mast, AE; Schmidt, HH; Sobey, CG. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: Role of estrogen. Stroke 2007, 38, 2142–2149. [Google Scholar]
- Zhang, QG; Raz, L; Wang, R; Han, D; De Sevilla, L; Yang, F; Vadlamudi, RK; Brann, DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J. Neurosci 2009, 29, 13823–13836. [Google Scholar]
- Kumar, S; Lata, K; Mukhopadhyay, S; Mukherjee, TK. Role of estrogen receptors in pro-oxidative and anti-oxidative actions of estrogens: A perspective. Biochim. Biophys. Acta 2010, 1800, 1127–1135. [Google Scholar]
- Sastre-Serra, J; Valle, A; Company, MM; Garau, I; Oliver, J; Roca, P. Estrogen down-regulates uncoupling proteins and increases oxidative stress in breast cancer. Free Radic. Biol. Med 2010, 48, 506–512. [Google Scholar]
- Felty, Q; Xiong, WC; Sun, D; Sarkar, S; Singh, KP; Parkash, J; Roy, D. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry 2005, 44, 6900–6909. [Google Scholar]
- Rempel, MA; Hester, B; Deharo, H; Hong, H; Wang, Y; Schlenk, D. Effects of 17beta-estradiol, and its metabolite, 4-hydroxyestradiol on fertilization, embryo development and oxidative DNA damage in sand dollar (Dendraster excentricus) sperm. Sci. Total Environ 2009, 407, 2209–2215. [Google Scholar]
- Symonds, DA; Merchenthaler, I; Flaws, JA. Methoxychlor and estradiol induce oxidative stress DNA damage in the mouse ovarian surface epithelium. Toxicol. Sci 2008, 105, 182–187. [Google Scholar]
- Pajovic, SB; Saicic, ZS; Spasic, MB; Petrovic, VM. The effect of ovarian hormones on antioxidant enzyme activities in the brain of male rats. Physiol. Res 2003, 52, 189–194. [Google Scholar]
- Genc, S; Gurdol, F; Oner-Iyidogan, Y; Suzme, R. Acute effects of estradiol and of diethylstilbestrol: Pro- or antioxidant potential? Res. Commun. Mol. Pathol. Pharmacol 1999, 105, 253–261. [Google Scholar]
- Ting, CM; Lee, YM; Wong, CK; Wong, AS; Lung, HL; Lung, ML; Lo, KW; Wong, RN; Mak, NK. 2-Methoxyestradiol induces endoreduplication through the induction of mitochondrial oxidative stress and the activation of MAPK signaling pathways. Biochem. Pharmacol 2010, 79, 825–841. [Google Scholar]
- Park, SA; Na, HK; Kim, EH; Cha, YN; Surh, YJ. 4-hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of IkappaB kinase: potential role of reactive oxygen species. Cancer Res 2009, 69, 2416–2424. [Google Scholar]
- She, MR; Li, JG; Guo, KY; Lin, W; Du, X; Niu, XQ. Requirement of reactive oxygen species generation in apoptosis of leukemia cells induced by 2-methoxyestradiol. Acta Pharmacol. Sin 2007, 28, 1037–1044. [Google Scholar]
- Jin, R; Yang, G; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol 2010, 87, 779–789. [Google Scholar]
- Barone, FC; Arvin, B; White, RF; Miller, A; Webb, CL; Willette, RN; Lysko, PG; Feuerstein, GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997, 28, 1233–1244. [Google Scholar]
- Yamasaki, Y; Matsuura, N; Shozuhara, H; Onodera, H; Itoyama, Y; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–680, discussion 681. [Google Scholar]
- Czlonkowska, A; Ciesielska, A; Gromadzka, G; Kurkowska-Jastrzebska, I. Gender differences in neurological disease: Role of estrogens and cytokines. Endocrine 2006, 29, 243–256. [Google Scholar]
- Mulcahy, NJ; Ross, J; Rothwell, NJ; Loddick, SA. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br. J. Pharmacol 2003, 140, 471–476. [Google Scholar]
- Relton, JK; Martin, D; Thompson, RC; Russell, DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp. Neurol 1996, 138, 206–213. [Google Scholar]
- Nawashiro, H; Martin, D; Hallenbeck, JM. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J. Cereb. Blood Flow Metab 1997, 17, 229–232. [Google Scholar]
- Martin-Villalba, A; Hahne, M; Kleber, S; Vogel, J; Falk, W; Schenkel, J; Krammer, PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ 2001, 8, 679–686. [Google Scholar]
- Loddick, SA; Rothwell, NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J. Cereb. Blood Flow Metab 1996, 16, 932–940. [Google Scholar]
