Genetic Association and Altered Gene Expression of Mir-155 in Multiple Sclerosis Patients


 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. miRNA Selection
- human miRNA genes mapping within loci previously associated with MS or EAE (experimental autoimmune encephalitis, the experimentally-induced form of MS in animal models) were extracted using the UCSC genome browser (reference sequence NCBI Build 36.1/hg18), crossing data extracted with the sno/miRNA track with those extracted either with the GAD View or with the RGD-Rat-QTL/ MGI-Mouse-QTL tracks;
- miRNAs known to be preferentially expressed in tissues/cells involved in MS were retrieved from the literature, as well as through publicly available miRNA expression repositories (i.e., the smiRNAdb database and the miRNA Map website [44]).
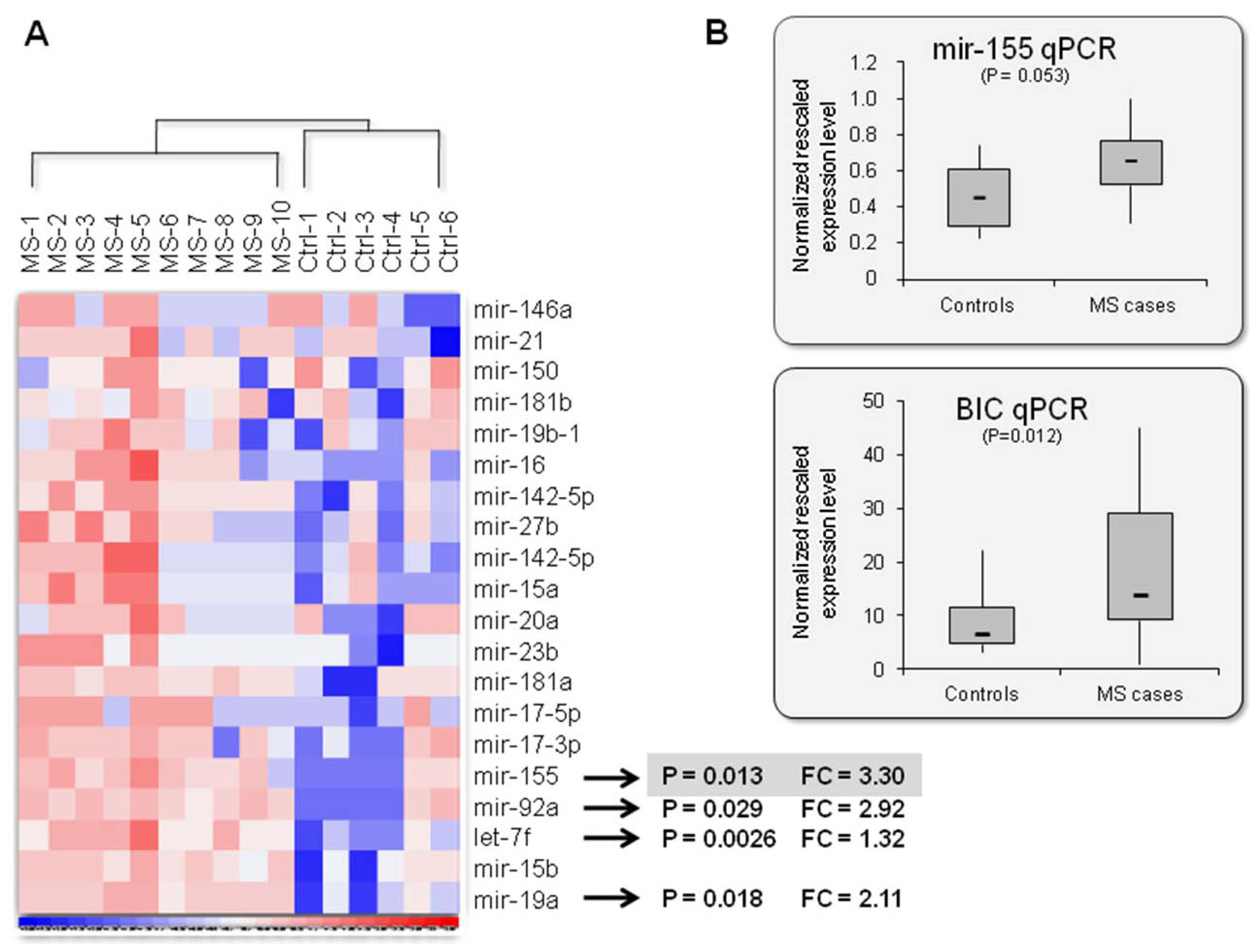
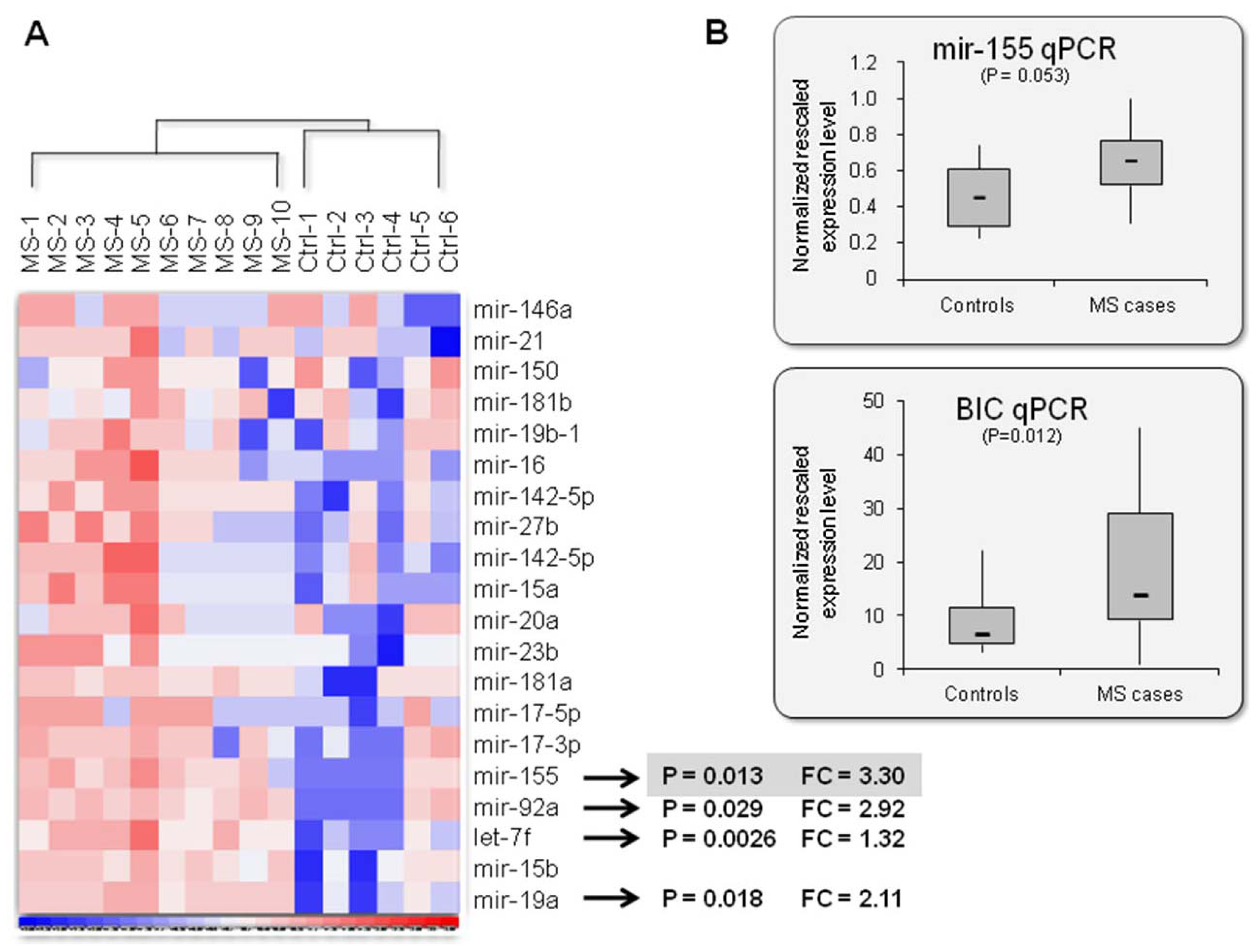
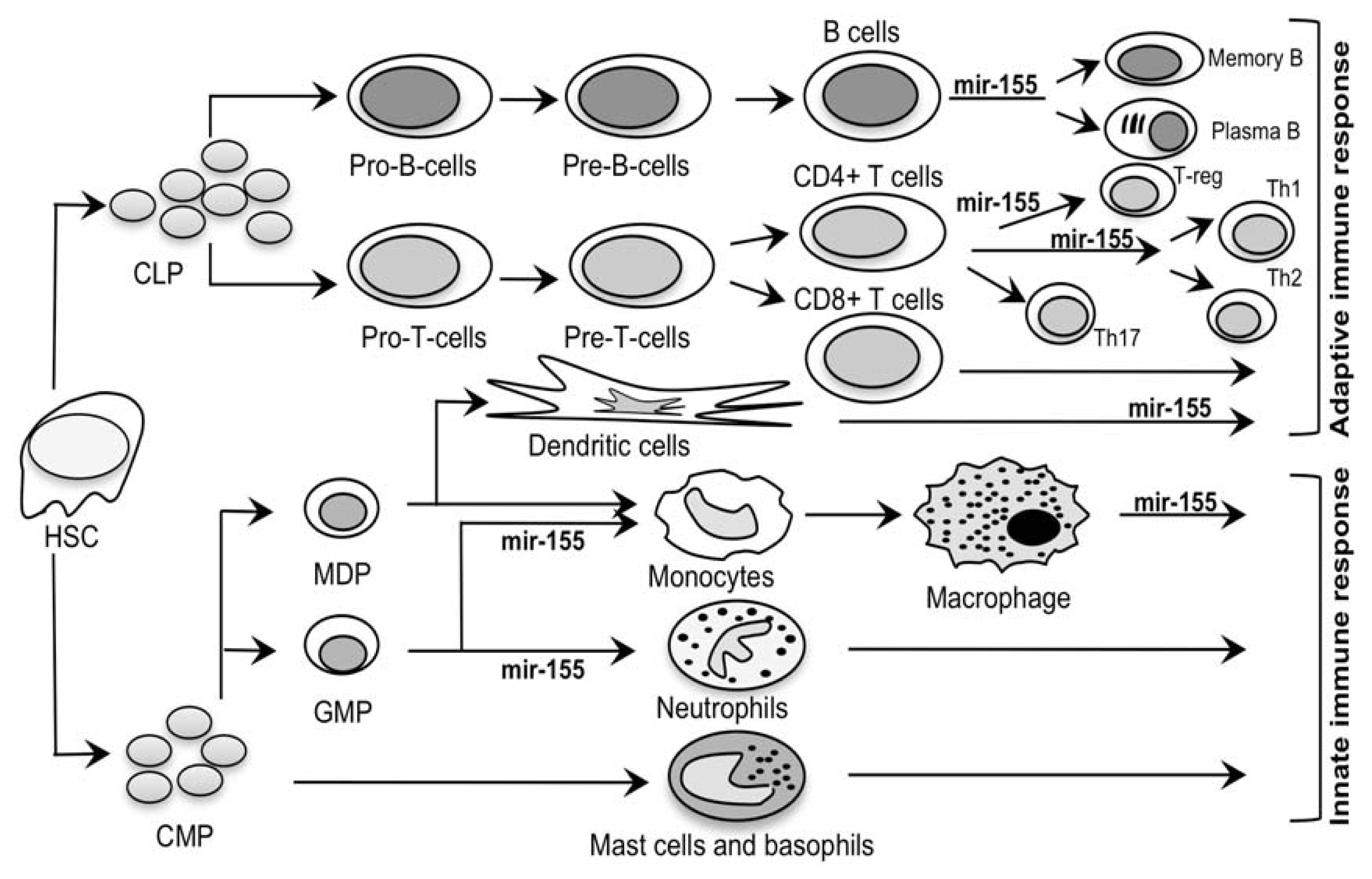
2.2. Mir-155: An MS Signature?
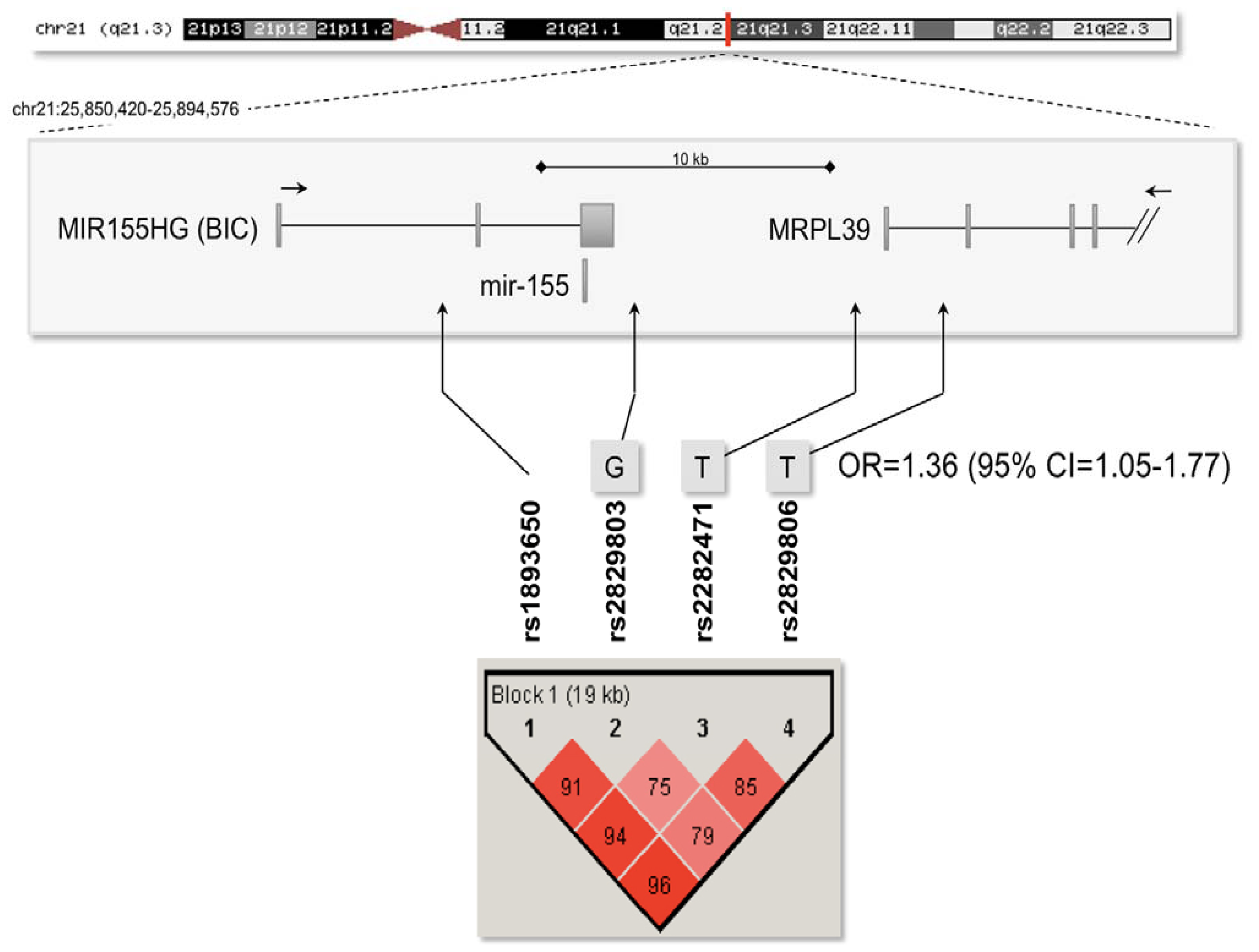
2.3. Association Analysis with the Mir-155 Locus
3. Experimental Section
3.1. Subjects
3.2. RNA Samples
3.3. MiRNA Expression Profiling By Microbead-Based Technology
3.4. Real-Time qRT-PCR Analyses
3.5. DNA Samples and Genotyping
3.6. Association Analysis
3.7. Web Resources
