The Role of the E2F Transcription Factor Family in UV-Induced Apoptosis

Abstract
:1. What is UV?
2. Mutagenic Effects of UV Radiation
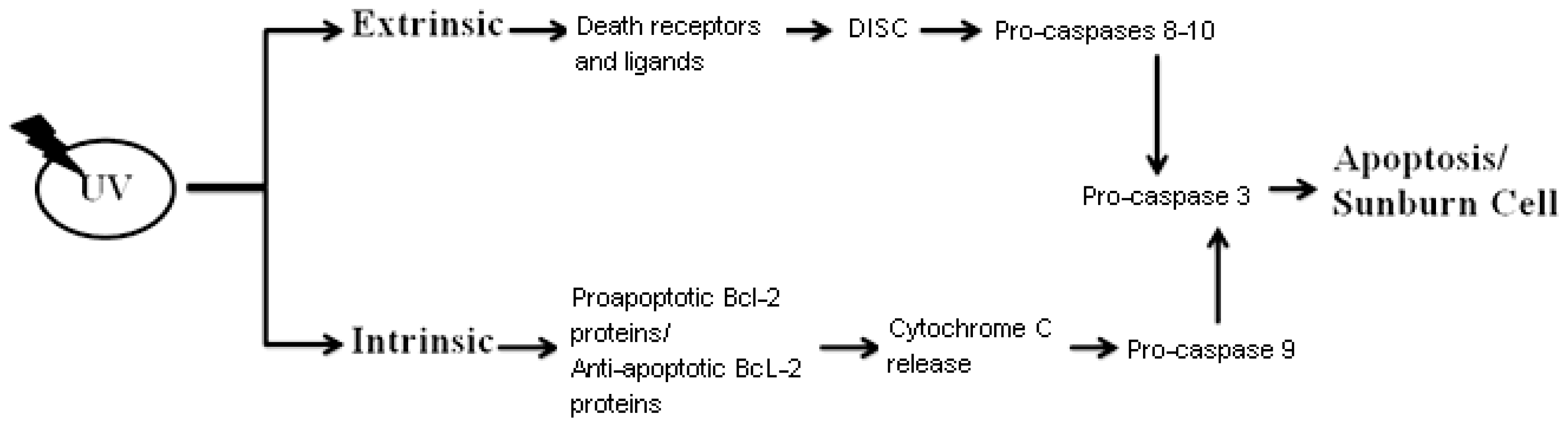
3. Sunburn Cells (UV-Induced Cell Death)
4. Role of UV in Skin Carcinogenesis
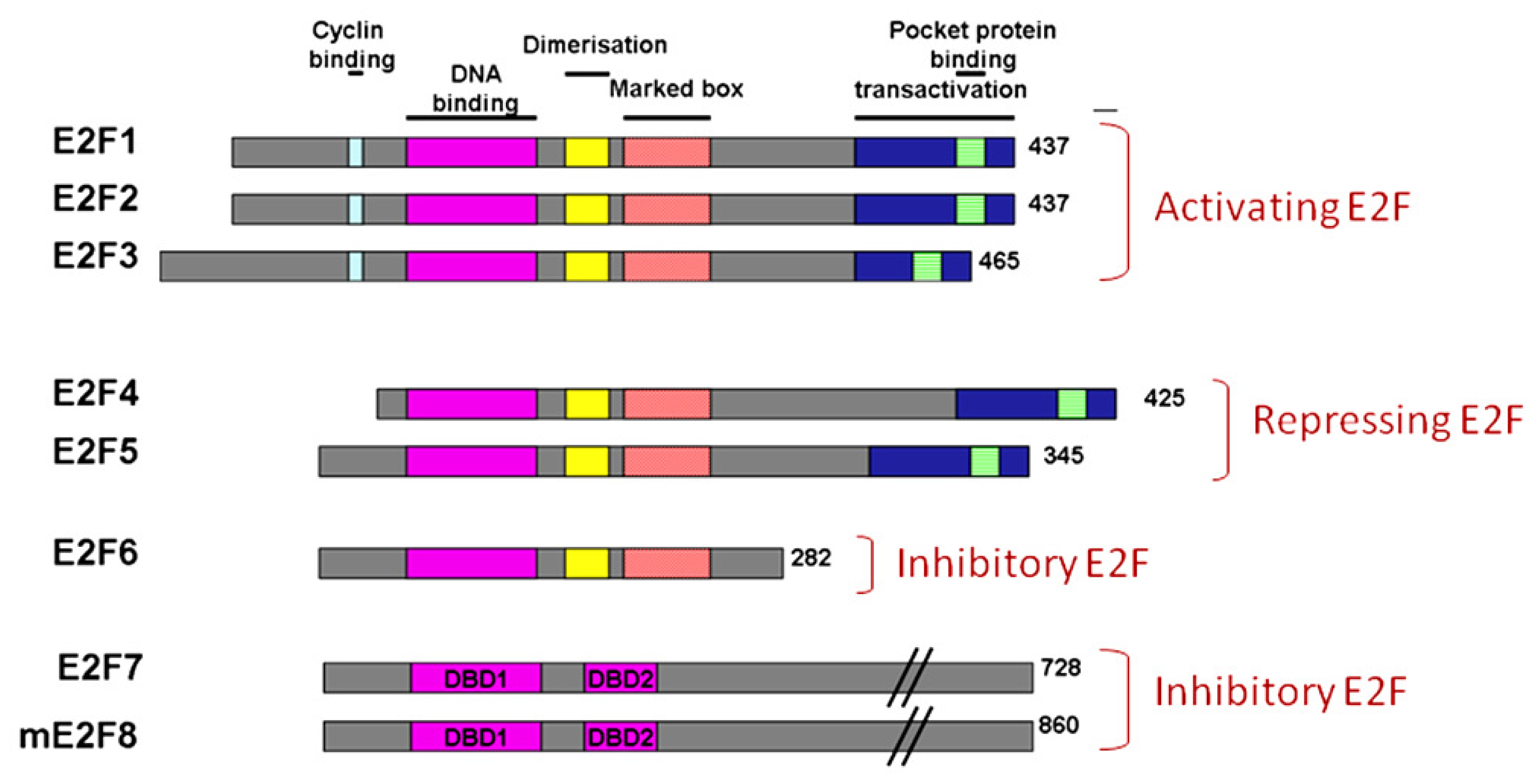
5. The E2F Family
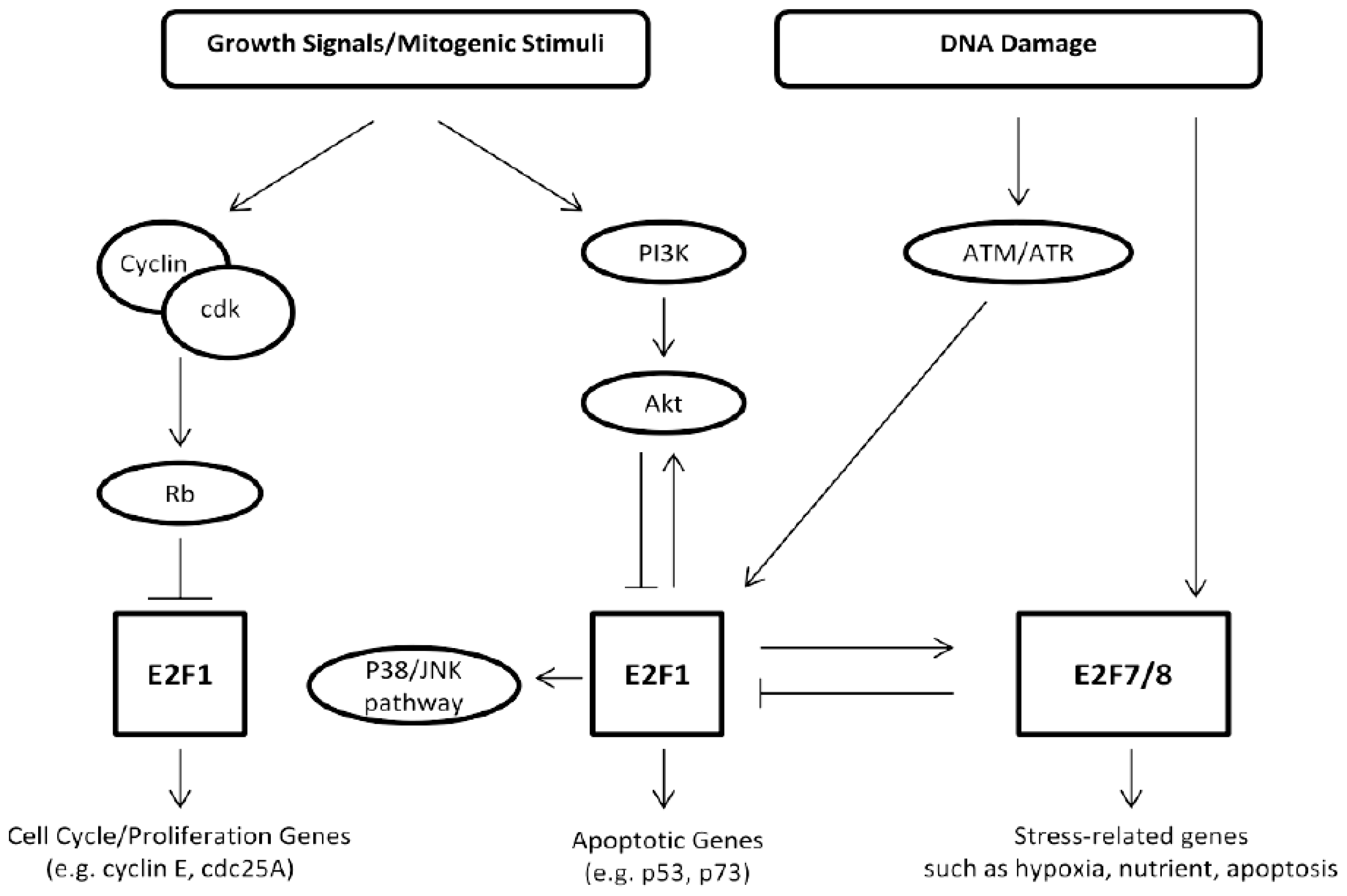
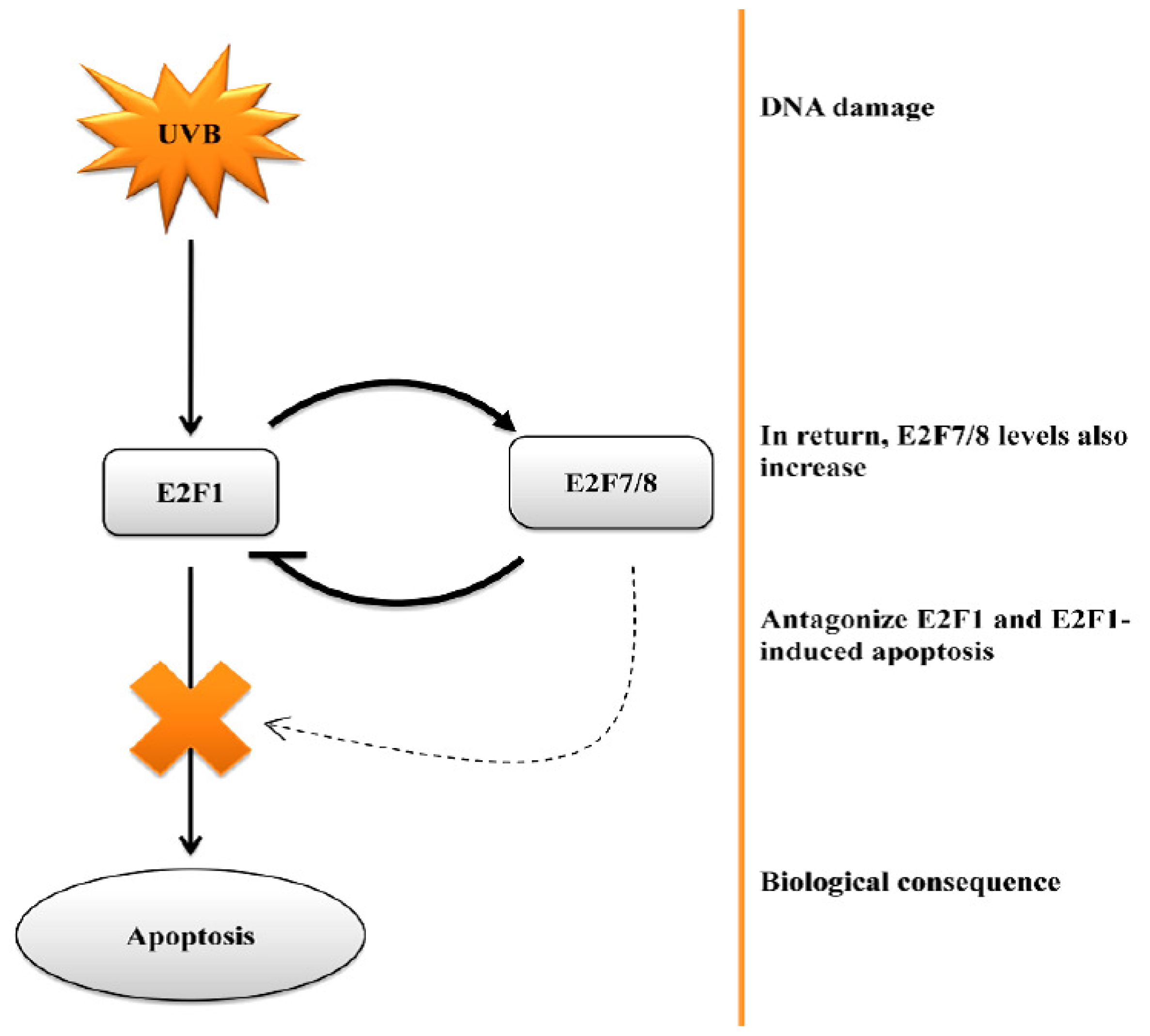
6. E2F-Induced Apoptosis and Skin Cancer Formation
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Urbach, F. Ultraviolet radiation and skin cancer of humans. J. Photochem. Photobiol. B 1997, 40, 3–7. [Google Scholar]
- Maverakis, E.; Miyamura, Y.; Bowen, M.P.; Correa, G.; Ono, Y.; Goodarzi, H. Light, including ultraviolet. J. Autoimmun 2010, 34, J247–J257. [Google Scholar]
- Nickoloff, B.J.; Qin, J.-Z.; Chaturvedi, V.; Bacon, P.; Panella, J.; Denning, M.F. Life and death signaling pathways contributing to skin cancer. J. Investig. Dermatol. Symp. Proc 2002, 7, 27–35. [Google Scholar]
- Hellweg, C.; Baumstark-Khan, C. Detection of UV-induced activation of nf-κb in a recombinant human cell line by means of enhanced green fluorescent protein (egfp). Radiat. Environ. Biophys 2007, 46, 269–279. [Google Scholar]
- Matsunaga, T.; Hieda, K.; Nikaido, O. Wavelength dependent formation of thymine dimers and (6–4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm. Photochem. Photobiol 1991, 54, 403–410. [Google Scholar]
- Nishigori, C. Cellular aspects of photocarcinogenesis. Photochem. Photobiol. Sci 2006, 5, 208–214. [Google Scholar]
- Lippens, S.; Hoste, E.; Vandenabeele, P.; Agostinis, P.; Declercq, W. Cell death in the skin. Apoptosis 2009, 14, 549–569. [Google Scholar]
- Marchese, C.; Maresca, V.; Cardinali, G.; Belleudi, F.; Ceccarelli, S.; Bellocci, M.; Frati, L.; Torrisi, M.R.; Picardo, M. UVB-induced activation and internalization of keratinocyte growth factor receptor. Oncogene 2003, 22, 2422–2431. [Google Scholar]
- Schwarz, T.; Schwarz, A. DNA repair and cytokine responses. J. Investig. Dermatol. Symp. Proc 2009, 14, 63–66. [Google Scholar]
