Zeranol Down-Regulates p53 Expression in Primary Cultured Human Breast Cancer Epithelial Cells through Epigenetic Modification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
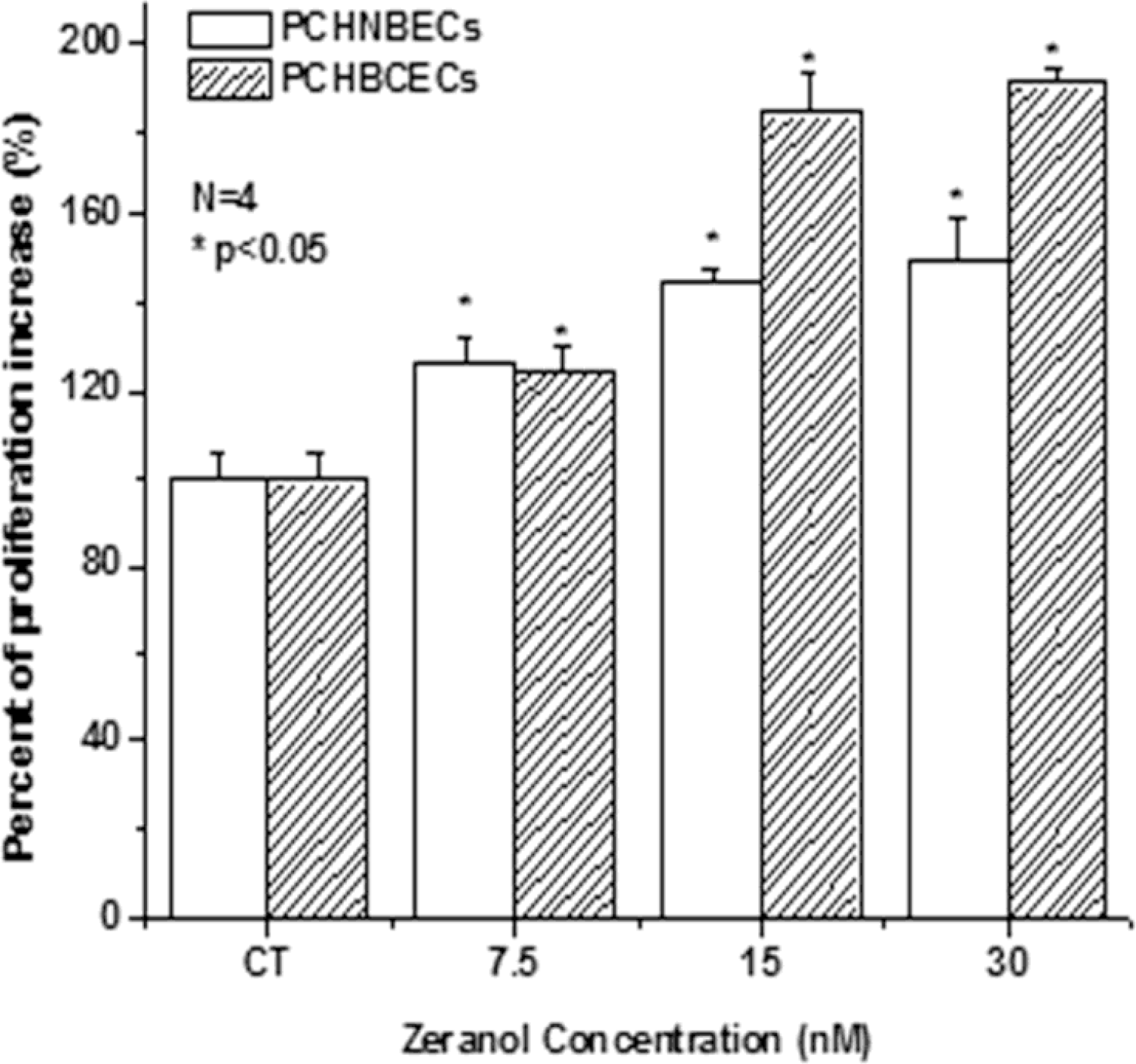
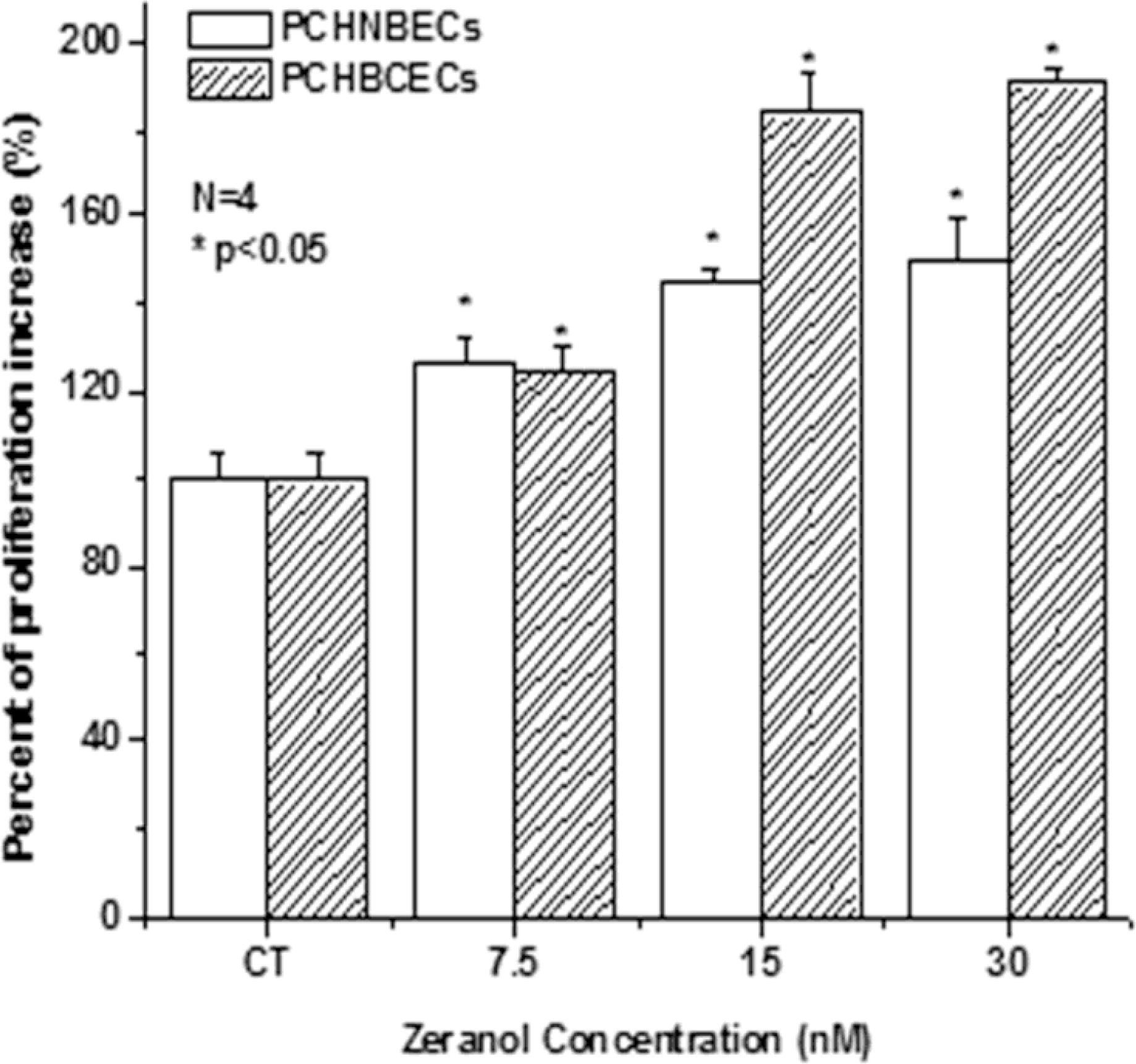
2.1. Z Increases the Proliferation of PCHNBECs and PCHBCECs
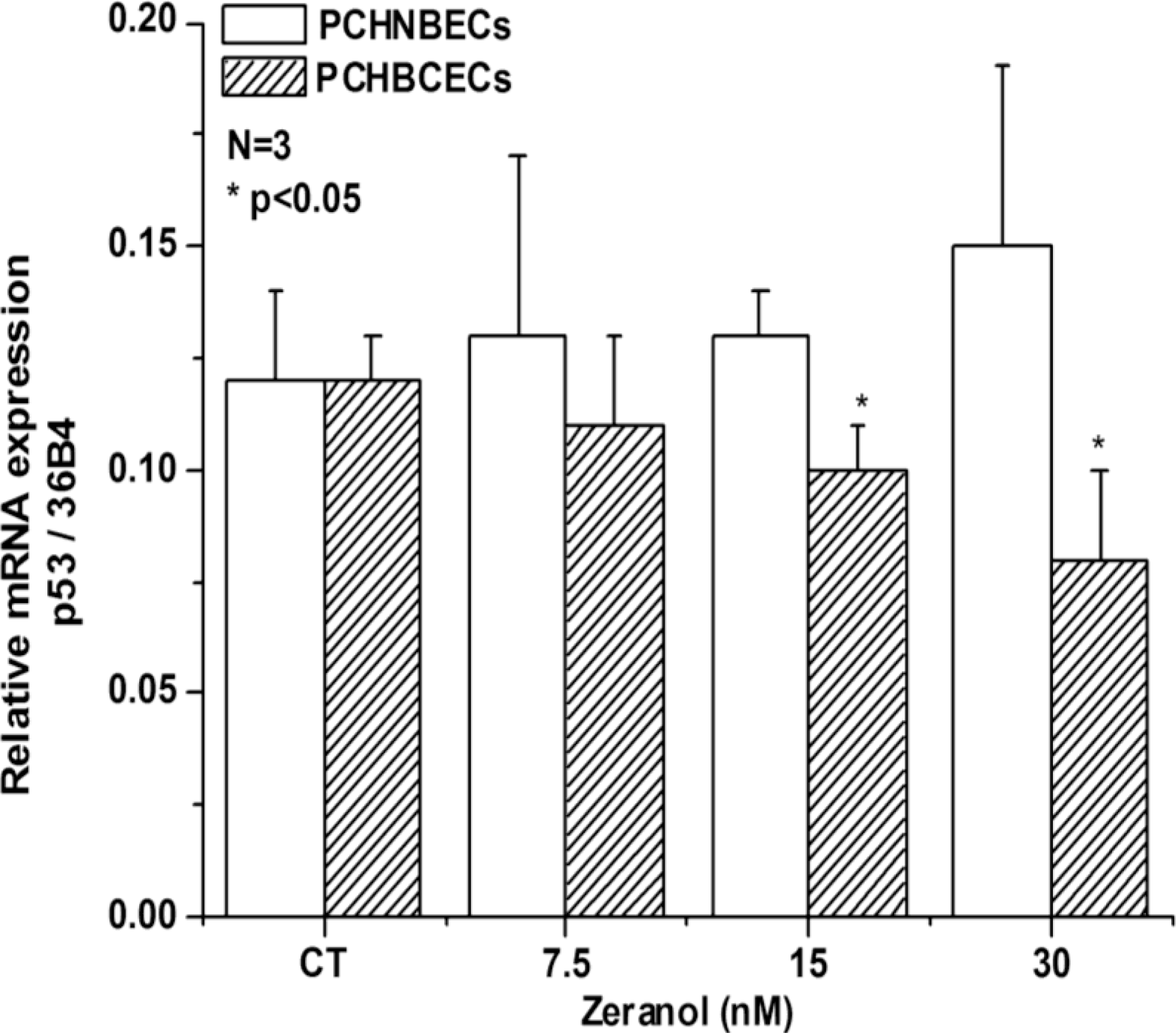
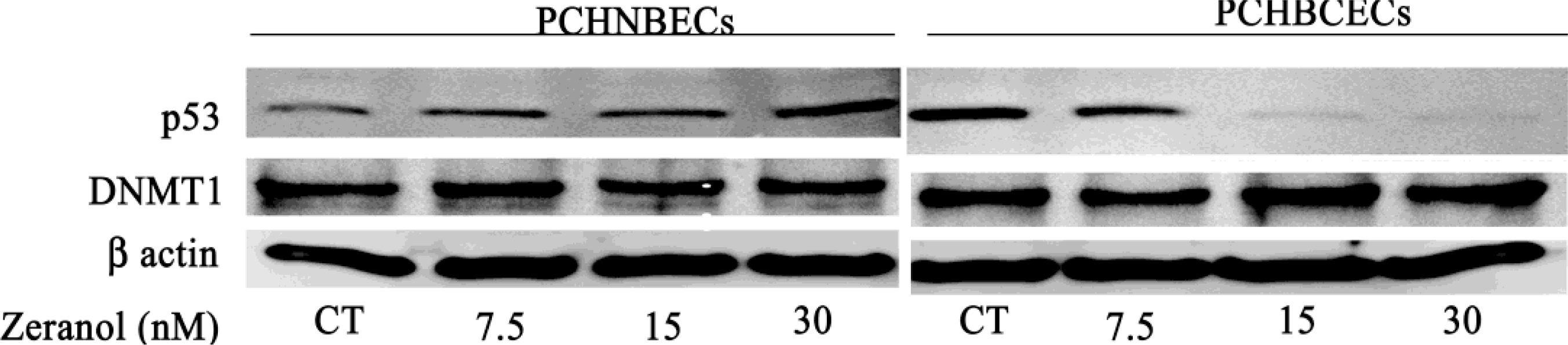
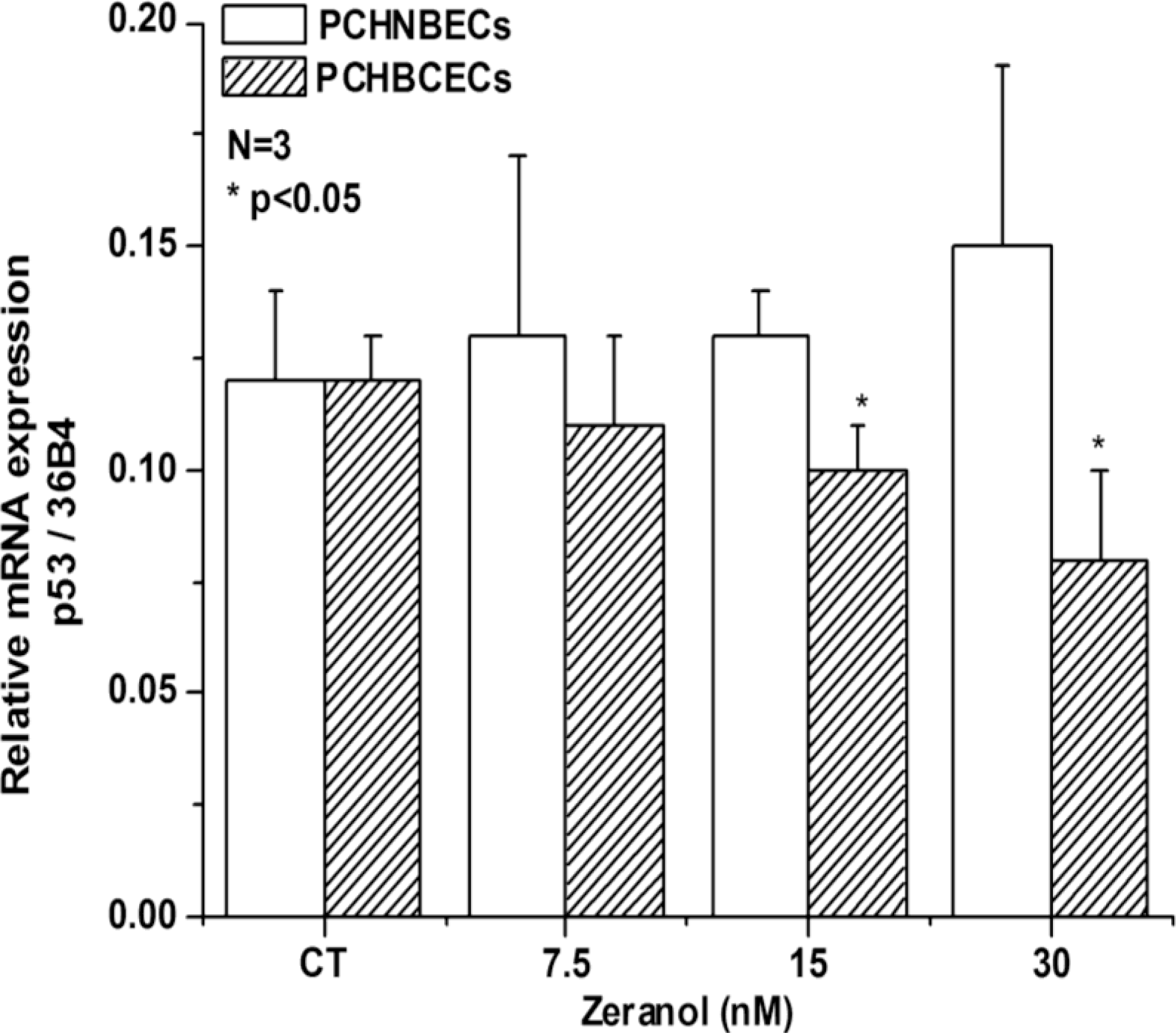
2.2. Regulatory Effects of Z on p53 mRNA and Protein Expression in PCHNBECs and PCHBCECs
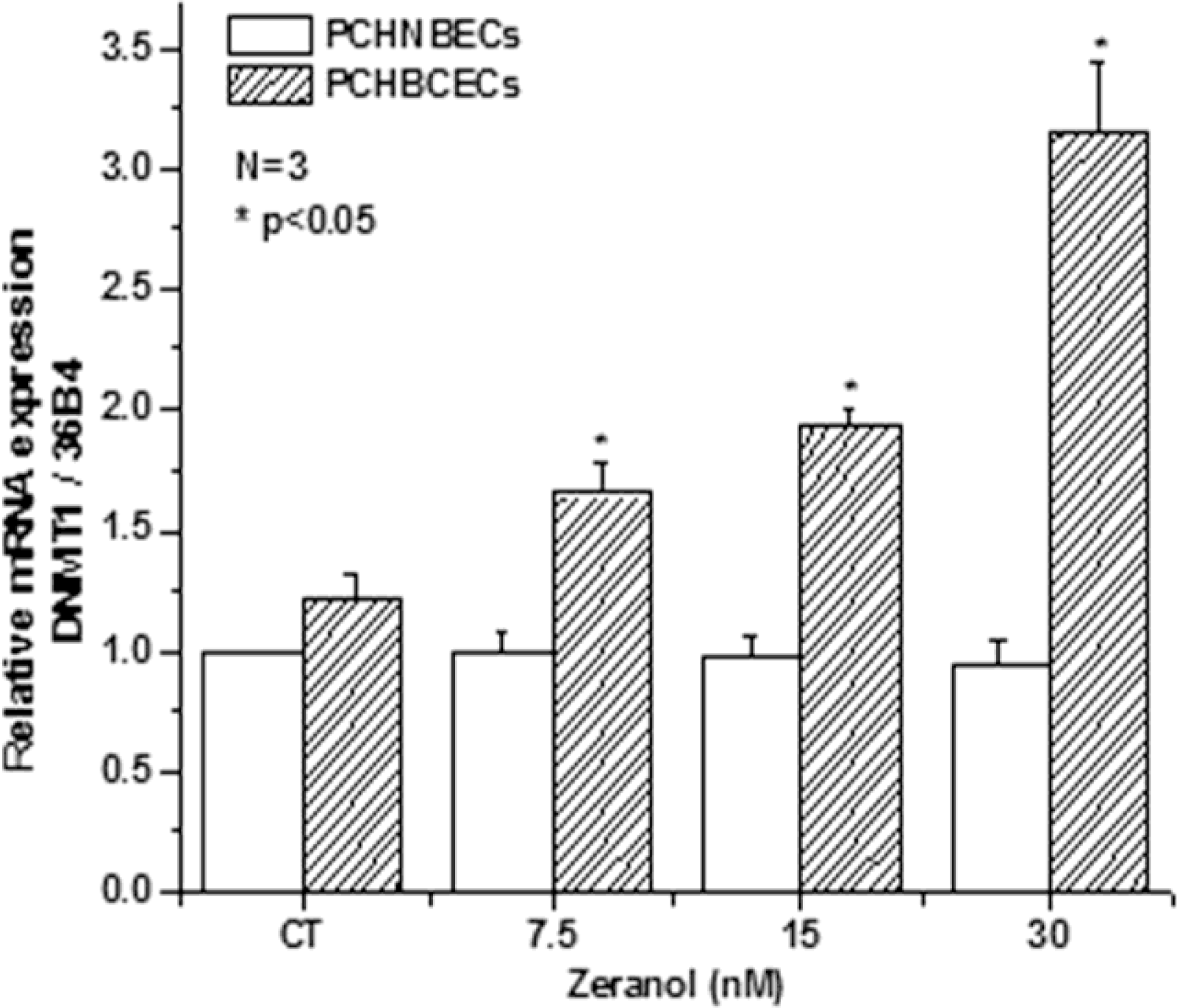
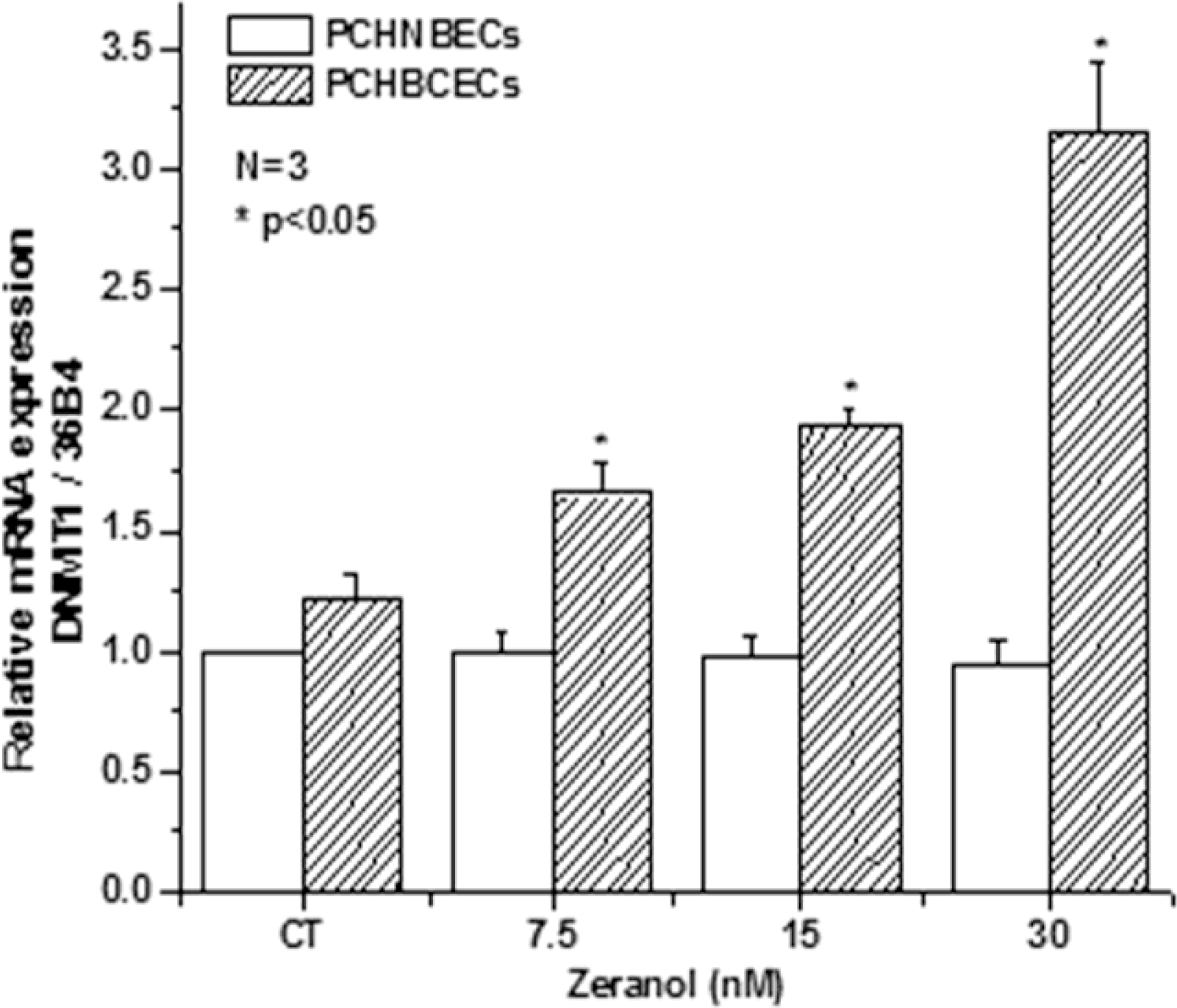
2.3. Z Regulates the Expression of DNMT1 mRNA and Protein in PCHNBECs and PCHBCECs
2.4. Discussion
3. Experimental Section
3.1. Isolation of Primary Cultured Human Normal Breast Epithelial Cells (PCHNBECs) and Cancer Epithelial Cells (PCHBCECs)
3.2. PCHBCECs and PCHNBECs Culture
3.3. Cell Proliferation Assay (MTT Assay)
3.4. Cell Treatment, RNA Isolation and cDNA Synthesis
3.5. Real Time PCR
3.6. Western Blotting Assay
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Jemal, A; Siegel, R; Xu, J; Ward, E. Cancer Statistics, 2010. CA Cancer J. Clin 2010, 60, 277–300. [Google Scholar]
- Chetrite, GS; Cortes-Prieto, J; Philippe, JC; Wright, F; Pasqualini, JR. Comparison of estrogen concentrations, estrone sulfatase and aromatase activities in normal, and in cancerous, human breast tissues. J. Steroid Biochem. Mol. Biol 2000, 72, 23–27. [Google Scholar]