- OMIM, Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/omim/
- The dChip software, http://www.dchip.org/
- The PLINK software, http://pngu.mgh.harvard.edu/~purcell/plink/
- The Haploview software, http://www.broad.mit.edu/mpg/haploview/
- The University of California Santa Cruz Genome Browser (UCSC), http://genome.ucsc.edu/
- The smiRNAdb repository, http://www.mirz.unibas.ch/cloningprofiles/
- The miRNA MAP website, http://mirnamap.mbc.nctu.edu.tw/
- The GeneCards website, http://www.genecards.org/
- The dbGaP database, http://www.ncbi.nlm.nih.gov/gap
4. Limitations and Conclusions
Acknowledgments
References
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med 2000, 343, 938–952. [Google Scholar]
- Greenstein, J.I. Current concepts of the cellular and molecular pathophysiology of multiple sclerosis. Dev. Neurobiol 2007, 67, 1248–1265. [Google Scholar]
- Hauser, S.L.; Oksenberg, J.R. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 2006, 52, 61–76. [Google Scholar]
- Dyment, D.A.; Ebers, G.C.; Sadovnick, A.D. Genetics of multiple sclerosis. Lancet Neurol 2004, 3, 104–110. [Google Scholar]
- Oksenberg, J.R.; Baranzini, S.E.; Sawcer, S.; Hauser, S.L. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat. Rev. Genet 2008, 9, 516–526. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862.