- Santizo, RA; Anderson, S; Ye, S; Koenig, HM; Pelligrino, DA. Effects of estrogen on leukocyte adhesion after transient forebrain ischemia. Stroke 2000, 31, 2231–2235. [Google Scholar]
- Mori, M; Tsukahara, F; Yoshioka, T; Irie, K; Ohta, H. Suppression by 17beta-estradiol of monocyte adhesion to vascular endothelial cells is mediated by estrogen receptors. Life Sci 2004, 75, 599–609. [Google Scholar]
- Nathan, L; Pervin, S; Singh, R; Rosenfeld, M; Chaudhuri, G. Estradiol inhibits leukocyte adhesion and transendothelial migration in rabbits in vivo: Possible mechanisms for gender differences in atherosclerosis. Circ. Res 1999, 85, 377–385. [Google Scholar]
- Suzuki, S; Brown, CM; Dela Cruz, CD; Yang, E; Bridwell, DA; Wise, PM. Timing of estrogen therapy after ovariectomy dictates the efficacy of its neuroprotective and antiinflammatory actions. Proc. Natl. Acad. Sci. USA 2007, 104, 6013–6018. [Google Scholar]
- Chiappetta, O; Gliozzi, M; Siviglia, E; Amantea, D; Morrone, LA; Berliocchi, L; Bagetta, G; Corasaniti, MT. Evidence to implicate early modulation of interleukin-1beta expression in the neuroprotection afforded by 17beta-estradiol in male rats undergone transient middle cerebral artery occlusion. Int. Rev. Neurobiol 2007, 82, 357–372. [Google Scholar]
- Nordell, VL; Scarborough, MM; Buchanan, AK; Sohrabji, F. Differential effects of estrogen in the injured forebrain of young adult and reproductive senescent animals. Neurobiol. Aging 2003, 24, 733–743. [Google Scholar]
- Tenenbaum, M; Azab, AN; Kaplanski, J. Effects of estrogen against LPS-induced inflammation and toxicity in primary rat glial and neuronal cultures. J. Endotoxin Res 2007, 13, 158–166. [Google Scholar]
- Vegeto, E; Bonincontro, C; Pollio, G; Sala, A; Viappiani, S; Nardi, F; Brusadelli, A; Viviani, B; Ciana, P; Maggi, A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci 2001, 21, 1809–1818. [Google Scholar]
- Lewis, DK; Johnson, AB; Stohlgren, S; Harms, A; Sohrabji, F. Effects of estrogen receptor agonists on regulation of the inflammatory response in astrocytes from young adult and middle-aged female rats. J. Neuroimmunol 2008, 195, 47–59. [Google Scholar]
- Ray, P; Ghosh, SK; Zhang, DH; Ray, A. Repression of interleukin-6 gene expression by 17beta-estradiol: Inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Lett 1997, 409, 79–85. [Google Scholar]
- Ospina, JA; Brevig, HN; Krause, DN; Duckles, SP. Estrogen suppresses IL-1beta-mediated induction of COX-2 pathway in rat cerebral blood vessels. Am. J. Physiol. Heart Circ. Physiol 2004, 286, H2010–2019. [Google Scholar]
- Vegeto, E; Belcredito, S; Etteri, S; Ghisletti, S; Brusadelli, A; Meda, C; Krust, A; Dupont, S; Ciana, P; Chambon, P; Maggi, A. Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc. Natl. Acad. Sci. USA 2003, 100, 9614–9619. [Google Scholar]
- Vegeto, E; Benedusi, V; Maggi, A. Estrogen anti-inflammatory activity in brain: A therapeutic opportunity for menopause and neurodegenerative diseases. Front. Neuroendocrinol 2008, 29, 507–519. [Google Scholar]
- Sunday, L; Osuna, C; Krause, DN; Duckles, SP. Age alters cerebrovascular inflammation and effects of estrogen. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2333–2340. [Google Scholar]
- Srivastava, S; Weitzmann, MN; Cenci, S; Ross, FP; Adler, S; Pacifici, R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J. Clin. Invest 1999, 104, 503–513. [Google Scholar]
- Yatkin, E; Bernoulli, J; Talvitie, EM; Santti, R. Inflammation and epithelial alterations in rat prostate: Impact of the androgen to oestrogen ratio. Int. J. Androl 2009, 32, 399–410. [Google Scholar]
- Salem, ML; Hossain, MS; Nomoto, K. Mediation of the immunomodulatory effect of beta-estradiol on inflammatory responses by inhibition of recruitment and activation of inflammatory cells and their gene expression of TNF-alpha and IFN-gamma. Int. Arch. Allergy Immunol 2000, 121, 235–245. [Google Scholar]
