A Greatly Under-Appreciated Fundamental Principle of Physical Organic Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
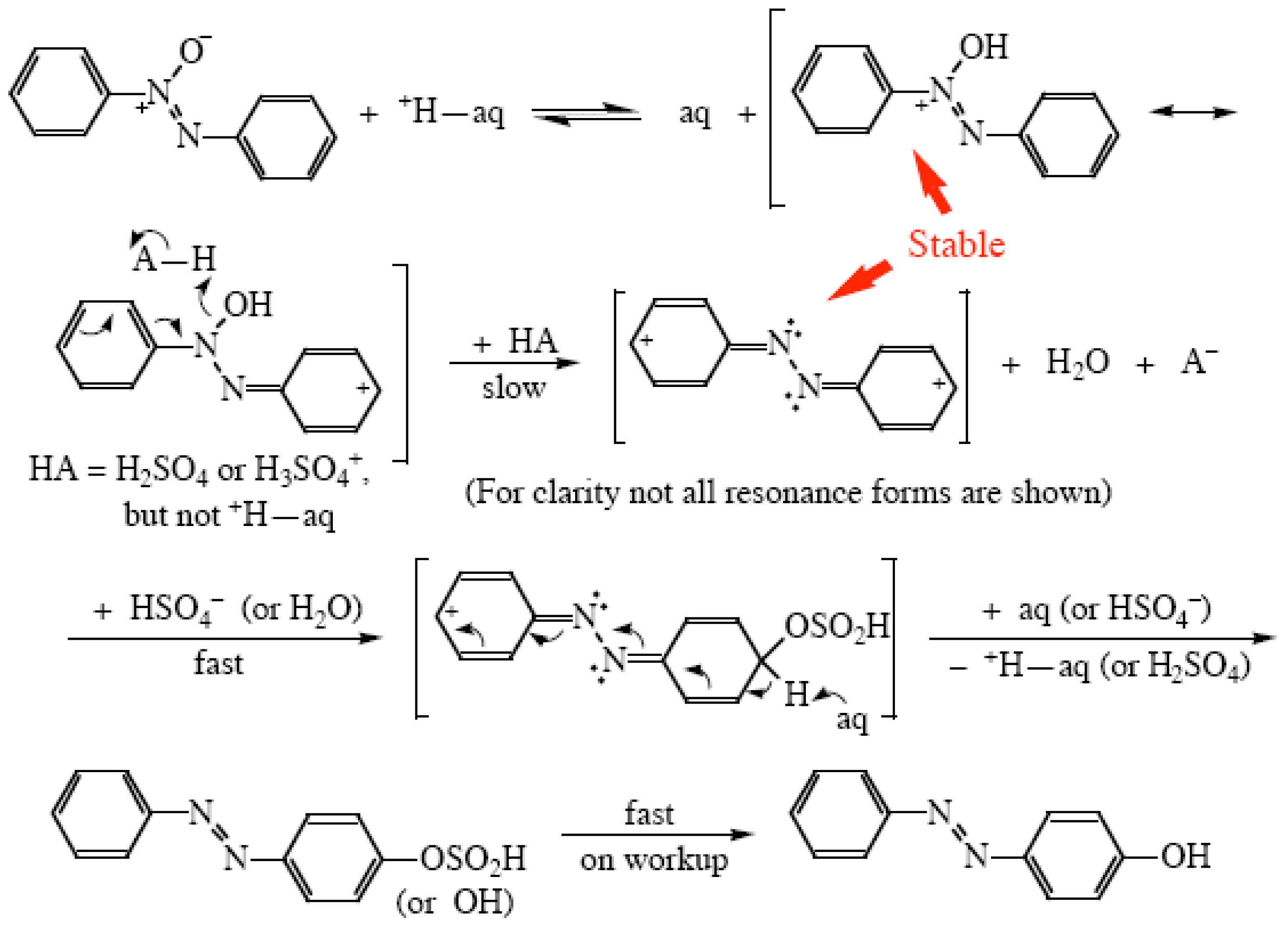{kind=link}
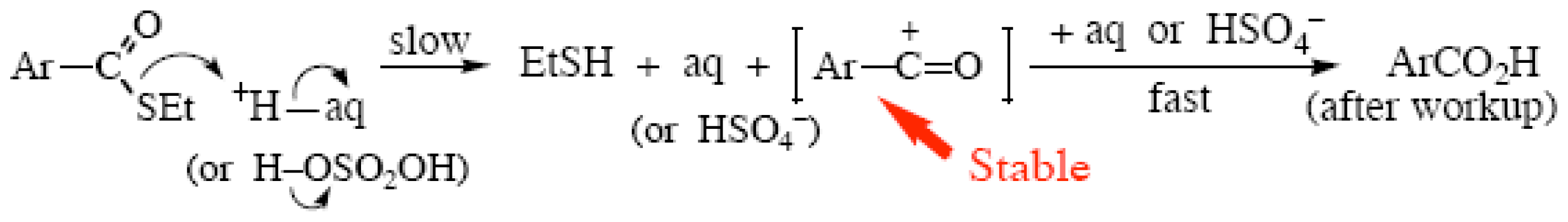{kind=link}
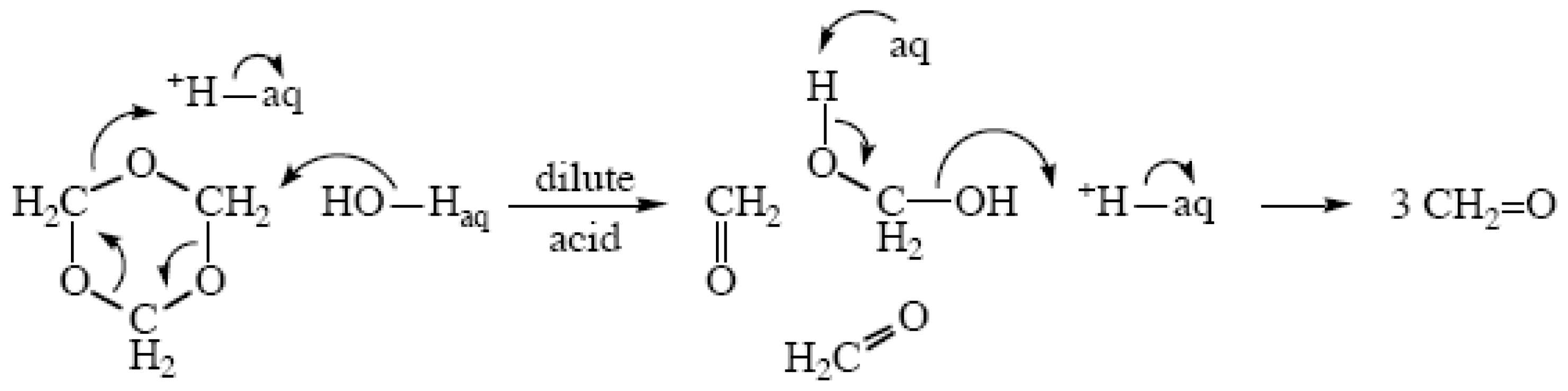{kind=link}
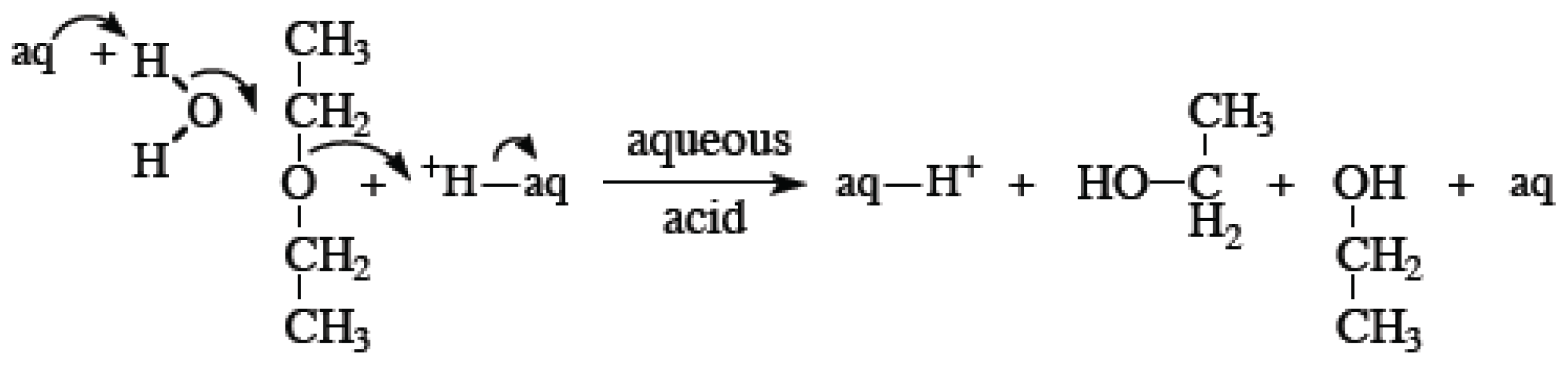{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. General Acid Catalysis in Strong Acid Media