- Haynes, BP; Straume, AH; Geisler, J; A'Hern, R; Helle, H; Smith, IE; Lonning, PE; Dowsett, M. Intratumoral estrogen disposition in breast cancer. Clin. Cancer Res 2010, 16, 1790–1801. [Google Scholar]
- Minervini, F; Giannoccaro, A; Cavallini, A; Visconti, A. Investigations on cellular proliferation induced by zearalenone and its derivatives in relation to the estrogenic parameters. Toxicol. Lett 2005, 159, 272–283. [Google Scholar]
- Massart, F; Meucci, V; Saggese, G; Soldani, G. High growth rate of girls with precocious puberty exposed to estrogenic mycotoxins. J. Pediatrics 2008, 152, 690–695. [Google Scholar]
- Takemura, H; Shim, JY; Sayama, K; Tsubura, A; Zhu, BT; Shimoi, K. Characterization of the estrogenic activities of zearalenone and zeranol in vivo and in vitro. J. Steroid Biochem. Mol. Biol 2007, 103, 170–177. [Google Scholar]
- Liu, S; Sugimoto, Y; Kulp, SK; Jiang, J; Chang, HL; Park, KY; Kashida, Y; Lin, YC. Estrogenic down-regulation of protein tyrosine phosphatase gamma (PTP gamma) in human breast is associated with estrogen receptor alpha. Anticancer Res 2002, 22, 3917–3923. [Google Scholar]
- Coe, JE; Ishak, KG; Ward, JM; Ross, MJ. Tamoxifen prevents induction of hepatic neoplasia by zeranol, an estrogenic food contaminant. Proc. Natl. Acad. Sci. USA 1992, 89, 1085–1089. [Google Scholar]
- Yuri, T; Tsukamoto, R; Miki, K; Uehara, N; Matsuoka, Y; Tsubura, A. Biphasic effects of zeranol on the growth of estrogen receptor-positive human breast carcinoma cells. Oncol. Rep 2006, 16, 1307–1312. [Google Scholar]
- Liu, S; Lin, YC. Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17beta. Breast J 2004, 10, 514–521. [Google Scholar]