- Wen, Y; Yang, S; Liu, R; Perez, E; Yi, KD; Koulen, P; Simpkins, JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res 2004, 1008, 147–154. [Google Scholar]
- Lei, DL; Long, JM; Hengemihle, J; O’Neill, J; Manaye, KF; Ingram, DK; Mouton, PR. Effects of estrogen and raloxifene on neuroglia number and morphology in the hippocampus of aged female mice. Neuroscience 2003, 121, 659–666. [Google Scholar]
- Brown, CM; Dela Cruz, CD; Yang, E; Wise, PM. Inducible nitric oxide synthase and estradiol exhibit complementary neuroprotective roles after ischemic brain injury. Exp. Neurol 2008, 210, 782–787. [Google Scholar]
- Park, EM; Cho, S; Frys, KA; Glickstein, SB; Zhou, P; Anrather, J; Ross, ME; Iadecola, C. Inducible nitric oxide synthase contributes to gender differences in ischemic brain injury. J. Cereb. Blood Flow Metab 2006, 26, 392–401. [Google Scholar]
- Xu, HL; Baughman, VL; Pelligrino, DA. Estrogen replacement treatment in diabetic ovariectomized female rats potentiates postischemic leukocyte adhesion in cerebral venules. Stroke 2004, 35, 1974–1978. [Google Scholar]
- Soucy, G; Boivin, G; Labrie, F; Rivest, S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J. Immunol 2005, 174, 6391–6398. [Google Scholar]
- Johnson, AB; Sohrabji, F. Estrogen’s effects on central and circulating immune cells vary with reproductive age. Neurobiol. Aging 2005, 26, 1365–1374. [Google Scholar]
- Marriott, LK; Hauss-Wegrzyniak, B; Benton, RS; Vraniak, PD; Wenk, GL. Long-term estrogen therapy worsens the behavioral and neuropathological consequences of chronic brain inflammation. Behav. Neurosci 2002, 116, 902–911. [Google Scholar]
- O’Donnell, ME; Lam, TI; Tran, LQ; Foroutan, S; Anderson, SE. Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J. Cereb. Blood Flow Metab 2006, 26, 1234–1249. [Google Scholar]
- Krause, DN; Duckles, SP; Pelligrino, DA. Influence of sex steroid hormones on cerebrovascular function. J. Appl. Physiol 2006, 101, 1252–1261. [Google Scholar]
- Wise, PM; Dubal, DB. Estradiol protects against ischemic brain injury in middle-aged rats. Biol. Reprod 2000, 63, 982–985. [Google Scholar]
- Dubal, DB; Wise, PM. Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology 2001, 142, 43–48. [Google Scholar]
- Toung, TJ; Chen, TY; Littleton-Kearney, MT; Hurn, PD; Murphy, SJ. Effects of combined estrogen and progesterone on brain infarction in reproductively senescent female rats. J. Cereb. Blood Flow Metab 2004, 24, 1160–1166. [Google Scholar]
- Alkayed, NJ; Murphy, SJ; Traystman, RJ; Hurn, PD; Miller, VM. Neuroprotective effects of female gonadal steroids in reproductively senescent female rats. Stroke 2000, 31, 161–168. [Google Scholar]
- Toung, TK; Hurn, PD; Traystman, RJ; Sieber, FE. Estrogen decreases infarct size after temporary focal ischemia in a genetic model of type 1 diabetes mellitus. Stroke 2000, 31, 2701–2706. [Google Scholar]
- Brouns, R; De Deyn, PP. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg 2009, 111, 483–495. [Google Scholar]
- Brann, DW; Zamorano, PL; Chorich, LP; Mahesh, VB. Steroid hormone effects on NMDA receptor binding and NMDA receptor mRNA levels in the hypothalamus and cerebral cortex of the adult rat. Neuroendocrinology 1993, 58, 666–672. [Google Scholar]
- Woolley, CS; Weiland, NG; McEwen, BS; Schwartzkroin, PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: Correlation with dendritic spine density. J. Neurosci 1997, 17, 1848–1859. [Google Scholar]
- Weiland, NG. Estradiol selectively regulates agonist binding sites on the N-methyl-D-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology 1992, 131, 662–668. [Google Scholar]
- Woolley, CS; McEwen, BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J. Comp. Neurol 1993, 336, 293–306. [Google Scholar]
- Woolley, CS; McEwen, BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J. Neurosci 1992, 12, 2549–2554. [Google Scholar]