- Baranzini, S.E.; Wang, J.; Gibson, R.A.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.W.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet 2009, 18, 767–778. [Google Scholar]
- Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene). Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009, 41, 824–828.
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet 2009, 41, 776–782. [Google Scholar]
- Jakkula, E.; Leppä, V.; Sulonen, A.M.; Varilo, T.; Kallio, S.; Kemppinen, A.; Purcell, S.; Koivisto, K.; Tienari, P.; Sumelahti, M.L.; et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet 2010, 86, 285–291. [Google Scholar]
- Sanna, S.; Pitzalis, M.; Zoledziewska, M.; Zara, I.; Sidore, C.; Murru, R.; Whalen, M.B.; Busonero, F.; Maschio, A.; Costa, G.; et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat. Genet 2010, 42, 495–497. [Google Scholar]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [Green Version]
- Vyse, T.J.; Todd, J.A. Genetic analysis of autoimmune disease. Cell 1996, 85, 311–318. [Google Scholar]
- Hoppenbrouwers, I.A.; Hintzen, R.Q. Genetics of multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 194–201. [Google Scholar]
- Stefani, G.; Slack, F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol 2008, 9, 219–230. [Google Scholar]
- Dai, R.; Ahmed, S.A. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl. Res 2011, 157, 163–179. [Google Scholar]
- Shlomchik, M.J.; Craft, J.E.; Mamula, M.J. From T to B and back again: Positive feedback in systemic autoimmune disease. Nat. Rev. Immunol 2001, 1, 147–153. [Google Scholar]
- Goodnow, C.C. Multistep pathogenesis of autoimmune disease. Cell 2007, 130, 25–35. [Google Scholar]
- Otaegui, D.; Mostafavi, S.; Bernard, C.C.; Lopez de Munain, A.; Mousavi, P.; Oksenberg, J.R.; Baranzini, S.E. Increased transcriptional activity of milk-related genes following the active phase of experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol 2007, 179, 4074–4082. [Google Scholar]
- Satoh, J.; Misawa, T.; Tabunoki, H.; Yamamura, T. Molecular network analysis of T-cell transcriptome suggests aberrant regulation of gene expression by NF-kappaB as a biomarker for relapse of multiple sclerosis. Dis. Markers 2008, 25, 27–35. [Google Scholar]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 2009, 132, 3342–3352. [Google Scholar]
- Keller, A.; Leidinger, P.; Lange, J.; Borries, A.; Schroers, H.; Scheffler, M.; Lenhof, H.P.; Ruprecht, K.; Meese, E. Multiple sclerosis: MicroRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS One 2009, 4, e7440:1–e7440:7. [Google Scholar]
- Otaegui, D.; Baranzini, S.E.; Armananzas, R.; Calvo, B.; Munoz-Culla, M.; Khankhanian, P.; Inza, I.; Lozano, J.A.; Castillo-Trivino, T.; Asensio, A.; et al. Differential micro RNA expression in PBMC from multiple sclerosis patients. PLoS One 2009, 4, e6309:1–e6309:9. [Google Scholar]
- Cox, M.B.; Cairns, M.J.; Gandhi, K.S.; Carroll, A.P.; Moscovis, S.; Stewart, G.J.; Broadley, S.; Scott, R.J.; Booth, D.R.; Lechner-Scott, J. ANZgene Multiple Sclerosis Genetics Consortium. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One 2010, 5, e12132:1–e12132:7. [Google Scholar]
- De Santis, G.; Ferracin, M.; Biondani, A.; Caniatti, L.; Rosaria Tola, M.; Castellazzi, M.; Zagatti, B.; Battistini, L.; Borsellino, G.; Fainardi, E.; et al. Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J. Neuroimmunol 2010, 226, 165–171. [Google Scholar]