- Ye, W; Xu, P; Threlfall, WR; Jen, R; Li, H; Lin, SH; Kuo, CT; Lin, YC. Zeranol enhances the proliferation of pre-adipocytes in beef heifers. Anticancer Res 2009, 29, 5045–5052. [Google Scholar]
- Ye, W; Xu, P; Zhong, S; Threlfall, WR; Frasure, C; Feng, E; Li, H; Lin, SH; Liu, JY; Lin, YC. Serum harvested from heifers one month post-zeranol implantation stimulates MCF-7 breast cancer growth. Exp. Ther. Med 2010, 1, 963–968. [Google Scholar]
- Ye, W; Xu, P; Zhong, S; Jen, R; Threlfall, WR; Frasure, CV; Feng, E; Li, H; Lin, SH; Liu, JY; Lin, YC. In vitro transformation of MCF-10A cells by sera harvested from heifers two months post-Zeranol implantation. Int. J. Oncol 2011, 38, 985–992. [Google Scholar]
- Xu, P; Ye, W; Jen, R; Lin, SH; Kuo, CT; Lin, YC. Mitogenic activity of zeranol in human breast cancer cells is enhanced by leptin and suppressed by gossypol. Anticancer Res 2009, 29, 4621–4628. [Google Scholar]
- Swan, SH; Liu, F; Overstreet, JW; Brazil, C; Skakkebaek, NE. Semen quality of fertile US males in relation to their mothers’ beef consumption during pregnancy. Hum. Reprod 2007, 22, 1497–1502. [Google Scholar]
- Linos, E; Willett, WC; Cho, E; Colditz, G; Frazier, LA. Red meat consumption during adolescence among premenopausal women and risk of breast cancer. Cancer Epidemiol. Biomarkers Prev 2008, 17, 2146–2151. [Google Scholar]
- Aw, TC; Smith, AB; Stephenson, RL; Glueck, CJ. Occupational exposure to zeranol, an animal growth promoter. Br. J. Ind. Med 1989, 46, 341–346. [Google Scholar]