- Schwarz, A.; Maeda, A.; Ständer, S.; van Steeg, H.; Schwarz, T. Il-18 reduces ultraviolet radiation-induced DNA damage and thereby affects photoimmunosuppression. J. Immunol 2006, 176, 2896–2901. [Google Scholar]
- Schwarz, A.; Maeda, A.; Kernebeck, K.; van Steeg, H.; Beissert, S.; Schwarz, T. Prevention of UV radiation–induced immunosuppression by il-12 is dependent on DNA repair. J. Exp. Med 2005, 201, 173–179. [Google Scholar]
- Chaturvedi, V.; Qin, J.-Z.; Denning, M.F.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J. Biol. Chem 1999, 274, 23358–23367. [Google Scholar]
- Qin, J.-Z.; Chaturvedi, V.; Denning, M.F.; Bacon, P.; Panella, J.; Choubey, D.; Nickoloff, B.J. Regulation of apoptosis by p53 in UV-irradiated human epidermis, psoriatic plaques and senescent keratinocytes. Oncogene 2002, 21, 2991–3002. [Google Scholar]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma—A role for antiapoptotic signalling pathways. Br. J. Dermatol 2009, 161, 107–115. [Google Scholar]
- Laethem, A.V.; Claerhout, S.; Garmyn, M.; Agostinis, P. The sunburn cell: Regulation of death and survival of the keratinocyte. Int. J. Biochem. Cell Biol 2005, 37, 1547–1553. [Google Scholar]
- Kulms, D.; Schwarz, T. Molecular mechanisms involved in UV-induced apoptotic cell death. Skin Pharmacol. Appl. Skin Physiol 2002, 15, 342–347. [Google Scholar]
- Aragane, Y.; Kulms, D.; Metze, D.; Wilkes, G.; Pöppelmann, B.; Luger, T.A.; Schwarz, T. Ultraviolet light induces apoptosis via direct activation of cd95 (fas/apo-1) independently of its ligand cd95l. J. Cell Biol 1998, 140, 171–182. [Google Scholar]
- Eberle, J.; Fecker, L.F.; Forschner, T.; Ulrich, C.; Röwert-Huber, J.; Stockfleth, E. Apoptosis pathways as promising targets for skin cancer therapy. Br. J. Dermatol 2007, 156, 18–24. [Google Scholar]
- Conney, A.H.; Kramata, P.; Lou, Y.-R.; Lu, Y.-P. Effect of caffeine on uvb-induced carcinogenesis, apoptosis, and the elimination of UVB-induced patches of p53 mutant epidermal cells in skh-1 mice. Photochem. Photobiol 2008, 84, 330–338. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med 2006, 12, 440–450. [Google Scholar]