- Pozzo-Miller, LD; Inoue, T; Murphy, DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J. Neurophysiol 1999, 81, 1404–1411. [Google Scholar]
- Buterbaugh, GG; Hudson, GM. Estradiol replacement to female rats facilitates dorsal hippocampal but not ventral hippocampal kindled seizure acquisition. Exp. Neurol 1991, 111, 55–64. [Google Scholar]
- Warren, SG; Humphreys, AG; Juraska, JM; Greenough, WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res 1995, 703, 26–30. [Google Scholar]
- Foy, MR; Xu, J; Xie, X; Brinton, RD; Thompson, RF; Berger, TW. 17Beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J. Neurophysiol 1999, 81, 925–929. [Google Scholar]
- Teyler, TJ; Vardaris, RM; Lewis, D; Rawitch, AB. Gonadal steroids: Effects on excitability of hippocampal pyramidal cells. Science 1980, 209, 1017–1018. [Google Scholar]
- Smith, SS; Waterhouse, BD; Woodward, DJ. Sex steroid effects on extrahypothalamic CNS. I. Estrogen augments neuronal responsiveness to iontophoretically applied glutamate in the cerebellum. Brain Res 1987, 422, 40–51. [Google Scholar]
- Sato, K; Matsuki, N; Ohno, Y; Nakazawa, K. Estrogens inhibit l-glutamate uptake activity of astrocytes via membrane estrogen receptor alpha. J. Neurochem 2003, 86, 1498–1505. [Google Scholar]
- Gu, Q; Moss, RL. 17Beta-Estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J. Neurosci 1996, 16, 3620–3629. [Google Scholar]
- Johansen, FF; Jorgensen, MB; Diemer, NH. Ischemic CA-1 pyramidal cell loss is prevented by preischemic colchicine destruction of dentate gyrus granule cells. Brain Res 1986, 377, 344–347. [Google Scholar]
- Onodera, H; Sato, G; Kogure, K. Lesions to Schaffer collaterals prevent ischemic death of CA1 pyramidal cells. Neurosci. Lett 1986, 68, 169–174. [Google Scholar]
- Wieloch, T; Lindvall, O; Blomqvist, P; Gage, FH. Evidence for amelioration of ischaemic neuronal damage in the hippocampal formation by lesions of the perforant path. Neurol. Res 1985, 7, 24–26. [Google Scholar]
- Ito, H; Watanabe, Y; Isshiki, A; Uchino, H. Neuroprotective properties of propofol and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiol. Scand 1999, 43, 153–162. [Google Scholar]
- Schwartz-Bloom, RD; McDonough, KJ; Chase, PJ; Chadwick, LE; Inglefield, JR; Levin, ED. Long-term neuroprotection by benzodiazepine full versus partial agonists after transient cerebral ischemia in the gerbil [corrected]. J. Cereb. Blood Flow Metab 1998, 18, 548–558. [Google Scholar]
- Goodman, Y; Bruce, AJ; Cheng, B; Mattson, MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J. Neurochem 1996, 66, 1836–1844. [Google Scholar]
- Azcoitia, I; Fernandez-Galaz, C; Sierra, A; Garcia-Segura, LM. Gonadal hormones affect neuronal vulnerability to excitotoxin-induced degeneration. J. Neurocytol 1999, 28, 699–710. [Google Scholar]
- Mendelowitsch, A; Ritz, MF; Ros, J; Langemann, H; Gratzl, O. 17beta-Estradiol reduces cortical lesion size in the glutamate excitotoxicity model by enhancing extracellular lactate: A new neuroprotective pathway. Brain Res 2001, 901, 230–236. [Google Scholar]
- DeGiorgio, LA; Attardi, B; Shimizu, Y; Ogata, M; Volpe, BT. 17Beta-estradiol treatment retards excitotoxic delayed degeneration in substantia nigra reticulata neurons. Brain Res 2002, 936, 15–20. [Google Scholar]
- Ritz, MF; Schmidt, P; Mendelowitsch, A. 17Beta-estradiol effect on the extracellular concentration of amino acids in the glutamate excitotoxicity model in the rat. Neurochem. Res 2002, 27, 1677–1683. [Google Scholar]
- Ritz, MF; Schmidt, P; Mendelowitsch, A. Acute effects of 17beta-estradiol on the extracellular concentration of excitatory amino acids and energy metabolites during transient cerebral ischemia in male rats. Brain Res 2004, 1022, 157–163. [Google Scholar]
- Zhang, H; Xie, M; Schools, GP; Feustel, PF; Wang, W; Lei, T; Kimelberg, HK; Zhou, M. Tamoxifen mediated estrogen receptor activation protects against early impairment of hippocampal neuron excitability in an oxygen/glucose deprivation brain slice ischemia model. Brain Res 2009, 1247, 196–211. [Google Scholar]