- Lindberg, R.L.; Hoffmann, F.; Mehling, M.; Kuhle, J.; Kappos, L. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur. J. Immunol 2010, 40, 888–898. [Google Scholar]
- Lindberg, R.L.P.; Hoffmann, F.; Kuhle, J.; Kappos, L. Circulating microRNAs as indicators for disease course of multiple sclerosis. Mult. Scler 2010, 16, S41–S196. [Google Scholar]
- Sievers, C.; Hoffmann, F.; Fontoura, P.; Kappos, L.; Lindberg, R.L.P. Effect of natalizumab on microRNA expression in B-lymphocytes of relapsing-remitting multiple sclerosis patients. Mult. Scler 2010, 16, S197–S352. [Google Scholar]
- Fenoglio, C.; Cantoni, C.; de Riz, M.; Ridolfi, E.; Cortini, F.; Serpente, M.; Villa, C.; Comi, C.; Monaco, F.; Mellesi, L.; Valzelli, S.; Bresolin, N.; Galimberti, D.; Scarpini, E. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci. Lett 2011, 504, 9–12. [Google Scholar]
- Fukuda, Y.; Kawasaki, H.; Taira, K. Exploration of human miRNA target genes in neuronal differentiation. Nucleic Acids Symp. Ser 2005, 49, 341–342. [Google Scholar]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet 2006, 38, 1375–1377. [Google Scholar]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.; Liu, C.G.; Rassenti, L.; et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 2006, 66, 11590–11593. [Google Scholar]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar]
- Cameron, J.E.; Yin, Q.; Fewell, C.; Lacey, M.; McBride, J.; Wang, X.; Lin, Z.; Schaefer, B.C.; Flemington, E.K. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J. Virol 2008, 82, 1946–1958. [Google Scholar]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell Biol 2007, 9, 775–787. [Google Scholar]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar]
- Li, Q.J.; Chau, J.; Ebert, P.J.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007, 129, 147–161. [Google Scholar]
- Neilson, J.R.; Zheng, G.X.; Burge, C.B.; Sharp, P.A. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev 2007, 21, 578–589. [Google Scholar]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar]
- Wu, H.; Neilson, J.R.; Kumar, P.; Manocha, M.; Shankar, P.; Sharp, P.A.; Manjunath, N. miRNA profiling of naïve, effector and memory CD8 T cells. PLoS One 2007, 2, e1020:1–e1020:11. [Google Scholar]
- Lodish, H.F.; Zhou, B.; Liu, G.; Chen, C.Z. Micromanagement of the immune system by microRNAs. Nat. Rev. Immunol 2008, 8, 120–130. [Google Scholar]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol 2008, 9, 405–414. [Google Scholar]
- Tufekci, K.U.; Oner, M.G.; Genc, S.; Genc, K. MicroRNAs and multiple sclerosis. Autoimmune Dis 2011, 2011, 807426:1–807426:27. [Google Scholar]
- Hsu, S.D.; Chu, C.H.; Tsou, A.P.; Chen, S.J.; Chen, H.C.; Hsu, P.W.; Wong, Y.H.; Chen, Y.H.; Chen, G.H.; Huang, H.D. miRNAMap 2.0: Genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res 2008, 36, D165–D169. [Google Scholar]
- Murugaiyan, G.; Beynon, V.; Mittal, A.; Joller, N.; Weiner, H.L. Silencing MicroRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol 2011, 187, 2213–2221. [Google Scholar]
- Stanczyk, J.; Pedrioli, D.M.; Brentano, F.; Sanchez-Pernaute, O.; Kolling, C.; Gay, R.E.; Detmar, M.; Gay, S.; Kyburz, D. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum 2008, 58, 1001–1009. [Google Scholar]
- Murata, K.; Yoshitomi, H.; Tanida, S.; Ishikawa, M.; Nishitani, K.; Ito, H.; Nakamura, T. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther 2010, 12, R86:1–R86:14. [Google Scholar]