- Schafer, Z.T.; Kornbluth, S. The apoptosome: Physiological, developmental, and pathological modes of regulation. Dev. Cell 2006, 10, 549–561. [Google Scholar]
- Black, H.S.; deGruijl, F.R.; Forbes, P.D.; Cleaver, J.E.; Ananthaswamy, H.N.; deFabo, E.C.; Ullrich, S.E.; Tyrrell, R.M. Photocarcinogenesis: An overview. J. Photochem. Photobiol. B 1997, 40, 29–47. [Google Scholar]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol 2003, 148, 913–922. [Google Scholar]
- Trakatelli, M.; Ulrich, C.; del Marmol, V.; Euvrard, S.; Stockfleth, E.; Abeni, D. Epidemiology of non-melanoma skin cancer (nmsc) in europe: Accurate and comparable data are needed for effective public monitoring and interventions. Br. J. Dermatol 2007, 156, 1–7. [Google Scholar]
- Clayman, G.L.; Lee, J.J.; Holsinger, F.C.; Zhou, X.; Duvic, M.; El-Naggar, A.K.; Prieto, V.G.; Altamirano, E.; Tucker, S.L.; Strom, S.S.; et al. Mortality risk from squamous cell skin cancer. J. Clin. Oncol 2005, 23, 759–765. [Google Scholar]
- De Gruijl, F.R.; van Kranen, H.J.; Mullenders, L.H.F. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B 2001, 63, 19–27. [Google Scholar]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar]
- Erb, P.; Ji, J.; Wernli, M.; Kump, E.; Glaser, A.; Büchner, S.A. Role of apoptosis in basal cell and squamous cell carcinoma formation. Immunol. Lett 2005, 100, 68–72. [Google Scholar]
- Walshe, J.; Serewko-Auret, M.M.; Teakle, N.; Cameron, S.; Minto, K.; Smith, L.; Burcham, P.C.; Russell, T.; Strutton, G.; Griffin, A.; et al. Inactivation of glutathione peroxidase activity contributes to UV-induced squamous cell carcinoma formation. Cancer Res 2007, 67, 4751–4758. [Google Scholar]
- Endo-Munoz, L.; Dahler, A.; Teakle, N.; Rickwood, D.; Hazar-Rethinam, M.; Abdul-Jabbar, I.; Sommerville, S.; Dickinson, I.; Kaur, P.; Paquet-Fifield, S.; et al. E2F7 can regulate proliferation, differentiation, and apoptotic responses in human keratinocytes: Implications for cutaneous squamous cell carcinoma formation. Cancer Res 2009, 69, 1800–1808. [Google Scholar]