- Zhang, F; Yin, W; Chen, J. Apoptosis in cerebral ischemia: Executional and regulatory signaling mechanisms. Neurol. Res 2004, 26, 835–845. [Google Scholar]
- Niizuma, K; Yoshioka, H; Chen, H; Kim, GS; Jung, JE; Katsu, M; Okami, N; Chan, PH. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar]
- Broughton, BR; Reutens, DC; Sobey, CG. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–339. [Google Scholar]
- Estus, S; Tucker, HM; van Rooyen, C; Wright, S; Brigham, EF; Wogulis, M; Rydel, RE. Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J. Neurosci 1997, 17, 7736–7745. [Google Scholar]
- Estus, S; Zaks, WJ; Freeman, RS; Gruda, M; Bravo, R; Johnson, EM, Jr. Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol 1994, 127, 1717–1727. [Google Scholar]
- Rau, SW; Dubal, DB; Bottner, M; Wise, PM. Estradiol differentially regulates c-Fos after focal cerebral ischemia. J. Neurosci 2003, 23, 10487–10494. [Google Scholar]
- Kitagawa, K; Matsumoto, M; Tsujimoto, Y; Ohtsuki, T; Kuwabara, K; Matsushita, K; Yang, G; Tanabe, H; Martinou, JC; Hori, M; Yanagihara, T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke 1998, 29, 2616–2621. [Google Scholar]
- Alkayed, NJ; Goto, S; Sugo, N; Joh, HD; Klaus, J; Crain, BJ; Bernard, O; Traystman, RJ; Hurn, PD. Estrogen and Bcl-2: Gene induction and effect of transgene in experimental stroke. J. Neurosci 2001, 21, 7543–7550. [Google Scholar]
- Krajewski, S; Mai, JK; Krajewska, M; Sikorska, M; Mossakowski, MJ; Reed, JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J. Neurosci 1995, 15, 6364–6376. [Google Scholar]
- Dubal, DB; Shughrue, PJ; Wilson, ME; Merchenthaler, I; Wise, PM. Estradiol modulates bcl-2 in cerebral ischemia: A potential role for estrogen receptors. J. Neurosci 1999, 19, 6385–6393. [Google Scholar]
- Jover, T; Tanaka, H; Calderone, A; Oguro, K; Bennett, MV; Etgen, AM; Zukin, RS. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J. Neurosci 2002, 22, 2115–2124. [Google Scholar]
- Pike, CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: Relevance to Alzheimer’s disease. J. Neurochem 1999, 72, 1552–1563. [Google Scholar]
- Singer, CA; Rogers, KL; Dorsa, DM. Modulation of Bcl-2 expression: A potential component of estrogen protection in NT2 neurons. Neuroreport 1998, 9, 2565–2568. [Google Scholar]
- Kandouz, M; Siromachkova, M; Jacob, D; Chretien Marquet, B; Therwath, A; Gompel, A. Antagonism between estradiol and progestin on Bcl-2 expression in breast-cancer cells. Int. J. Cancer 1996, 68, 120–125. [Google Scholar]
- Yune, TY; Park, HG; Lee, JY; Oh, TH. Estrogen-induced Bcl-2 expression after spinal cord injury is mediated through phosphoinositide-3-kinase/Akt-dependent CREB activation. J. Neurotrauma 2008, 25, 1121–1131. [Google Scholar]
- Fan, L; Pandey, SC; Cohen, RS. Estrogen affects levels of Bcl-2 protein and mRNA in medial amygdala of ovariectomized rats. J. Neurosci. Res 2008, 86, 3655–3664. [Google Scholar]
- Sharma, K; Mehra, RD. Long-term administration of estrogen or tamoxifen to ovariectomized rats affords neuroprotection to hippocampal neurons by modulating the expression of Bcl-2 and Bax. Brain Res 2008, 1204, 1–15. [Google Scholar]
- Gollapudi, L; Oblinger, MM. Estrogen and NGF synergistically protect terminally differentiated, ERalpha-transfected PC12 cells from apoptosis. J. Neurosci. Res 1999, 56, 471–481. [Google Scholar]
- Honda, K; Shimohama, S; Sawada, H; Kihara, T; Nakamizo, T; Shibasaki, H; Akaike, A. Nongenomic antiapoptotic signal transduction by estrogen in cultured cortical neurons. J. Neurosci. Res 2001, 64, 466–475. [Google Scholar]
- Lee, SY; Andoh, T; Murphy, DL; Chiueh, CC. 17Beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. FASEB J 2003, 17, 947–948. [Google Scholar]