- Takagi, T.; Naito, Y.; Mizushima, K.; Hirata, I.; Yagi, N.; Tomatsuri, N.; Ando, T.; Oyamada, Y.; Isozaki, Y.; Hongo, H.; et al. Increased expression of microRNA in the inflamed colonic mucosa of patients with active ulcerative colitis. J. Gastroenterol. Hepatol 2010, 25, S129–S133. [Google Scholar]
- Dai, R.; Zhang, Y.; Khan, D.; Heid, B.; Caudell, D.; Crasta, O.; Ahmed, S.A. Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS One 2010, 5, e14302:1–e14302:8. [Google Scholar]
- Divekar, A.A.; Dubey, S.; Gangalum, P.R.; Singh, R.R. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: An apparent paradox in the setting of an inflammatory milieu. J. Immunol 2011, 186, 924–930. [Google Scholar]
- Leng, R.X.; Pan, H.F.; Qin, W.Z.; Chen, G.M.; Ye, D.Q. Role of microRNA-155 in autoimmunity. Cytokine Growth Factor Rev 2011, 22, 141–147. [Google Scholar]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol 2007, 8, 913–919. [Google Scholar]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci 2008, 31, 247–269. [Google Scholar]
- Li, C.; Wong, W.H. Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 2001, 98, 31–36. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002, 3, RESEARCH0034:1–RESEARCH0034:12. [Google Scholar]
- Polman, C.H.; Reingold, S.C.; Edan, G.; Filippi, M.; Hartung, H.P.; Kappos, L.; Lublin, F.D.; Metz, L.M.; McFarland, H.F.; O’Connor, P.W.; et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann. Neurol 2005, 58, 840–846. [Google Scholar]
- Lublin, F.D. Clinical features and diagnosis of multiple sclerosis. Neurol. Clin 2005, 23, 1–15. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 2007, 81, 559–575. [Google Scholar]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Genomic location 1 | Expression 2 | Known targets 2, 3 | Function2 | Association 1 | ||
|---|---|---|---|---|---|---|---|
| MS | EAE | ||||||
| 1 | let-7f | chr9q22 chrXp11 | CD8+ T cells (ubiquitous) | TLR4 | innate immune response | no | yes |
| 2 | mir-15a | chr13q14 | thymus, spleen | DMP1/DMTF1 | unknown | no | |
| 3 | mir-15b | 3q26 | CD8+ T cells, lymphocyte development | no validated targets | mutations in mir16–15 cause autoimmune and B lymphoproliferative disease in mice | ||
| 4 | mir-16 | chr3q26 chr13q14 | BCL2, TPPP3 | ||||
| 5 | mir-17-3p | chr13q31 | lymphocytes, lymphoid tissues | PTEN, Bim, AML1 | mir-17–92 cluster higher expression in lymphocytes causes autoimmune disease in mice | ||
| 6 | mir-17-5p | chr13q32 | myeloid cells | no validated targets | mir-17–92 cluster higher expression in lymphocytes causes autoimmune disease in mice; monocyte proliferation and differentiation | ||
| 7 | mir-18a | chr13q31 | placenta, spleen, kidney, thymus | no validated targets | mir-17–92 cluster higher expression in lymphocytes causes autoimmune disease in mice | ||
| 8 | mir-19a | chr13q31 chrXq26 | lymphocytes, lymphoid tissues | PTEN, Bim | |||
| 9 | mir-19b-1 | chr13q31 | thymus, ovary, prostate, spleen | no validated targets | |||
| 10 | mir-20a | lymphocyte development; bladder, lung, thymus | AML1 | ||||
| 11 | mir-21 | chr17q23 | CD8+ T cells, lymphocyte development | PTEN, Pdcd4, TPM1, IL12a | macrophage activation | no 4 | |
| 12 | mir-23b | chr9q22 | hematopoietic cells | Notch1 | neural development | no | yes |
| 13 | mir-27b | lung, UBC-EPC, MVEC, CTCL | CYP1B1, Notch1 | neural development | |||
| 14 | mir-92a | chr13q31 Xq26 | lymphocytes, lymphoid tissues | PTEN, Bim | mir-17–92 cluster higher expression in lymphocytes causes autoimmune disease in mice | no | |
| 15 | mir-105 | Xq28 | nervous and reproductive system | no validated targets | unknown | yes | |