- Pierce, A.M.; Gimenez-Conti, I.B.; Schneider-Broussard, R.; Martinez, L.A.; Conti, C.J.; Johnson, D.G. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc. Natl. Acad. Sci. USA 1998, 95, 8858–8863. [Google Scholar]
- Pierce, A.M.; Schneider-Broussard, R.; Gimenez-Conti, I.B.; Russell, J.L.; Conti, C.J.; Johnson, D.G. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol. Cell. Biol 1999, 19, 6408–6414. [Google Scholar]
- Dicker, A.J.; Popa, C.; Dahler, A.L.; Serewko, M.M.; Hilditch-Maguire, P.A.; Frazer, I.H.; Saunders, N.A. E2F-1 induces proliferation-specific genes and suppresses squamous differentiation-specific genes in human epidermal keratinocytes. Oncogene 2000, 19, 2887–2894. [Google Scholar]
- Wong, C.F.; Barnes, L.M.; Dahler, A.L.; Smith, L.; Serewko-Auret, M.M.; Popa, C.; Abdul-Jabbar, I.; Saunders, N.A. E2F modulates keratinocyte squamous differentiation. J. Biol. Chem 2003, 278, 28516–28522. [Google Scholar]
- DeGregori, J.; Johnson, D.G. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med 2006, 6, 739–748. [Google Scholar]
- Dyson, N. The regulation of E2F by prb-family proteins. Genes Dev 1998, 12, 2245–2262. [Google Scholar]
- Einspahr, J.G.; Alberts, D.S.; Wameke, J.A.; Bozzo, P.; Basye, J.; Grogan, T.M.; Nelson, M.A.; Bowden, G.T. Relationship of p53 mutations to epidermal cell proliferation and apoptosis in human uv-induced skin carcinogenesis. Neoplasia 1999, 1, 468–475. [Google Scholar]
- Sherr, C.J.; McCormick, F. The rb and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar]
- Martínez-Carpio, P.A.; Trelles, M.A. Cutaneous epidermal growth factor receptor system following ultraviolet irradiation: Exploring the role of molecular mechanisms. Photodermatol. Photoimmunol. Photomed 2010, 26, 250–256. [Google Scholar]
- Wrone-Smith, T.; Bergstrom, J.; Quevedo, M.E.; Reddy, V.; Gutierrez-Steil, C.; Nickoloff, B.J. Differential expression of cell survival and cell cycle regulatory proteins in cutaneous squamoproliferative lesions. J. Dermatol. Sci 1999, 19, 53–67. [Google Scholar]