- Zhang, Y; Bhavnani, BR. Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci 2005, 6, 13. [Google Scholar]
- Teixeira, C; Reed, JC; Pratt, MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res 1995, 55, 3902–3907. [Google Scholar]
- Funakoshi, T; Yanai, A; Shinoda, K; Kawano, MM; Mizukami, Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun 2006, 346, 904–910. [Google Scholar]
- Simpkins, JW; Wang, J; Wang, X; Perez, E; Prokai, L; Dykens, JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr. Drug. Targets CNS Neurol. Disord 2005, 4, 69–83. [Google Scholar]
- Hansen, TM; Moss, AJ; Brindle, NP. Vascular endothelial growth factor and angiopoietins in neurovascular regeneration and protection following stroke. Curr. Neurovasc. Res 2008, 5, 236–245. [Google Scholar]
- Garcia-Segura, LM; Azcoitia, I; DonCarlos, LL. Neuroprotection by estradiol. Prog. Neurobiol 2001, 63, 29–60. [Google Scholar] [Green Version]
- Jones, KJ. Gonadal steroids as promoting factors in axonal regeneration. Brain Res. Bull 1993, 30, 491–498. [Google Scholar]
- Garcia-Segura, LM; Sanz, A; Mendez, P. Cross-talk between IGF-I and estradiol in the brain: Focus on neuroprotection. Neuroendocrinology 2006, 84, 275–279. [Google Scholar]
- Duenas, M; Luquin, S; Chowen, JA; Torres-Aleman, I; Naftolin, F; Garcia-Segura, LM. Gonadal hormone regulation of insulin-like growth factor-I-like immunoreactivity in hypothalamic astroglia of developing and adult rats. Neuroendocrinology 1994, 59, 528–538. [Google Scholar]
- Jezierski, MK; Sohrabji, F. Region- and peptide-specific regulation of the neurotrophins by estrogen. Brain Res. Mol. Brain Res 2000, 85, 77–84. [Google Scholar]
- Gibbs, RB. Treatment with estrogen and progesterone affects relative levels of brain-derived neurotrophic factor mRNA and protein in different regions of the adult rat brain. Brain Res 1999, 844, 20–27. [Google Scholar]
- Sohrabji, F; Miranda, RC; Toran-Allerand, CD. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11110–11114. [Google Scholar]
- Dhandapani, KM; Wade, FM; Mahesh, VB; Brann, DW. Astrocyte-derived transforming growth factor-{beta} mediates the neuroprotective effects of 17{beta}-estradiol: Involvement of nonclassical genomic signaling pathways. Endocrinology 2005, 146, 2749–2759. [Google Scholar]
- Mueller, MD; Vigne, JL; Minchenko, A; Lebovic, DI; Leitman, DC; Taylor, RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors alpha and beta. Proc. Natl. Acad. Sci. USA 2000, 97, 10972–10977. [Google Scholar]
- El-Ashry, D; Chrysogelos, SA; Lippman, ME; Kern, FG. Estrogen induction of TGF-alpha is mediated by an estrogen response element composed of two imperfect palindromes. J. Steroid Biochem. Mol. Biol 1996, 59, 261–269. [Google Scholar]
- Ferreira, A; Caceres, A. Estrogen-enhanced neurite growth: Evidence for a selective induction of Tau and stable microtubules. J. Neurosci 1991, 11, 392–400. [Google Scholar]
- Singh, M; Setalo, G, Jr; Guan, X; Frail, DE; Toran-Allerand, CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J. Neurosci 2000, 20, 1694–1700. [Google Scholar]
- Mendez, P; Azcoitia, I; Garcia-Segura, LM. Estrogen receptor alpha forms estrogen-dependent multimolecular complexes with insulin-like growth factor receptor and phosphatidylinositol 3-kinase in the adult rat brain. Brain Res. Mol. Brain Res 2003, 112, 170–176. [Google Scholar]
- Quesada, A; Micevych, PE. Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J. Neurosci. Res 2004, 75, 107–116. [Google Scholar]
- Azcoitia, I; Sierra, A; Garcia-Segura, LM. Neuroprotective effects of estradiol in the adult rat hippocampus: Interaction with insulin-like growth factor-I signalling. J. Neurosci. Res 1999, 58, 815–822. [Google Scholar]
- Jover-Mengual, T; Zukin, RS; Etgen, AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology 2007, 148, 1131–1143. [Google Scholar]