| 16 | mir-142-3p | chr17q22 | CD8+ T cells, lymphocyte development | no validated targets | unknown | no 4 | |
| 17 | mir-142-5p | ||||||
| 18 | mir-146a | chr5q33 | B cells, monocytes | IRF7, TRAF6, IRAK1, IRAK2 | innate immune response, induced by EBV, TLR signaling | no | yes |
| 19 | mir-150 | chr19q13 | CD8+ T cells, spleen, thymus, lymphocyte development | Myb, AID, BACH1, CEBPB, CSFR | B-cell development, T-cell activation, innate and adaptive immune response | no | |
| 20 | mir-155 | chr21q21 | B, T, and dendritic cells, monocytes, spleen, thymus, lung | MAF, AGTR1, FADD, IKK, JARID2, PU.1, Ripk1, SOCS1, TAB2, CD47 | macrophages germinal center response, IgG class switch, peripheral T-cell development | ||
| 21 | mir-181a | chr1q32.1 chr9q33.3 | lymphocyte maturation (highest in CD4+CD8+ T cells) | SHP2, PTPN22, AID, DUSP5-6, CD69, BCL2, TCR alpha | lymphocyte maturation, both positive and negative selection | yes | yes |
| 22 | mir-181b | brain, thymus, lymphocytes | Tcl1, AID | B-cell class switch | |||
| SNP | Minor allele | Minor allele frequency in MS cases (%) | Minor allele frequency in controls (%) | P value | OR | 95% CI |
|---|---|---|---|---|---|---|
| rs1893650 | T | 29.4 | 28.1 | 0.53 | 1.07 | 0.86–1.32 |
| rs2829803 | G | 26.9 | 24.0 | 0.15 | 1.17 | 0.94–1.44 |
| rs2282471 | T | 16.6 | 14.6 | 0.28 | 1.16 | 0.89–1.51 |
| rs2829806 | T | 28.1 | 24.4 | 0.076 | 1.22 | 0.98–1.52 |
| SNP | Minor allele | Major allele | Genotype frequencies in MS cases (%) | Genotype frequencies in controls (%) | P value |
|---|---|---|---|---|---|
| rs1893650 | T | C | 7.5/44.0/48.5 | 8.9/38.4/52.7 | 0.24 |
| rs2829803 | G | A | 7.2/39.5/53.3 | 6.5/35.1/58.4 | 0.30 |
| rs2282471 | T | C | 2.0/29.2/68.8 | 3.1/23.1/73.8 | 0.099 |
| rs2829806 | T | G | 6.1/44.2/49.7 | 5.1/38.5/56.4 | 0.16 |
| SNPs | Haplotype | Haplotype frequency in MS cases (%) | Haplotype frequency in controls (%) | P value |
|---|---|---|---|---|
| rs1893650 rs2829803 rs2282471 | TGT | 13.4 | 11.7 | 0.28 |
| TAT | 2.4 | 2.3 | 0.99 | |
| TGC | 10.9 | 11.4 | 0.70 | |
| CGC | 1.1 | 1.3 | 0.70 | |
| TAC | 3.3 | 2.8 | 0.55 | |
| CAC | 68.9 | 70.4 | 0.51 | |
| rs2829803 rs2282471 rs2829806 | GTT | 13.5 | 10.3 | 0.035 |
| ATT | 2.3 | 2.4 | 0.88 | |
| GCT | 10.6 | 9.7 | 0.52 | |
| ACT | 2.3 | 2.0 | 0.61 | |
| GTG | 0.3 | 1.6 | 0.016 | |
| GCG | 1.4 | 2.9 | 0.039 | |
| ACG | 69.5 | 71.2 | 0.46 | |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paraboschi, E.M.; Soldà, G.; Gemmati, D.; Orioli, E.; Zeri, G.; Benedetti, M.D.; Salviati, A.; Barizzone, N.; Leone, M.; Duga, S.; et al. Genetic Association and Altered Gene Expression of Mir-155 in Multiple Sclerosis Patients. Int. J. Mol. Sci. 2011, 12, 8695-8712. https://doi.org/10.3390/ijms12128695
Paraboschi EM, Soldà G, Gemmati D, Orioli E, Zeri G, Benedetti MD, Salviati A, Barizzone N, Leone M, Duga S, et al. Genetic Association and Altered Gene Expression of Mir-155 in Multiple Sclerosis Patients. International Journal of Molecular Sciences. 2011; 12(12):8695-8712. https://doi.org/10.3390/ijms12128695
Chicago/Turabian StyleParaboschi, Elvezia Maria, Giulia Soldà, Donato Gemmati, Elisa Orioli, Giulia Zeri, Maria Donata Benedetti, Alessandro Salviati, Nadia Barizzone, Maurizio Leone, Stefano Duga, and et al. 2011. "Genetic Association and Altered Gene Expression of Mir-155 in Multiple Sclerosis Patients" International Journal of Molecular Sciences 12, no. 12: 8695-8712. https://doi.org/10.3390/ijms12128695