- Raj, D.; Brash, D.E.; Grossman, D. Keratinocyte apoptosis in epidermal development and disease. J. Invest. Dermatol 2006, 126, 243–257. [Google Scholar]
- Serewko, M.M.; Popa, C.; Dahler, A.L.; Smith, L.; Strutton, G.M.; Coman, W.; Dicker, A.J.; Saunders, N.A. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res 2002, 62, 3759–3765. [Google Scholar]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol 2002, 3, 11–20. [Google Scholar]
- Stevens, C.; La Thangue, N.B. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair 2004, 3, 1071–1079. [Google Scholar]
- Panagiotis Zalmas, L.; Zhao, X.; Graham, A.L.; Fisher, R.; Reilly, C.; Coutts, A.S.; La Thangue, N.B. DNA-damage response control of E2F7 and E2F8. EMBO Rep 2008, 9, 252–259. [Google Scholar]
- Moon, N.-S.; Dyson, N. E2F7 and E2F8 keep the E2F family in balance. Dev. Cell 2008, 14, 1–3. [Google Scholar]
- Helin, K.; Lees, J.A.; Vidal, M.; Dyson, N.; Harlow, E.; Fattaey, A. A cdna encoding a prb-binding protein with properties of the transcription factor E2F. Cell 1992, 70, 337–350. [Google Scholar]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar]
- Bashari, D.; Hacohen, D.; Ginsberg, D. Jnk activation is regulated by E2F and promotes E2F1-induced apoptosis. Cell. Signal 2011, 23, 65–70. [Google Scholar]
- Chaussepied, M.; Ginsberg, D. E2F and signal transduction pathways. Cell Cycle 2005, 4, 392–396. [Google Scholar]
- Nevins, J. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 1998, 9, 585–593. [Google Scholar]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar]
- Johnson, D.G.; Ohtani, K.; Nevins, J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev 1994, 8, 1514–1525. [Google Scholar]
- Iaquinta, P.J.; Lees, J.A. Life and death decisions by the E2F transcription factors. Curr. Opin. Cell Biol 2007, 19, 649–657. [Google Scholar]
- Wong, C.F.; Barnes, L.M.; Smith, L.; Popa, C.; Serewko-Auret, M.M.; Saunders, N.A. E2F6: A member of the E2F family that does not modulate squamous differentiation. Biochem. Biophys. Res. Commun 2004, 324, 497–503. [Google Scholar]