- Traub, ML; De Butte-Smith, M; Zukin, RS; Etgen, AM. Oestradiol and insulin-like growth factor-1 reduce cell loss after global ischaemia in middle-aged female rats. J. Neuroendocrinol 2009, 21, 1038–1044. [Google Scholar]
- Shughrue, PJ; Dorsa, DM. Estrogen modulates the growth-associated protein GAP-43 (Neuromodulin) mRNA in the rat preoptic area and basal hypothalamus. Neuroendocrinology 1993, 57, 439–447. [Google Scholar]
- Teter, B; Harris-White, ME; Frautschy, SA; Cole, GM. Role of apolipoprotein E and estrogen in mossy fiber sprouting in hippocampal slice cultures. Neuroscience 1999, 91, 1009–1016. [Google Scholar]
- Scoville, SA; Bufton, SM; Liuzzi, FJ. Estrogen regulates neurofilament gene expression in adult female rat dorsal root ganglion neurons. Exp. Neurol 1997, 146, 596–599. [Google Scholar]
- Salom, JB; Burguete, MC; Perez-Asensio, FJ; Torregrosa, G; Alborch, E. Relaxant effects of 17-beta-estradiol in cerebral arteries through Ca(2+) entry inhibition. J. Cereb. Blood Flow Metab 2001, 21, 422–429. [Google Scholar]
- Ono, H; Sasaki, Y; Bamba, E; Seki, J; Giddings, JC; Yamamoto, J. Cerebral thrombosis and microcirculation of the rat during the oestrous cycle and after ovariectomy. Clin. Exp. Pharmacol. Physiol 2002, 29, 73–78. [Google Scholar]
- Ospina, JA; Krause, DN; Duckles, SP. 17Beta-estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase-1 and prostacyclin synthase. Stroke 2002, 33, 600–605. [Google Scholar]
- Ospina, JA; Duckles, SP; Krause, DN. 17Beta-estradiol decreases vascular tone in cerebral arteries by shifting COX-dependent vasoconstriction to vasodilation. Am. J. Physiol. Heart Circ. Physiol 2003, 285, H241–250. [Google Scholar]
- Momoi, H; Ikomi, F; Ohhashi, T. Estrogen-induced augmentation of endothelium-dependent nitric oxide-mediated vasodilation in isolated rat cerebral small arteries. Jpn. J. Physiol 2003, 53, 193–203. [Google Scholar]
- Geary, GG; McNeill, AM; Ospina, JA; Krause, DN; Korach, KS; Duckles, SP. Selected contribution: Cerebrovascular nos and cyclooxygenase are unaffected by estrogen in mice lacking estrogen receptor-alpha. J Appl Physiol 2001, 91, 2391–2399, discussion 2389–2390. [Google Scholar]
- McNeill, AM; Kim, N; Duckles, SP; Krause, DN; Kontos, HA. Chronic estrogen treatment increases levels of endothelial nitric oxide synthase protein in rat cerebral microvessels. Stroke 1999, 30, 2186–2190. [Google Scholar]
- Geary, GG; Krause, DN; Duckles, SP. Gonadal hormones affect diameter of male rat cerebral arteries through endothelium-dependent mechanisms. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H610–618. [Google Scholar]
- Pelligrino, DA; Ye, S; Tan, F; Santizo, RA; Feinstein, DL; Wang, Q. Nitric-oxide-dependent pial arteriolar dilation in the female rat: Effects of chronic estrogen depletion and repletion. Biochem. Biophys. Res. Commun 2000, 269, 165–171. [Google Scholar]
- Stirone, C; Chu, Y; Sunday, L; Duckles, SP; Krause, DN. 17Beta-estradiol increases endothelial nitric oxide synthase mRNA copy number in cerebral blood vessels: Quantification by real-time polymerase chain reaction. Eur. J. Pharmacol 2003, 478, 35–38. [Google Scholar]
- Geary, GG; Krause, DN; Duckles, SP. Estrogen reduces myogenic tone through a nitric oxide-dependent mechanism in rat cerebral arteries. Am. J. Physiol 1998, 275, H292–300. [Google Scholar]
- Geary, GG; Krause, DN; Duckles, SP. Estrogen reduces mouse cerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H511–519. [Google Scholar]
- Skarsgard, P; van Breemen, C; Laher, I. Estrogen regulates myogenic tone in pressurized cerebral arteries by enhanced basal release of nitric oxide. Am. J. Physiol 1997, 273, H2248–2256. [Google Scholar]
- Sobey, CG; Weiler, JM; Boujaoude, M; Woodman, OL. Effect of short-term phytoestrogen treatment in male rats on nitric oxide-mediated responses of carotid and cerebral arteries: Comparison with 17beta-estradiol. J. Pharmacol. Exp. Ther 2004, 310, 135–140. [Google Scholar]