- Vogelstein, B; Lane, D; Levine, AJ. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar]
- Golubovskaya, VM; Conway-Dorsey, K; Edmiston, SN; Tse, CK; Lark, AA; Livasy, CA; Moore, D; Millikan, RC; Cance, WG. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int. J. Cancer 2009, 125, 1735–1738. [Google Scholar]
- Dinu, I; Potter, JD; Mueller, T; Liu, Q; Adewale, AJ; Jhangri, GS; Einecke, G; Famulski, KS; Halloran, P; Yasui, Y. Gene-set analysis and reduction. Briefings Bioinf 2009, 10, 24–34. [Google Scholar]
- Hassan, NM; Tada, M; Hamada, J; Kashiwazaki, H; Kameyama, T; Akhter, R; Yamazaki, Y; Yano, M; Inoue, N; Moriuchi, T. Presence of dominant negative mutation of TP53 is a risk of early recurrence in oral cancer. Cancer Lett 2008, 270, 108–119. [Google Scholar]
- Baker, L; Quinlan, PR; Patten, N; Ashfield, A; Birse-Stewart-Bell, LJ; McCowan, C; Bourdon, JC; Purdie, CA; Jordan, LB; Dewar, JA; Wu, L; Thompson, AM. p53 mutation, deprivation and poor prognosis in primary breast cancer. Br. J. Cancer 2010, 102, 719–726. [Google Scholar]
- Lai, H; Lin, L; Nadji, M; Lai, S; Trapido, E; Meng, L. Mutations in the p53 tumor suppressor gene and early onset breast cancer. Cancer Biol. Ther 2002, 1, 31–36. [Google Scholar]
- Xu, X; Wagner, KU; Larson, D; Weaver, Z; Li, C; Ried, T; Hennighausen, L; Wynshaw-Boris, A; Deng, CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet 1999, 22, 37–43. [Google Scholar]