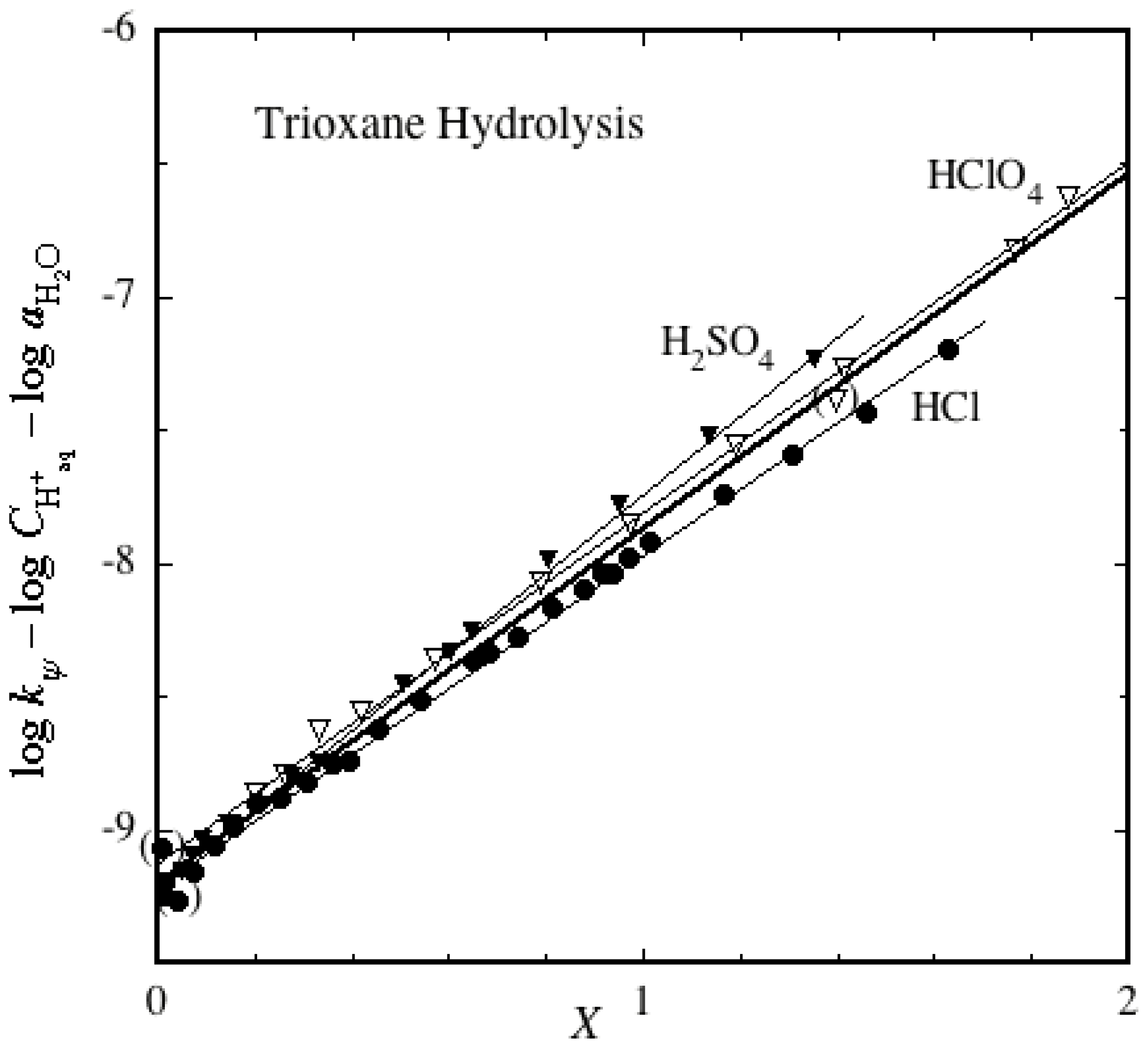
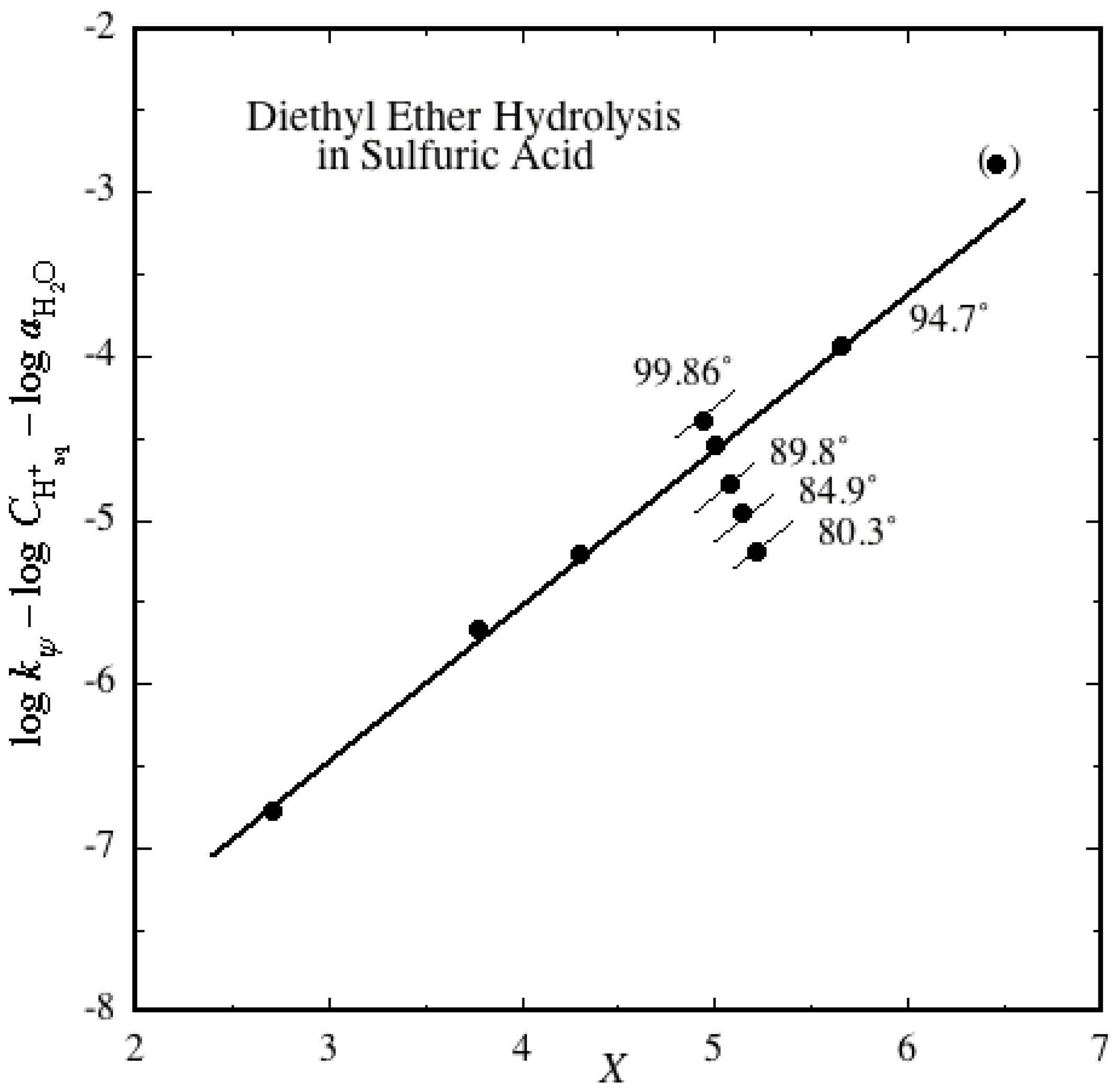
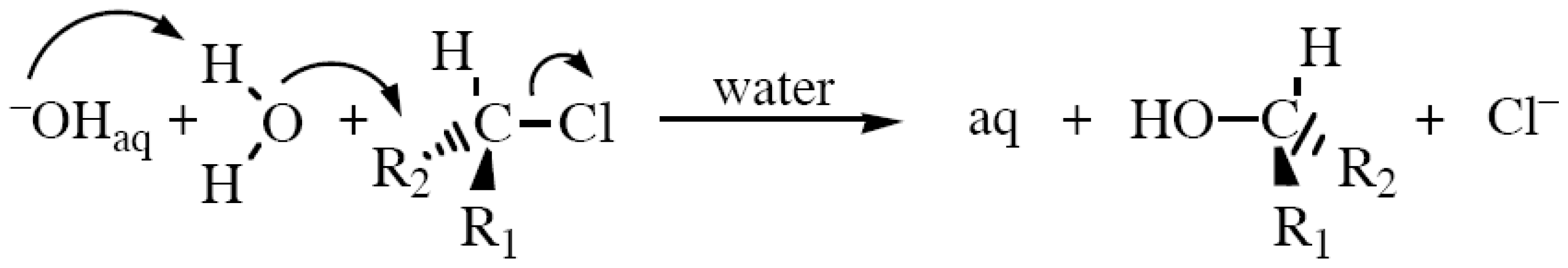
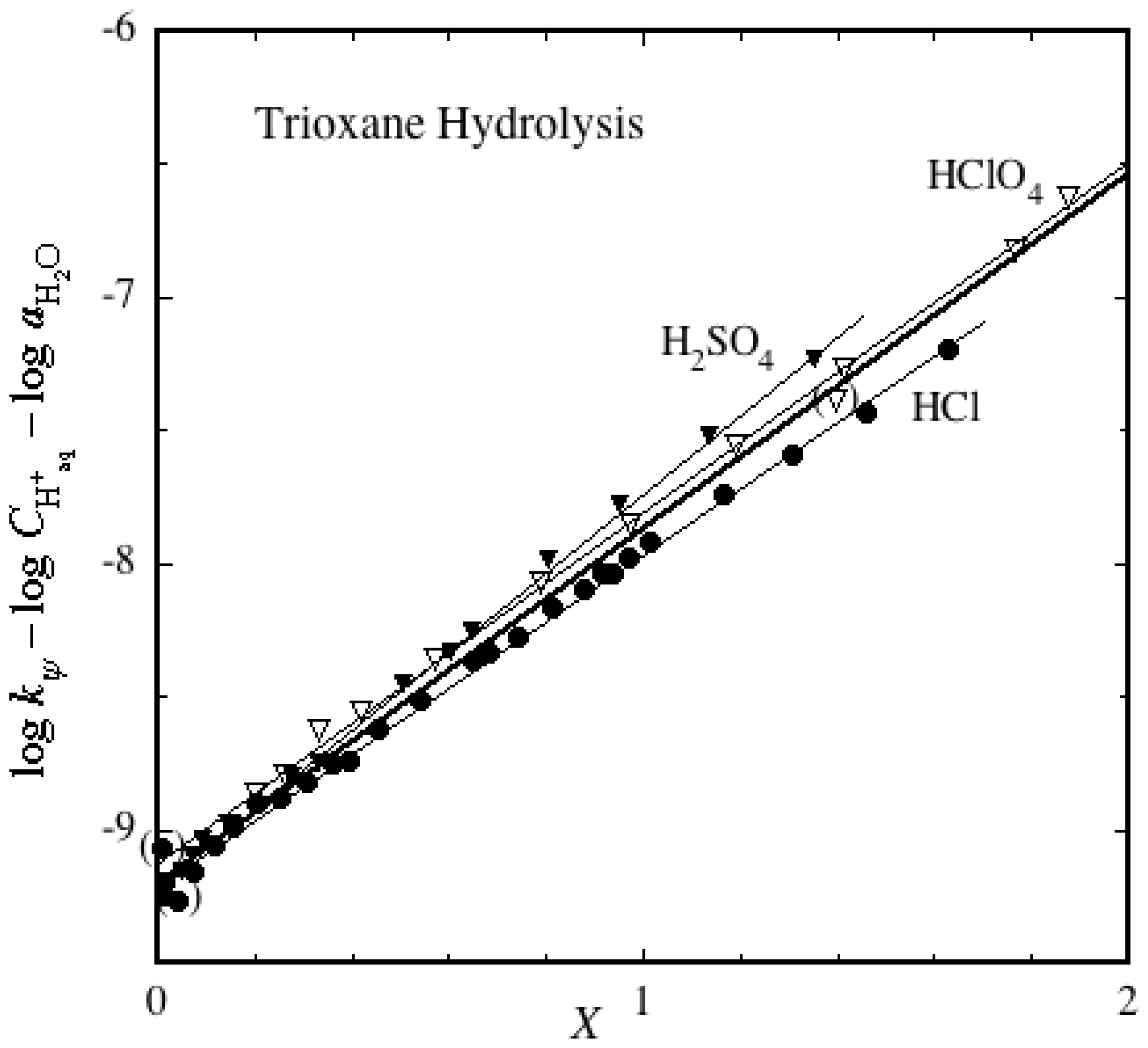
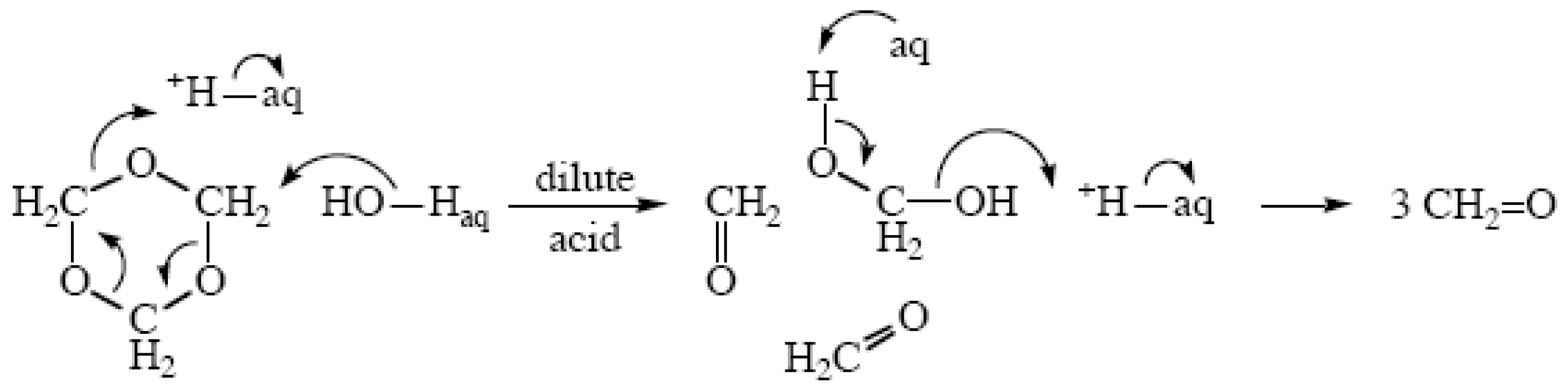
2.2. Ether Hydrolyses
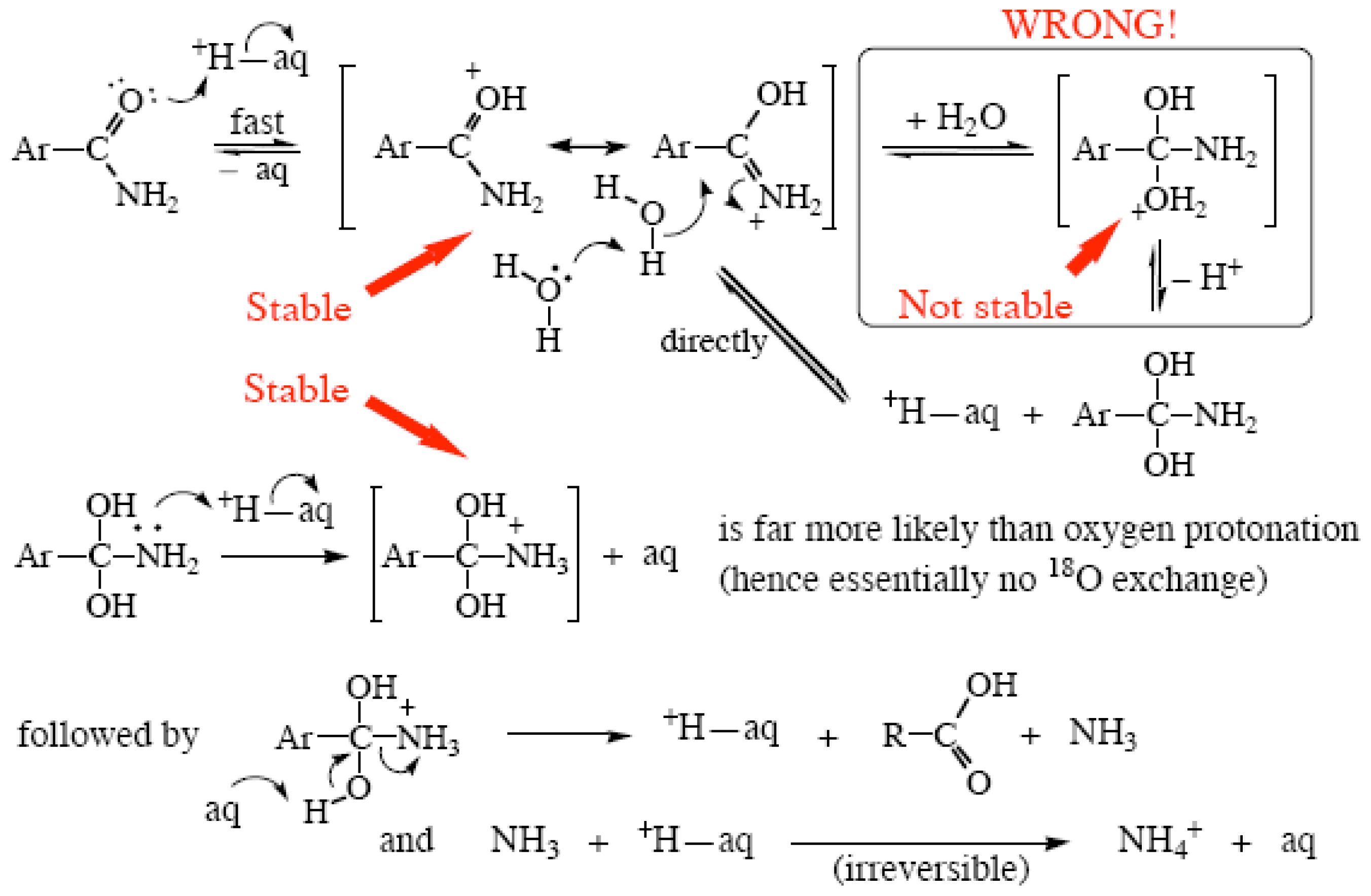
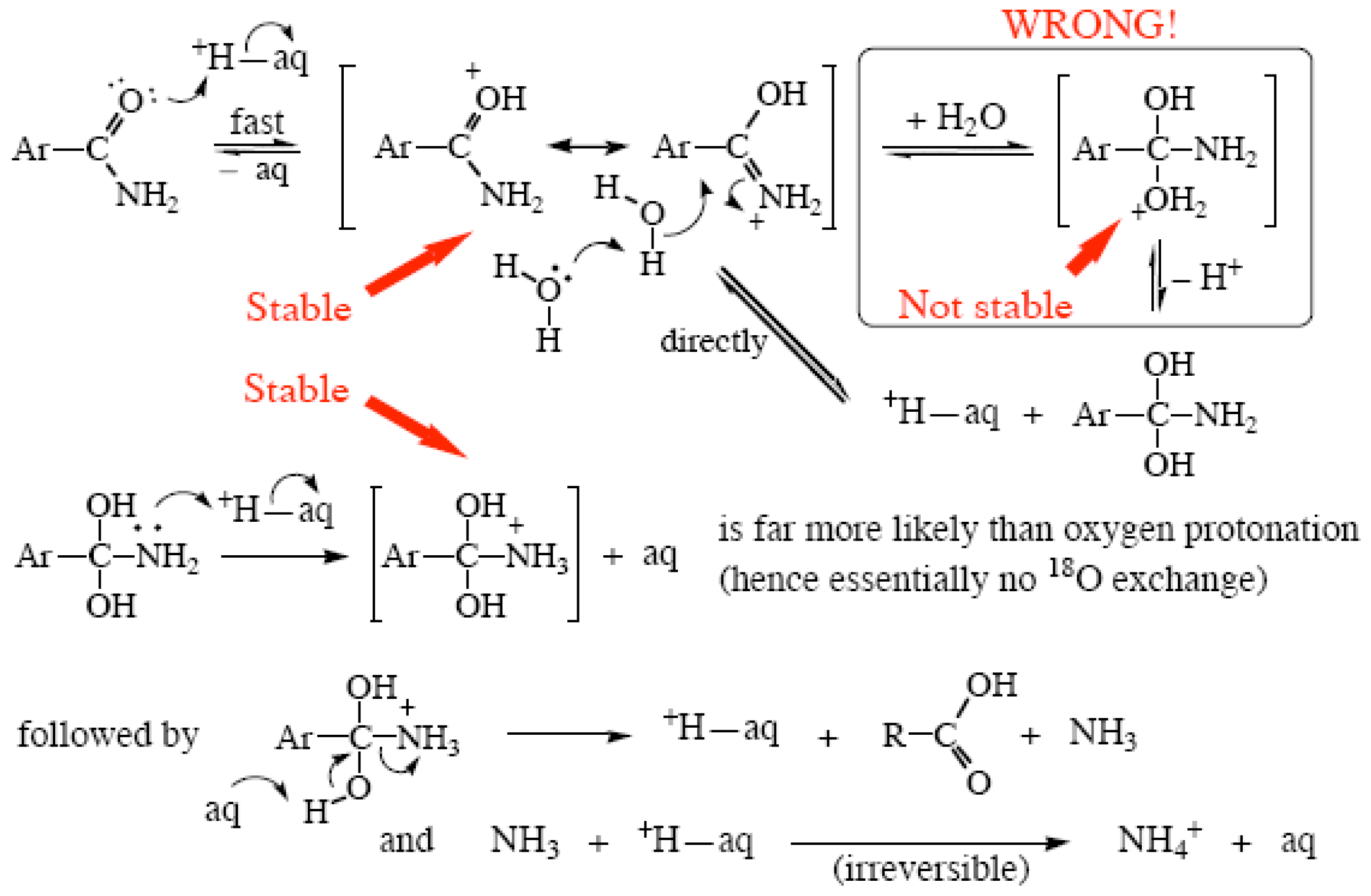