- Berton, T.R.; Mitchell, D.L.; Guo, R.; Johnson, D.G. Regulation of epidermal apoptosis and DNA repair by E2F1 in response to ultraviolet b radiation. Oncogene 2005, 24, 2449–2460. [Google Scholar]
- Leone, G.; DeGregori, J.; Yan, Z.; Jakoi, L.; Ishida, S.; Williams, R.S.; Nevins, J.R. E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev 1998, 12, 2120–2130. [Google Scholar]
- Hazar-Rethinam, M.; Cameron, S.R.; Dahler, A.L.; Endo-Munoz, L.B.; Smith, L.; Rickwood, D.; Saunders, N.A. Loss of E2F7 expression is an early event in squamous differentiation and causes derepression of the key differentiation activator sp1. J. Invest. Dermatol 2011, 131, 1077–1084. [Google Scholar]
- Lammens, T.; Li, J.; Leone, G.; de Veylder, L. Atypical E2Fs: New players in the E2F transcription factor family. Trends Cell Biol 2009, 19, 111–118. [Google Scholar]
- Stevens, C.; la Thangue, N.B. E2F and cell cycle control: A double-edged sword. Arch. Biochem. Biophys 2003, 412, 157–169. [Google Scholar]
- De Bruin, A.; Maiti, B.; Jakoi, L.; Timmers, C.; Buerki, R.; Leone, G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem 2003, 278, 42041–42049. [Google Scholar]
- Di Stefano, L.; Jensen, M.R.; Helin, K. E2F7, a novel E2F featuring dp-independent repression of a subset of E2F-regulated genes. EMBO J 2003, 22, 6289–6298. [Google Scholar]
- Logan, N.; Delavaine, L.; Graham, A.; Reilly, C.; Wilson, J.; Brummelkamp, T.R.; Hijmans, E.M.; Bernards, R.; La Thangue, N.B. E2F-7: A distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene 2004, 23, 5138–5150. [Google Scholar]
- Logan, N.; Graham, A.; Zhao, X.; Fisher, R.; Maiti, B.; Leone, G.; Thangue, N.B.L. E2F-8: An E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene 2005, 24, 5000–5004. [Google Scholar]
- Maiti, B.; Li, J.; de Bruin, A.; Gordon, F.; Timmers, C.; Opavsky, R.; Patil, K.; Tuttle, J.; Cleghorn, W.; Leone, G. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem 2005, 280, 18211–18220. [Google Scholar]
- Cam, H.; Dynlacht, B.D. Emerging roles for E2F: Beyond the g1/s transition and DNA replication. Cancer Cell 2003, 3, 311–316. [Google Scholar]