- Ting, AH; Jair, KW; Schuebel, KE; Baylin, SB. Differential requirement for DNA methyltransferase 1 in maintaining human cancer cell gene promoter hypermethylation. Cancer Res 2006, 66, 729–735. [Google Scholar]
- Xu, XL; Yu, J; Zhang, HY; Sun, MH; Gu, J; Du, X; Shi, DR; Wang, P; Yang, ZH; Zhu, JD. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J. Gastroenterol 2004, 10, 3441–3454. [Google Scholar]
- Datta, J; Ghoshal, K; Denny, WA; Gamage, SA; Brooke, DG; Phiasivongsa, P; Redkar, S; Jacob, ST. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res 2009, 69, 4277–4285. [Google Scholar]
- Kang, JH; Kim, SJ; Noh, DY; Park, IA; Choe, KJ; Yoo, OJ; Kang, HS. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab. Invest 2001, 81, 573–579. [Google Scholar]
- Girault, I; Tozlu, S; Lidereau, R; Bieche, I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res 2003, 9, 4415–4422. [Google Scholar]
- Agoston, AT; Argani, P; de Marzo, AM; Hicks, JL; Nelson, WG. Retinoblastoma pathway dysregulation causes DNA methyltransferase 1 overexpression in cancer via MAD2-mediated inhibition of the anaphase-promoting complex. Am. J. Pathol 2007, 170, 1585–1593. [Google Scholar]
- Weiss, RA. Multistage carcinogenesis. Br. J. Cancer 2004, 91, 1981–1982. [Google Scholar]
- Hartmann, A; Blaszyk, H; Kovach, JS; Sommer, SS. The molecular epidemiology of p53 gene mutations in human breast cancer. Trends Genet 1997, 13, 27–33. [Google Scholar]
- McGill, G; Fisher, DE. p53 and cancer therapy: a double-edged sword. J. Clin. Invest 1999, 104, 223–225. [Google Scholar]