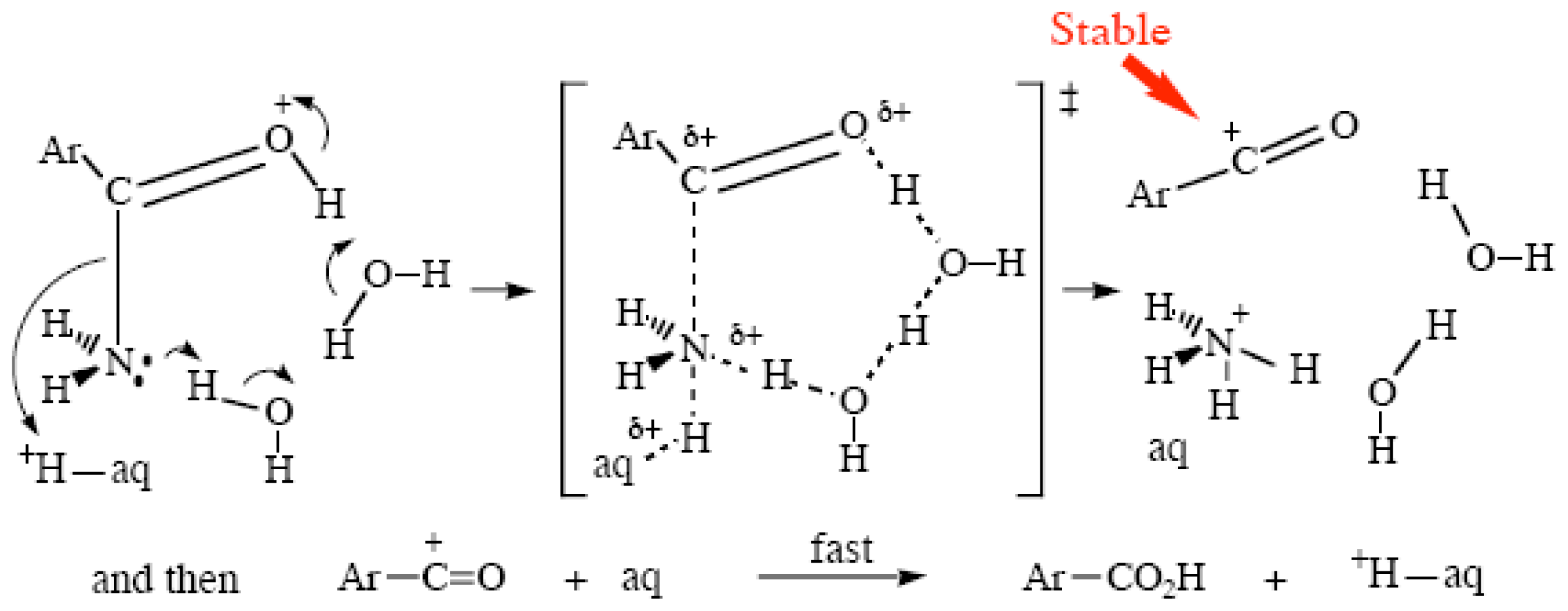
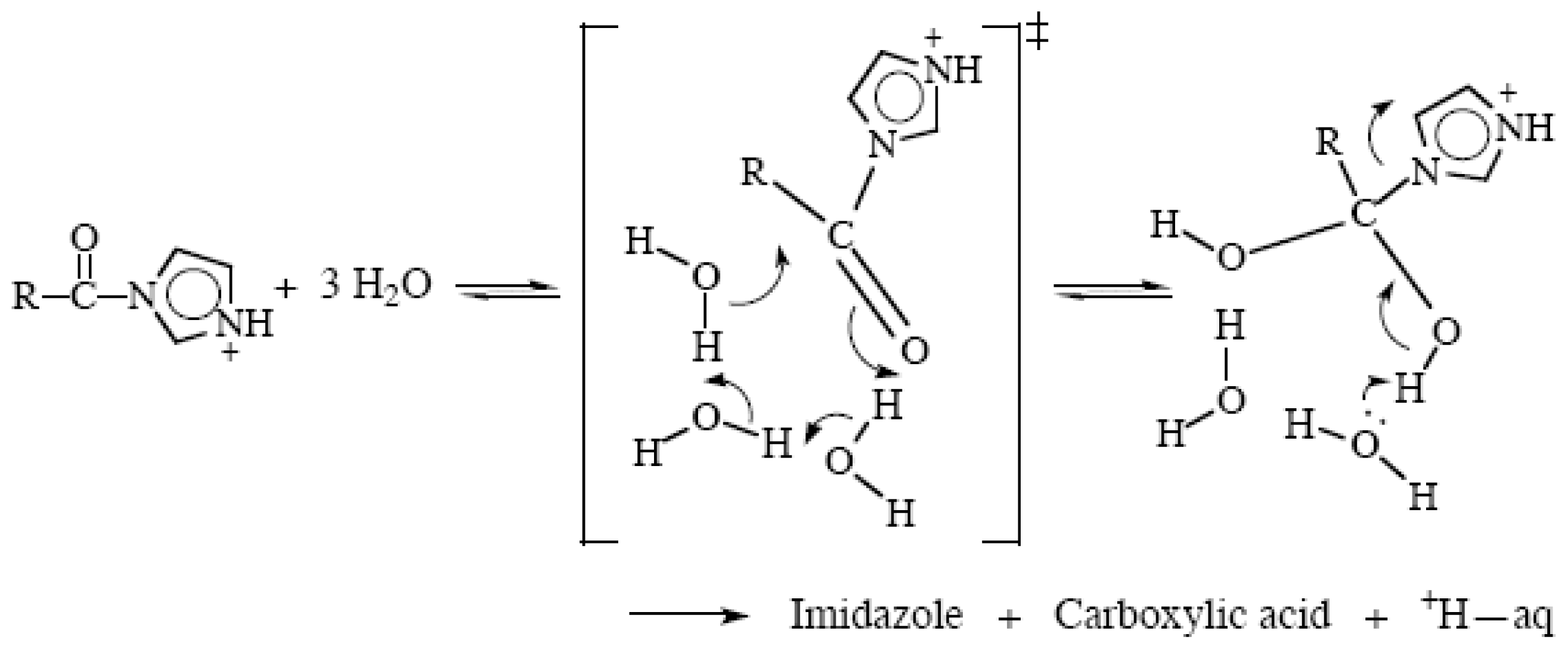
2.3. Amide Hydrolyses
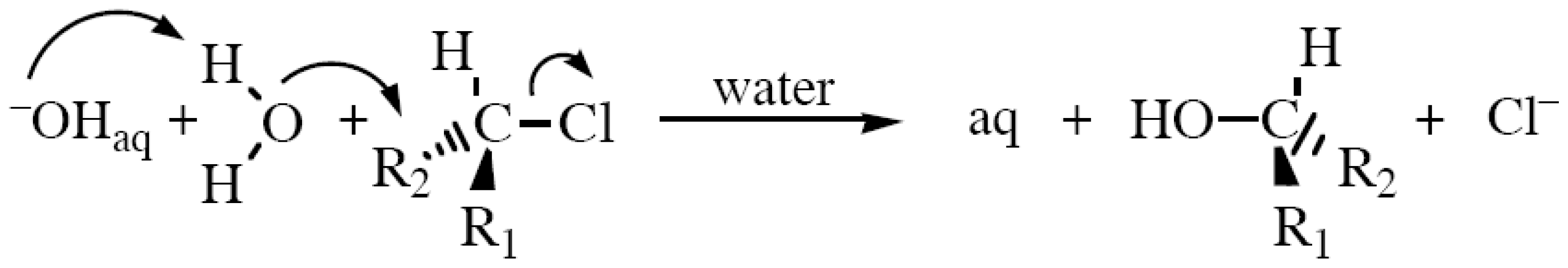
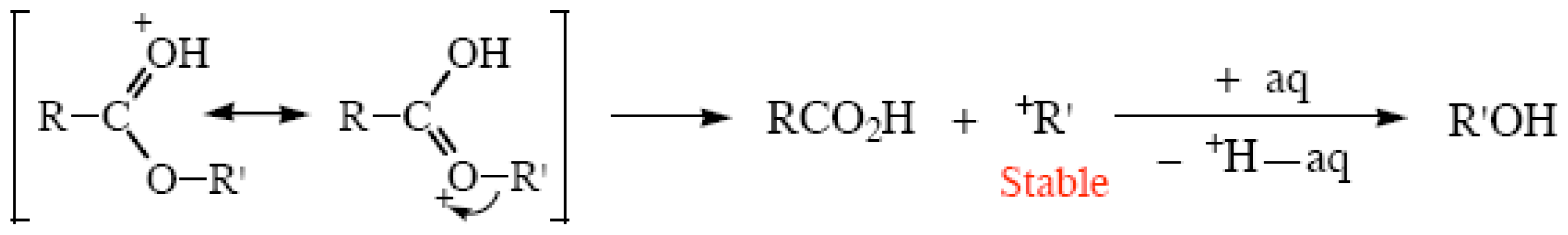
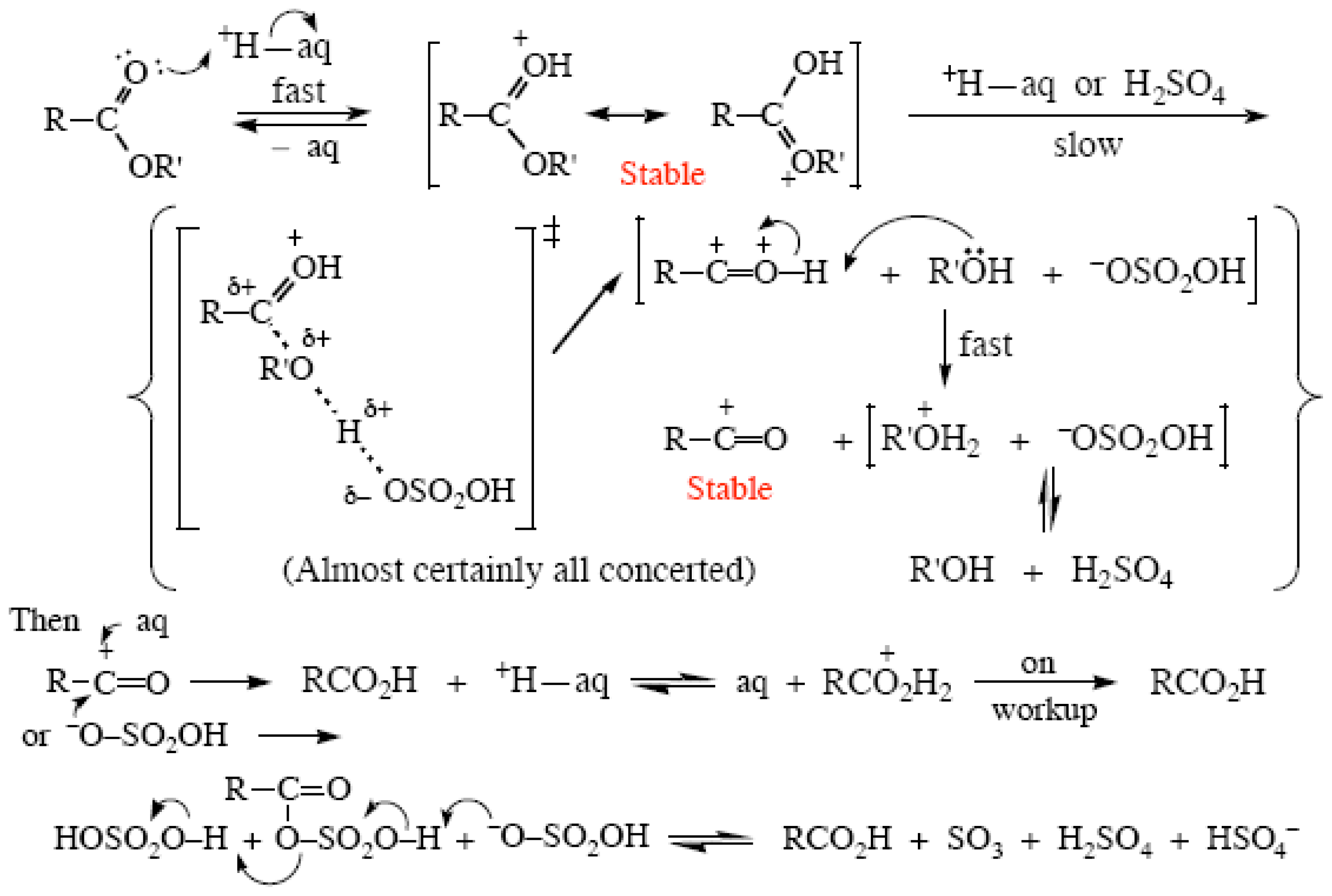
2.4. Ester Hydrolyses
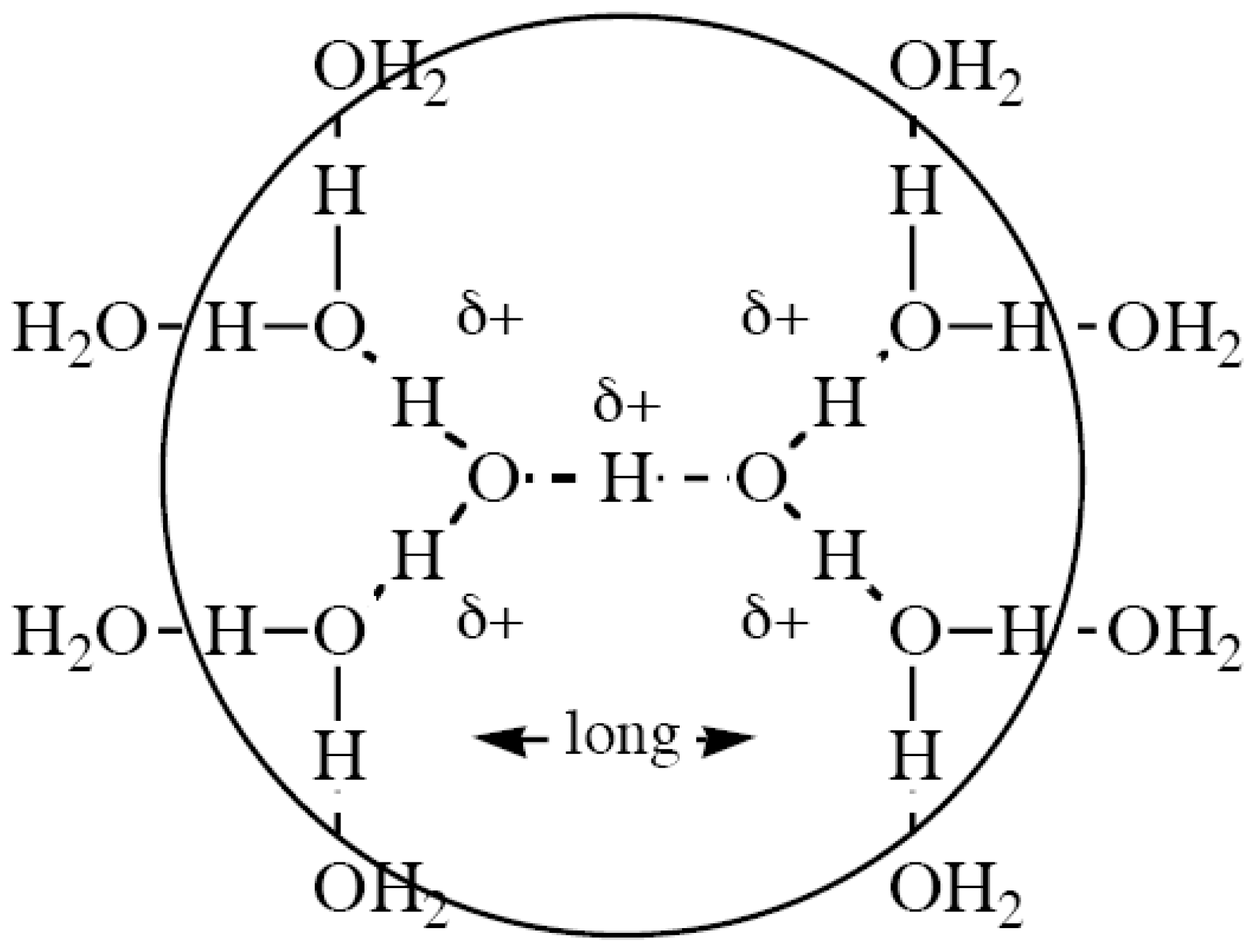
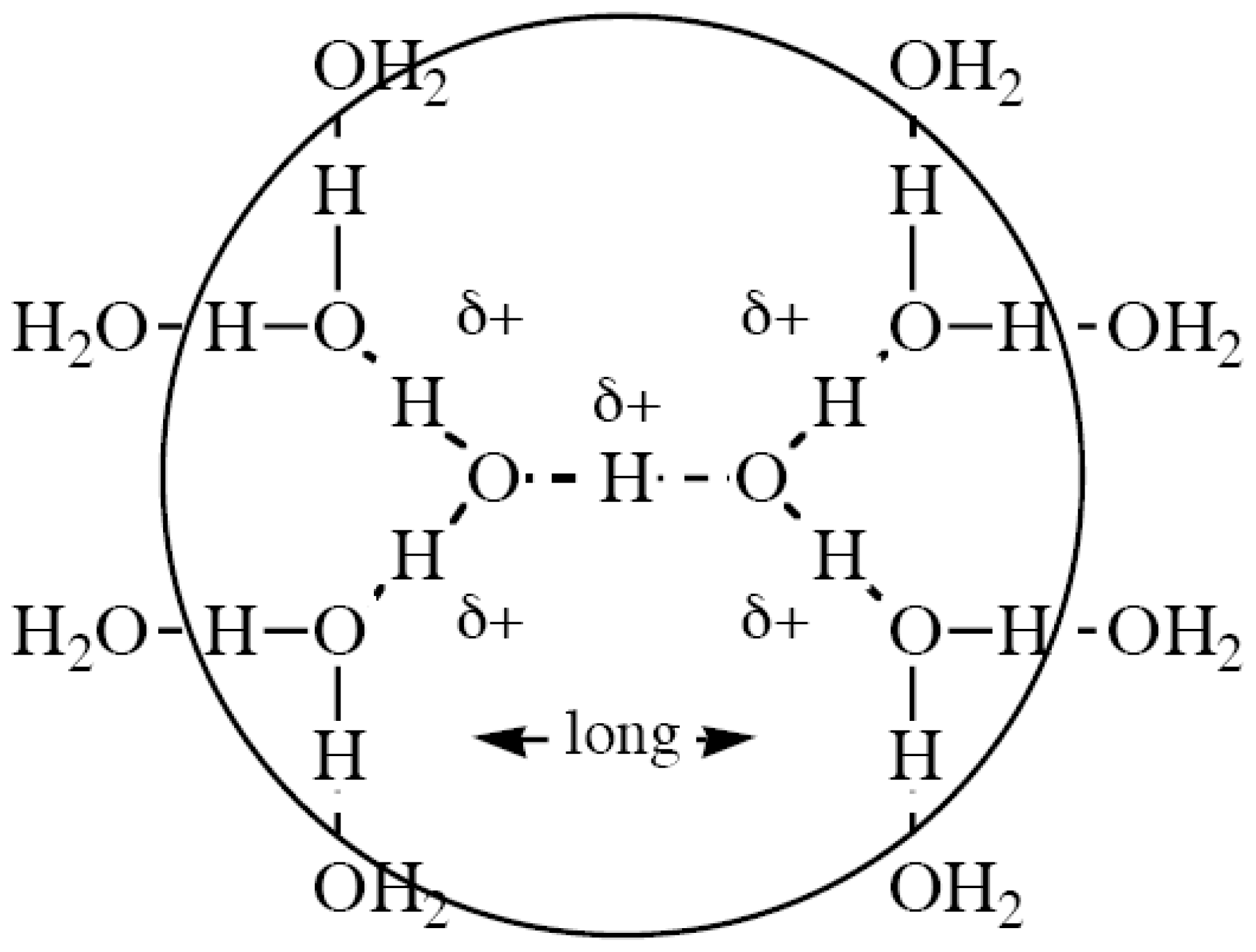
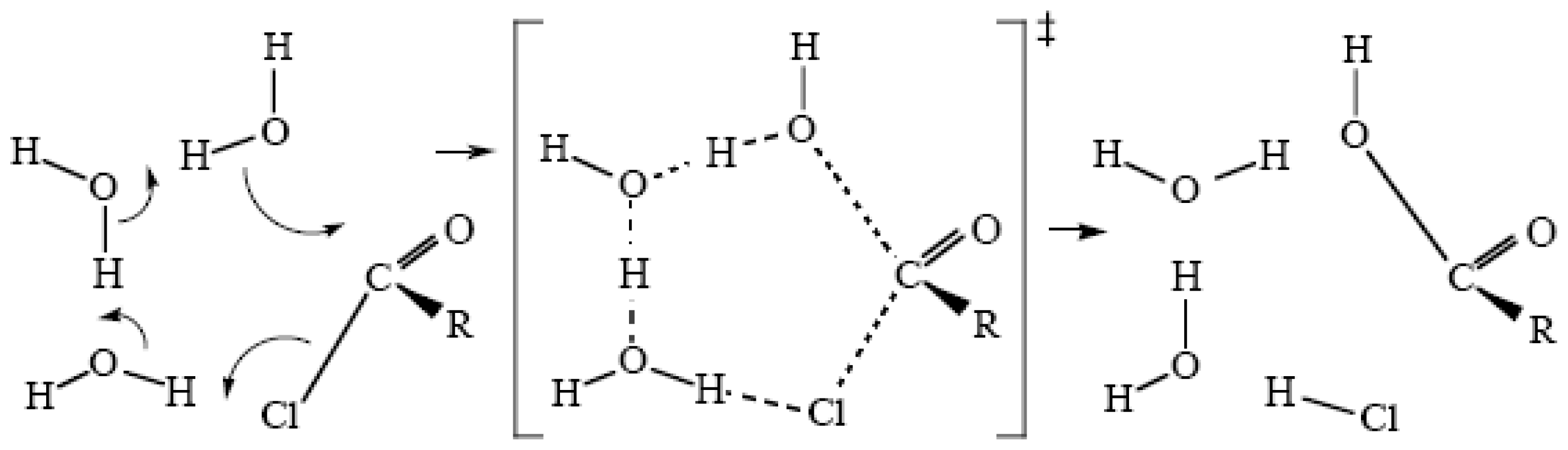
2.5. Mechanisms Involving Chains of Water Molecules
3. Conclusions
- If a species does not have a finite lifetime in the solution in which the reaction is performed it cannot be a reaction intermediate. No primary or secondary carbocations in aqueous media; only T0, no T+, T−, T± or T2− tetrahedral intermediates.
- Positive or negative charge, if present, will be as delocalized as possible during the reaction, especially in reaction intermediates, often into the aqueous solvent. A highly electronegative atom like oxygen is simply not going to support a positive charge all by itself. O+ is almost as unlikely as F+!
- Also, reactions will be unimolecular, as far as possible, for entropic reasons (SN1 favored over SN2); however, mechanisms involving chains of water molecules are favored in aqueous media thanks to the highly structured nature of water and the Grotthuss process.
Acknowledgments
- Conflict of InterestThe author declares no conflict of interest.
References and Notes
- For instance, the many reviews given in the series Reviews of Chemical Intermediates; Elsevier: Amsterdam, The Netherlands., commencing in1981.