- Alkayed, NJ; Harukuni, I; Kimes, AS; London, ED; Traystman, RJ; Hurn, PD. Gender-linked brain injury in experimental stroke. Stroke 1998, 29, 159–165, discussion 166. [Google Scholar]
- Krejza, J; Siemkowicz, J; Sawicka, M; Szylak, A; Kochanowicz, J; Mariak, Z; Lewko, J; Spektor, V; Babikian, V; Bert, R. Oscillations of cerebrovascular resistance throughout the menstrual cycle in healthy women. Ultrasound Obstet. Gynecol 2003, 22, 627–632. [Google Scholar]
- McCullough, LD; Alkayed, NJ; Traystman, RJ; Williams, MJ; Hurn, PD. Postischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke 2001, 32, 796–802. [Google Scholar]
- Watanabe, Y; Littleton-Kearney, MT; Traystman, RJ; Hurn, PD. Estrogen restores postischemic pial microvascular dilation. Am. J. Physiol. Heart Circ. Physiol 2001, 281, H155–160. [Google Scholar]
- Coma, M; Guix, FX; Uribesalgo, I; Espuna, G; Sole, M; Andreu, D; Munoz, FJ. Lack of oestrogen protection in amyloid-mediated endothelial damage due to protein nitrotyrosination. Brain 2005, 128, 1613–1621. [Google Scholar]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar]
- Wang, Q; Santizo, R; Baughman, VL; Pelligrino, DA; Iadecola, C. Estrogen provides neuroprotection in transient forebrain ischemia through perfusion-independent mechanisms in rats. Stroke 1999, 30, 630–637. [Google Scholar]
- Carswell, HV; Anderson, NH; Morton, JJ; McCulloch, J; Dominiczak, AF; Macrae, IM. Investigation of estrogen status and increased stroke sensitivity on cerebral blood flow after a focal ischemic insult. J. Cereb. Blood Flow Metab 2000, 20, 931–936. [Google Scholar]
- Shi, J; Bui, JD; Yang, SH; He, Z; Lucas, TH; Buckley, DL; Blackband, SJ; King, MA; Day, AL; Simpkins, JW. Estrogens decrease reperfusion-associated cortical ischemic damage: An MRI analysis in a transient focal ischemia model. Stroke 2001, 32, 987–992. [Google Scholar]
- Rusa, R; Alkayed, NJ; Crain, BJ; Traystman, RJ; Kimes, AS; London, ED; Klaus, JA; Hurn, PD. 17Beta-estradiol reduces stroke injury in estrogen-deficient female animals. Stroke 1999, 30, 1665–1670. [Google Scholar]
- Choi, JM; Romeo, RD; Brake, WG; Bethea, CL; Rosenwaks, Z; McEwen, BS. Estradiol increases pre- and post-synaptic proteins in the CA1 region of the hippocampus in female rhesus macaques (Macaca mulatta). Endocrinology 2003, 144, 4734–4738. [Google Scholar]

{kind=link}
| Administration method | Pellet dose/silastic capsule concentration/injection dose ranges | |
|---|---|---|
| Neuroprotection | Neurodamage | |
| Slow-release pellets, subcutaneous | 0.025–0.25 mg [19,20] | 0.025–1.5 mg [6,8] |
| Silastic capsules filled with 17β-estradiol dissolved in oil, subcutaneous | 180–4000 μg/mL [21,22] | Not reported |
| Injections, subcutaneous | 10–5000 μg/kg BW [23,24] | Not reported |
| Injection, intravenous | 10–1000 μg/kg BW [2,25] | Not reported |
| Injection, intraperitoneal | 100–20,000 μg/kg BW [26,27] | Not reported |
| Injection, intramuscular | 100 μg/kg BW [28] | Not reported |
| Infusion, intraventricular | 50–150 μg [29,30] | Not reported |
| Oral administration | 10 μg/kg BW [31] | Not reported |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Strom, J.O.; Theodorsson, A.; Theodorsson, E. Mechanisms of Estrogens’ Dose-Dependent Neuroprotective and Neurodamaging Effects in Experimental Models of Cerebral Ischemia. Int. J. Mol. Sci. 2011, 12, 1533-1562. https://doi.org/10.3390/ijms12031533
Strom JO, Theodorsson A, Theodorsson E. Mechanisms of Estrogens’ Dose-Dependent Neuroprotective and Neurodamaging Effects in Experimental Models of Cerebral Ischemia. International Journal of Molecular Sciences. 2011; 12(3):1533-1562. https://doi.org/10.3390/ijms12031533
Chicago/Turabian StyleStrom, Jakob O., Annette Theodorsson, and Elvar Theodorsson. 2011. "Mechanisms of Estrogens’ Dose-Dependent Neuroprotective and Neurodamaging Effects in Experimental Models of Cerebral Ischemia" International Journal of Molecular Sciences 12, no. 3: 1533-1562. https://doi.org/10.3390/ijms12031533