- Toillon, RA; Chopin, V; Jouy, N; Fauquette, W; Boilly, B; Le Bourhis, X. Normal breast epithelial cells induce p53-dependent apoptosis and p53-independent cell cycle arrest of breast cancer cells. Breast Cancer Res. Treat 2002, 71, 269–280. [Google Scholar]
- Blaszyk, H; Hartmann, A; Cunningham, JM; Schaid, D; Wold, LE; Kovach, JS; Sommer, SS. A prospective trial of midwest breast cancer patients: A p53 gene mutation is the most important predictor of adverse outcome. Int. J. Cancer 2000, 89, 32–38. [Google Scholar]
- Baylin, SB; Herman, JG; Graff, JR; Vertino, PM; Issa, JP. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res 1998, 72, 141–196. [Google Scholar]
- Ai, L; Tao, Q; Zhong, S; Fields, CR; Kim, WJ; Lee, MW; Cui, Y; Brown, KD; Robertson, KD. Inactivation of Wnt inhibitory factor-1 (WIF1) expression by epigenetic silencing is a common event in breast cancer. Carcinogenesis 2006, 27, 1341–1348. [Google Scholar]
- Rivenbark, AG; Jones, WD; Risher, JD; Coleman, WB. DNA methylation-dependent epigenetic regulation of gene expression in MCF-7 breast cancer cells. Epigenetics 2006, 1, 32–44. [Google Scholar]
- Razin, A; Riggs, AD. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar]
- Surani, MA. Imprinting and the initiation of gene silencing in the germ line. Cell 1998, 93, 309–312. [Google Scholar]
- Baylin, SB; Herman, JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000, 16, 168–174. [Google Scholar]
- Huang, TH; Perry, MR; Laux, DE. Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet 1999, 8, 459–470. [Google Scholar]