- Olah, G.A.; White, A.M. Stable carbonium ions. XCI. Carbon-13 nuclear magnetic resonance spectroscopic study of carbonium ions. J. Am. Chem. Soc 1969, 91, 5801–5810. [Google Scholar]
- Olah, G.A. My search for carbocations and their role in chemistry. Angew. Chem. Int. Ed. Engl 1995, 34, 1393–1405, and references therein. [Google Scholar]
- Olah, G.A.; Dunne, K.; Kelly, D.P.; Mo, Y.K. Stable carbocations. CXXIX. Mechanism of the Benzidine and Wallach rearrangements based on direct observation of dicationic reaction intermediates and related model compounds. J. Am. Chem. Soc 1972, 94, 7438–7447. [Google Scholar]
- Moore, W.J. Physical Chemistry, 4th ed.; Prentice Hall: Englewood Cliffs, NJ, USA, 1972; p. 769. [Google Scholar]
- Jencks, W.P. When is an intermediate not an intermediate? Enforced mechanisms of general acid-base catalyzed, carbocation, carbanion, and ligand exchange reactions. Acc. Chem. Res 1980, 13, 161–169. [Google Scholar]
- Richard, J.P.; Amyes, T.L.; Toteva, M.M. Formation and stability of carbocations and carbanions in water and intrinsic barriers to their reactions. Acc. Chem. Res 2001, 34, 981–988, and references therein. [Google Scholar]
- Vorob’eva, E.N.; Kuznetsov, L.L.; Gidaspov, B.V. Kinetics of decomposition of primary aliphatic N-nitroamines in aqueous sulfuric acid. Zh. Org. Khim 1983, 19, 698–704. [Google Scholar]Russ. J. Org. Chem 1983, 19, 615–620.
- Dietze, P.E.; Jencks, W.P. Oxygen exchange into 2-butanol and hydration of 1-butene do not proceed through a common carbocation intermediate. J. Am. Chem. Soc 1987, 109, 2057–2062. [Google Scholar]
- Dietze, P.E.; Wojciechowski, M. Oxygen scrambling and stereochemistry during the trifluoroethanolysis of optically active 2-butyl 4-bromobenzenesulfonate. J. Am. Chem. Soc 1990, 112, 5240–5244. [Google Scholar]
- Bruice, P.Y. Organic Chemistry, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2001; pp. 380–381. [Google Scholar]
- Ingold, C.K. Structure and Mechanism in Organic Chemistry, 2nd ed.; Cornell University Press: Ithaca, NY, USA, 1969; p. 430. [Google Scholar]
- Murphy, T.J. Absence of SN1 involvement in the solvolysis of secondary alkyl compounds. J. Chem. Educ 2009, 86, 519–524. [Google Scholar]
- Bentley, T.W.; Schleyer, P.v.R. The SN2-SN1 spectrum. 1. Role of nucleophilic solvent assistance and nucleophilically solvated ion pair intermediates in solvolyses of primary and secondary arenesulfonates. J. Am. Chem. Soc 1976, 98, 7658–7666. [Google Scholar]
- Bunton, C.A.; Konasiewicz, A.; Llewellyn, D.R. Oxygen exchange and the Walden inversion in sec-butyl alcohol. J. Chem. Soc 1955, 604–607. [Google Scholar]
- Bunton, C.A.; Llewellyn, D. R. Tracer studies on alcohols. Part II. The exchange of oxygen-18 between sec-butyl alcohol and water. J. Chem. Soc 1957, 3402–3407. [Google Scholar]
- Mata-Segreda, J.F. Hydroxide as a general base in the saponification of ethyl acetate. J. Am. Chem. Soc 2002, 124, 2259–2262. [Google Scholar]
- Haeffner, F.; Hu, C.-H.; Brinck, T.; Norin, T. The catalytic effect of water in basic hydrolysis of methyl acetate: A theoretical study. J. Mol. Struct. (Theochem.) 1999, 459, 85–93. [Google Scholar]
- Hori, K.; Hashitani, Y.; Kaku, Y.; Ohkubo, K. Theoretical study on oxygen exchange accompanying alkaline hydrolysis of esters and amides. J. Mol. Struct. (Theochem.) 1999, 461–462, 589–596. [Google Scholar]
- Cox, R.A. Scarborough, ON, Canada, Unpublished work; 2011.
- Dolman, D.; Stewart, R. Strongly basic systems. VIII. The H− function for dimethyl sulfoxide-water-tetramethylammonium hydroxide. Can. J. Chem 1967, 45, 911–924. [Google Scholar]
- Eigen, M. Proton transfer, acid-base catalysis, and enzymatic hydrolysis. Part 1. elementary processes. Angew. Chem. Int. Ed. Engl 1964, 3, 1–19. [Google Scholar]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys 2000, 111, 1–217, and many earlier papers. [Google Scholar]
- Niedner-Schatteburg, G. Infrared spectroscopy and ab initio theory of isolated H5O2+: from buckets of water to the Schrödinger equation and back. Angew. Chem. Int. Ed. Engl 2008, 47, 1008–1011. [Google Scholar]
- Librovich, N.B.; Maiorov, V.D.; Savel’ev, V.A. The H5O2+ ion in the vibrational spectra of aqueous solutions of strong acids.
- Bascombe, K.N.; Bell, R.P. Properties of concentrated acid solutions. Discuss. Faraday Soc 1957, 24, 158–161. [Google Scholar]
- Robertson, E.B.; Dunford, H.B. The state of the proton in aqueous sulfuric acid. J. Am. Chem. Soc 1964, 86, 5080–5089. [Google Scholar]
- Ault, A. Telling it like it is: Teaching mechanisms in organic chemistry. J. Chem. Educ 2010, 87, 922–923. [Google Scholar]
- Silverstein, T.P. The solvated proton is NOT H3O+! J. Chem. Educ 2011, 88, 875. [Google Scholar]
- Roberts, S.T.; Ramasesha, K.; Petersen, P.B.; Mandal, A.; Tokmakoff, A. Proton transfer in concentrated aqueous hydroxide visualized using ultrafast infrared spectroscopy. J. Phys. Chem. A 2011, 115, 3957–3972. [Google Scholar]
- Marx, D.; Chandra, A.; Tuckerman, M.E. Aqueous basic solutions: hydroxide solvation, structural diffusion, and comparison to the hydrated proton. Chem. Rev 2010, 110, 2174–2216. [Google Scholar]
- Tuckerman, M.E.; Chandra, A.; Marx, D. Structure and dynamics of HOaq−. Acc. Chem. Res 2006, 39, 151–158. [Google Scholar]
- Stoyanov, E.S.; Stoyanova, I.V.; Reed, C.A. The structure of the hydrogen ion (Haq+) in water. J. Am. Chem. Soc 2010, 132, 1484–1486. [Google Scholar]
- Stoyanov, E.S.; Stoyanova, I.V.; Reed, C.A. The unique nature of H+ in water. Chem. Sci 2011, 2, 462–472. [Google Scholar]
- Jiang, J.-C.; Wang, Y.-S.; Chang, H.-C.; Lin, S.H.; Lee, Y.T.; Niedner-Schatteburg, G.; Chang, H.-C. Infrared spectra of H+(H2O)5–8 clusters: evidence for symmetric proton hydration. J. Am. Chem. Soc 2000, 122, 1398–1410. [Google Scholar]
- Stoyanov, E.S.; Reed, C.A. Private communication, Department of Chemistry, University of California: CA, USA, 2011.
- Shevkunov, S.V. Computer simulation of molecular complexes H3O+(H2O)n under conditions of thermal fluctuation. II. Work of formation and structure. Zh. Obshch. Khim 2004, 74, 1585–1592. [Google Scholar]Russ. J. Gen. Chem 2004, 74, 1471–1477.
- Grotthuss, C.J.T. Sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'électricité galvanique. Ann. Chim 1806, LVIII, 54–74. [Google Scholar]
- Marcus, Y. Effect of ions on the structure of water: structure making and breaking. Chem. Rev 2009, 109, 1346–1370, and references therein. [Google Scholar]
- Aqueous HCl is only usable up to about 38 wt%, when the water is saturated with gaseous HCl, and aqueous perchloric acid only up to 78 wt% or so, when the solution solidifies at 25 °C. Nitric acid has problems and is not normally used; it is considerably weaker, it is an oxidizing agent, as is strong perchloric acid, and it can give NO2+ and related species at higher concentrations. Aqueous HF is not often used; it is very weak at high dilution, and if concentrated it can dissolve glassware. Trifluoromethanesulfonic acid would probably be useful, but it is very expensive. Methanesulfonic acid is not used much. Trifluoroacetic and the other carboxylic acid variants are too weak to be useful.