- Li, J.; Ran, C.; Li, E.; Gordon, F.; Comstock, G.; Siddiqui, H.; Cleghorn, W.; Chen, H.-Z.; Kornacker, K.; Liu, C.-G.; et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev. Cell 2008, 14, 62–75. [Google Scholar]
- Takahashi, Y.; Rayman, J.B.; Dynlacht, B.D. Analysis of promoter binding by the E2F and prb families in vivo: Distinct E2F proteins mediate activation and repression. Genes Dev 2000, 14, 804–816. [Google Scholar]
- Attwooll, C.; Denchi, E.L.; Helin, K. The E2F family: Specific functions and overlapping interests. EMBO J 2004, 23, 4709–4716. [Google Scholar]
- Christensen, J.; Cloos, P.; Toftegaard, U.; Klinkenberg, D.; Bracken, A.P.; Trinh, E.; Heeran, M.; di Stefano, L.; Helin, K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res 2005, 33, 5458–5470. [Google Scholar]
- Qin, X.Q.; Livingston, D.M.; Kaelin, W.G.; Adams, P.D. Deregulated transcription factor E2F-1 expression leads to s-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 1994, 91, 10918–10922. [Google Scholar]
- Holmberg, C.; Helin, K.; Sehested, M.; Karlström, O. E2F-1 induced p53-independent apoptosis apoptosis in transgenic mice. Oncogene 1998, 17, 143–155. [Google Scholar]
- Wang, D.; Russell, J.L.; Johnson, D.G. E2F4 and E2F1 have similar proliferative properties but different apoptotic and oncogenic properties in vivo. Mol. Cell. Biol 2000, 20, 3417–3424. [Google Scholar]
- Field, S.J.; Tsai, F.-Y.; Kuo, F.; Zubiaga, A.M.; Kaelin, W.G.; Livingston, D.M.; Orkin, S.H.; Greenberg, M.E. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996, 85, 549–561. [Google Scholar]
- Yamasaki, L.; Jacks, T.; Bronson, R.; Goillot, E.; Harlow, E.; Dyson, N.J. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 1996, 85, 537–548. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Pediconi, N.; Ianari, A.; Costanzo, A.; Belloni, L.; Gallo, R.; Cimino, L.; Porcellini, A.; Screpanti, I.; Balsano, C.; Alesse, E.; et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat. Cell Biol 2003, 5, 552–558. [Google Scholar]
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis.