- Muller, HM; Widschwendter, A; Fiegl, H; Ivarsson, L; Goebel, G; Perkmann, E; Marth, C; Widschwendter, M. DNA methylation in serum of breast cancer patients: An independent prognostic marker. Cancer Res 2003, 63, 7641–7645. [Google Scholar]
- Razin, A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J 1998, 17, 4905–4908. [Google Scholar]
- Leonhardt, H; Page, AW; Weier, HU; Bestor, TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [Green Version]
- Bestor, TH; Gundersen, G; Kolsto, AB; Prydz, H. CpG islands in mammalian gene promoters are inherently resistant to de novo methylation. Genet. Anal. Tech. Appl 1992, 9, 48–53. [Google Scholar]
- Issa, JP; Vertino, PM; Wu, J; Sazawal, S; Celano, P; Nelkin, BD; Hamilton, SR; Baylin, SB. Increased cytosine DNA-methyltransferase activity during colon cancer progression. J. Natl. Cancer Inst 1993, 85, 1235–1240. [Google Scholar]
- Robertson, KD; Uzvolgyi, E; Liang, G; Talmadge, C; Sumegi, J; Gonzales, FA; Jones, PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999, 27, 2291–2298. [Google Scholar]
- Ahluwalia, A; Hurteau, JA; Bigsby, RM; Nephew, KP. DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol. Oncol 2001, 82, 299–304. [Google Scholar]
- Laird, PW; Jackson-Grusby, L; Fazeli, A; Dickinson, SL; Jung, WE; Li, E; Weinberg, RA; Jaenisch, R. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995, 81, 197–205. [Google Scholar]
- MacLeod, AR; Szyf, M. Expression of antsense to DNA methyltransferase mRNA induces DNA demethylation and inhibits tumorigenesis. J. Biol. Chem 1995, 270, 8037–8043. [Google Scholar]