- Cox, R.A. Mechanistic studies in strong acids. I. General considerations. Catalysis by individual acid species in sulfuric acid. J. Am. Chem. Soc 1974, 96, 1059–1063. [Google Scholar]
- Cox, R.A.; Fung, D.Y.K.; Csizmadia, I.G.; Buncel, E. An ab initio molecular orbital study of the geometry of the dicationic Wallach rearrangement intermediate. Can. J. Chem 2003, 81, 535–541. [Google Scholar]
- Buncel, E.; Keum, S.-R.; Rajagopal, S.; Cox, R.A. Rearrangement mechanisms for azoxypyridines and axoxypyridine N-oxides in the 100% H2SO4 region—the Wallach rearrangement story comes full circle. Can. J. Chem 2009, 87, 1127–1134. [Google Scholar]
- Cox, R.A.; Buncel, E. Rearrangements of Hydrazo, Azoxy and Azo CompoundsThe Chemistry of the Hydrazo, Azo and Azoxy Groups; Patai, S., Ed.; Wiley: London, UK, 1975; Volume 1, pp. 775–859. [Google Scholar]
- Cox, R.A.; Buncel, E. Rearrangements of Hydrazo, Azoxy and Azo Compounds: Kinetic, Product and Isotope StudiesThe Chemistry of the Hydrazo, Azo and Azoxy Groups; Patai, S., Ed.; Wiley: London, UK, 1997; Volume 2, pp. 569–602. [Google Scholar]
- Cox, R.A.; Buncel, E.; Bolduc, R. Department of Chemistry, Queen’s University: Kingston, Canada, Unpublished observations; 1971.
- Cox, R.A.; Yates, K. Mechanistic studies in strong acids. VIII. Hydrolysis mechanisms for some thiobenzoic acids and esters in aqueous sulfuric acid, determined using the excess acidity method. Can. J. Chem 1982, 60, 3061–3070. [Google Scholar]
- Cox, R.A. Excess acidities. Adv. Phys. Org. Chem 2000, 35, 1–66. [Google Scholar]
- Bell, R.P.; Bascombe, K.N.; McCoubrey, J.C. Kinetics of the depolymerization of trioxane in aqueous acids, and the acidic properties of aqueous hydrogen fluoride. J. Chem. Soc 1956, 1286–1291. [Google Scholar]
- Giauque, W.F.; Hornung, E.W.; Kunzler, J.E.; Rubin, T.R. The thermodynamic properties of aqueous sulfuric acid solutions and hydrates from 15 to 300 K. J. Am. Chem. Soc 1960, 82, 62–70. [Google Scholar]
- Zeleznik, F.J. Thermodynamic properties of the aqueous sulfuric acid system to 350 K. J. Phys. Chem. Ref. Data 1991, 20, 1157–1200. [Google Scholar]
- Randall, M.; Young, L.E. The calomel and silver chloride electrodes in acid and neutral solutions. The activity coefficient of aqueous hydrochloric acid and the single potential of the deci-molal calomel electrode. J. Am. Chem. Soc 1928, 50, 989–1004. [Google Scholar]
- Åkerlöf, G.; Teare, J.W. Thermodynamics of concentrated aqueous solutions of hydrochloric acid. J. Am. Chem. Soc 1937, 59, 1855–1868. [Google Scholar]
- Liu, Y.; Grén, U.; Theliander, H.; Rasmuson, A. Simultaneous correlation of activity coefficient and partial thermal properties for electrolyte solutions using a model with ion-specific parameters. Fluid Phase Equilibria 1993, 83, 243–251. [Google Scholar]
- Pearce, J.N.; Nelson, A.F. The vapor pressures and activity coefficients of aqueous solutions of perchloric acid at 25°. J. Am. Chem. Soc 1933, 55, 3075–3081. [Google Scholar]
- Robinson, R.A.; Baker, O.J. The vapor pressures of perchloric acid solutions at 25°. Trans. Proc. R. Soc. N. Z 1946, 76, 250–254. [Google Scholar]
- Wai, H.; Yates, K. Determination of the activity of water in highly concentrated perchloric acid solutions. Can. J. Chem 1969, 47, 2326–2328. [Google Scholar]
- Bidinosti, D.R.; Biermann, W.J. A redetermination of the relative enthalpies of aqueous perchloric acid solutions from 1 to 24 molal. Can. J. Chem 1956, 34, 1591–1595. [Google Scholar]
- Bell, R.P.; Brown, A.H. Kinetics of the depolymerization of paraldehyde in aqueous solution. J. Chem. Soc 1954, 774–778. [Google Scholar]
- Hamer, D.; Leslie, J. The Hammett acidity function in reactions catalyzed by carboxylic acids. The hydrolysis of methylal and the depolymerization of trioxane. J. Chem. Soc 1960, 4198–4202. [Google Scholar]
- Jaques, D.; Leisten, J.A. Acid-catalysed ether fission. Part II. Diethyl ether in aqueous acids. J. Chem. Soc 1964, 2683–2689. [Google Scholar]
- Ruiz Pernía, J.J.; Tuñón, I.; Williams, I.H. Computational simulation of the lifetime of methoxymethyl cation in water. A simple model for a glycosyl cation: When is an intermediate an intermediate? J. Phys. Chem. B 2010, 114, 5769–5774. [Google Scholar]
- Cox, R.A. Benzamide hydrolysis in strong acids—the last word. Can. J. Chem 2008, 86, 290–297. [Google Scholar]
- Cox, R.A. A comparison of the mechanism of hydrolysis of benzimidates, esters, and amides in sulfuric acid media. Can. J. Chem 2005, 83, 1391–1399. [Google Scholar]
- Bender, M.L. Oxygen exchange as evidence for the existence of an intermediate in ester hydrolysis. J. Am. Chem. Soc 1951, 73, 1626–1629. [Google Scholar]
- McClelland, R.A. Benzamide oxygen exchange concurrent with acid hydrolysis. J. Am. Chem. Soc 1975, 97, 5281. [Google Scholar]
- Yates, K.; McClelland, R.A. Mechanisms of ester hydrolysis in aqueous sulfuric acids. J. Am. Chem. Soc 1967, 89, 2686–2692. [Google Scholar]
- Yates, K. Kinetics of ester hydrolysis in concentrated acid. Acc. Chem. Res 1971, 4, 136–144. [Google Scholar]
- Marlier, J.F. Heavy-atom isotope effects on the alkaline hydrolysis of methyl formate. The role of hydroxide ion in ester hydrolysis. J. Am. Chem. Soc 1993, 115, 5953–5956. [Google Scholar]
- Bender, M.L.; Ginger, R.D.; Unik, J.P. Activation energies of the hydrolysis of esters and amides involving carbonyl oxygen exchange. J. Am. Chem. Soc 1958, 80, 1044–1048. [Google Scholar]
- Shain, S.A.; Kirsch, J.F. Absence of carbonyl oxygen exchange concurrent with the alkaline hydrolysis of substituted methyl benzoates. J. Am. Chem. Soc 1968, 90, 5848–5854. [Google Scholar]
- Cox, R.A. The mechanism of the hydrolysis of acylimidazoles in aqueous mineral acids. The excess acidity method for reactions that are not acid catalyzed. Can. J. Chem 1997, 75, 1093–1098. [Google Scholar]
- Cox, R.A. The acid catalyzed decomposition of nitramide. Can. J. Chem 1996, 74, 1779–1783. [Google Scholar]
- Eckert-Maksic, M.; Maskill, H.; Zrinski, I. Acidic and basic properties of nitramide, and the catalyzed decomposition of nitramide and related compounds; an ab initio theoretical investigation. J. Chem. Soc. Perkin Trans 2001, 2, 2147–2154. [Google Scholar]
- Bentley, T.W.; Harris, H.C. Solvolyses of para-substituted benzoyl chlorides in trifluoroethanol and in highly aqueous media. J. Chem. Soc. Perkin Trans 1986, 2, 619–624. [Google Scholar]
- Williams, A. Concerted mechanisms of acyl group transfer reactions in solution. Acc. Chem. Res 1989, 22, 387–392. [Google Scholar]
- Bentley, T.W.; Ebdon, D.N.; Kim, E.-J.; Koo, I.S. Solvent polarity and organic reactivity in mixed solvents: Evidence using a reactive molecular probe to assess the role of preferential solvation in aqueous alcohols. J. Org. Chem 2005, 70, 1647–1653. [Google Scholar]
- Ruff, F.; Farkas, Ö. Concerted SN2 mechanism for the hydrolysis of acid chlorides: comparisons of reactivities calculated by the density functional theory with experimental data. J. Phys. Org. Chem. 2011, 24, 480–491. [Google Scholar]
- Ji, P.; Atherton, J.; Page, M.I. Liquid ammonia as a dipolar aprotic solvent for aliphatic nucleophilic substitution reactions. J. Org. Chem 2011, 76, 1425–1435. [Google Scholar]
- Ji, P.; Atherton, J.H.; Page, M.I. The kinetics and mechanisms of aromatic nuclear substitution reactions in liquid ammonia. J. Org. Chem 2011, 76, 3286–3295. [Google Scholar]







© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cox, R.A. A Greatly Under-Appreciated Fundamental Principle of Physical Organic Chemistry. Int. J. Mol. Sci. 2011, 12, 8316-8332. https://doi.org/10.3390/ijms12128316
Cox RA. A Greatly Under-Appreciated Fundamental Principle of Physical Organic Chemistry. International Journal of Molecular Sciences. 2011; 12(12):8316-8332. https://doi.org/10.3390/ijms12128316
Chicago/Turabian StyleCox, Robin A. 2011. "A Greatly Under-Appreciated Fundamental Principle of Physical Organic Chemistry" International Journal of Molecular Sciences 12, no. 12: 8316-8332. https://doi.org/10.3390/ijms12128316
APA StyleCox, R. A. (2011). A Greatly Under-Appreciated Fundamental Principle of Physical Organic Chemistry. International Journal of Molecular Sciences, 12(12), 8316-8332. https://doi.org/10.3390/ijms12128316