- Moroni, M.C.; Hickman, E.S.; Denchi, E.L.; Caprara, G.; Colli, E.; Cecconi, F.; Muller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol 2001, 3, 552–558. [Google Scholar]
- Rogoff, H.A.; Pickering, M.T.; Frame, F.M.; Debatis, M.E.; Sanchez, Y.; Jones, S.; Kowalik, T.F. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and atm/nbs1/chk2. Mol. Cell. Biol 2004, 24, 2968–2977. [Google Scholar]
- Carcagno, A.L.; Ogara, M.F.; Sonzogni, S.V.; Marazita, M.C.; Sirkin, P.F.; Ceruti, J.M.; Cánepa, E.T. E2F1 transcription is induced by genotoxic stress through atm/atr activation. IUBMB Life 2009, 61, 537–543. [Google Scholar]
- Wikonkal, N.M.; Remenyik, E.; Knezevic, D.; Zhang, W.; Liu, M.; Zhao, H.; Berton, T.R.; Johnson, D.G.; Brash, D.E. Inactivating E2F1 reverts apoptosis resistance and cancer sensitivity in trp53-deficient mice. Nat. Cell Biol 2003, 5, 655–660. [Google Scholar]
- Maser, R.S.; Mirzoeva, O.K.; Wells, J.; Olivares, H.; Williams, B.R.; Zinkel, R.A.; Farnham, P.J.; Petrini, J.H.J. Mre11 complex and DNA replication: Linkage to E2F and sites of DNA synthesis. Mol. Cell. Biol 2001, 21, 6006–6016. [Google Scholar]
- Liu, K.; Lin, F.-T.; Ruppert, J.M.; Lin, W.-C. Regulation of E2F1 by brct domain-containing protein topbp1. Mol. Cell. Biol 2003, 23, 3287–3304. [Google Scholar]
- Polager, S.; Kalma, Y.; Berkovich, E.; Ginsberg, D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 2002, 21, 437–446. [Google Scholar]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and g2/m checkpoints. Genes Dev 2002, 16, 245–256. [Google Scholar]
- Abu-Yousif, A.O.; Smith, K.A.; Getsios, S.; Green, K.J.; van Dross, R.T.; Pelling, J.C. Enhancement of uvb-induced apoptosis by apigenin in human keratinocytes and organotypic keratinocyte cultures. Cancer Res 2008, 68, 3057–3065. [Google Scholar]
- Lin, W.-C.; Lin, F.-T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by atm-dependent phosphorylation. Genes Dev 2001, 15, 1833–1844. [Google Scholar]
- Stevens, C.; Smith, L.; la Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol 2003, 5, 401–409. [Google Scholar]
- Yang, W.W.; Wang, Z.H.; Zhu, Y.; Yang, H.T. E2F6 negatively regulates ultraviolet-induced apoptosis via modulation of brca1. Cell Death Differ 2006, 14, 807–817. [Google Scholar]
- Lyons, T.E.; Salih, M.; Tuana, B.S. Activating E2Fs mediate transcriptional regulation of human E2F6 repressor. Am. J. Physiol 2006, 290, C189–C199. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UVA | UVB | |
|---|---|---|
| Wavelength (nm) | 320–400 | 280–320 |
| Chromophores | Photosensitizers | DNA |
| Site of damage | ROS | Pyrimidine dimers (CDP) 6–4 photoproducts |
| Mechanism | Indirect | Direct |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hazar-Rethinam, M.; Endo-Munoz, L.; Gannon, O.; Saunders, N. The Role of the E2F Transcription Factor Family in UV-Induced Apoptosis. Int. J. Mol. Sci. 2011, 12, 8947-8960. https://doi.org/10.3390/ijms12128947
Hazar-Rethinam M, Endo-Munoz L, Gannon O, Saunders N. The Role of the E2F Transcription Factor Family in UV-Induced Apoptosis. International Journal of Molecular Sciences. 2011; 12(12):8947-8960. https://doi.org/10.3390/ijms12128947
Chicago/Turabian StyleHazar-Rethinam, Mehlika, Liliana Endo-Munoz, Orla Gannon, and Nicholas Saunders. 2011. "The Role of the E2F Transcription Factor Family in UV-Induced Apoptosis" International Journal of Molecular Sciences 12, no. 12: 8947-8960. https://doi.org/10.3390/ijms12128947