- Shu, ST; Sugimoto, Y; Liu, S; Chang, HL; Ye, W; Wang, LS; Huang, YW; Yan, P; Lin, YC. Function and regulatory mechanisms of the candidate tumor suppressor receptor protein tyrosine phosphatase gamma (PTPRG) in breast cancer cells. Anticancer Res 2010, 30, 1937–1946. [Google Scholar]
- Liu, S; Kulp, SK; Sugimoto, Y; Jiang, J; Chang, HL; Lin, Y. Involvement of breast epithelial-stromal interactions in the regulation of protein tyrosine phosphatase-γ (PTP γ) mRNA expression by estrogenically active agents. Breast Cancer Res. Treat 2002, 71, 21–35. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ye, W.; Xu, P.; Jen, R.; Feng, E.; Zhong, S.; Li, H.; Lin, S.-H.; Liu, J.-Y.; Lin, Y.C. Zeranol Down-Regulates p53 Expression in Primary Cultured Human Breast Cancer Epithelial Cells through Epigenetic Modification. Int. J. Mol. Sci. 2011, 12, 1519-1532. https://doi.org/10.3390/ijms12031519
Ye W, Xu P, Jen R, Feng E, Zhong S, Li H, Lin S-H, Liu J-Y, Lin YC. Zeranol Down-Regulates p53 Expression in Primary Cultured Human Breast Cancer Epithelial Cells through Epigenetic Modification. International Journal of Molecular Sciences. 2011; 12(3):1519-1532. https://doi.org/10.3390/ijms12031519
Chicago/Turabian StyleYe, Weiping, Pingping Xu, Robert Jen, Eric Feng, Saiyi Zhong, Hong Li, Shu-Hong Lin, Jie-Yu Liu, and Young C. Lin. 2011. "Zeranol Down-Regulates p53 Expression in Primary Cultured Human Breast Cancer Epithelial Cells through Epigenetic Modification" International Journal of Molecular Sciences 12, no. 3: 1519-1532. https://doi.org/10.3390/ijms12031519