Chromatin Structure Following UV-Induced DNA Damage—Repair or Death?

Abstract
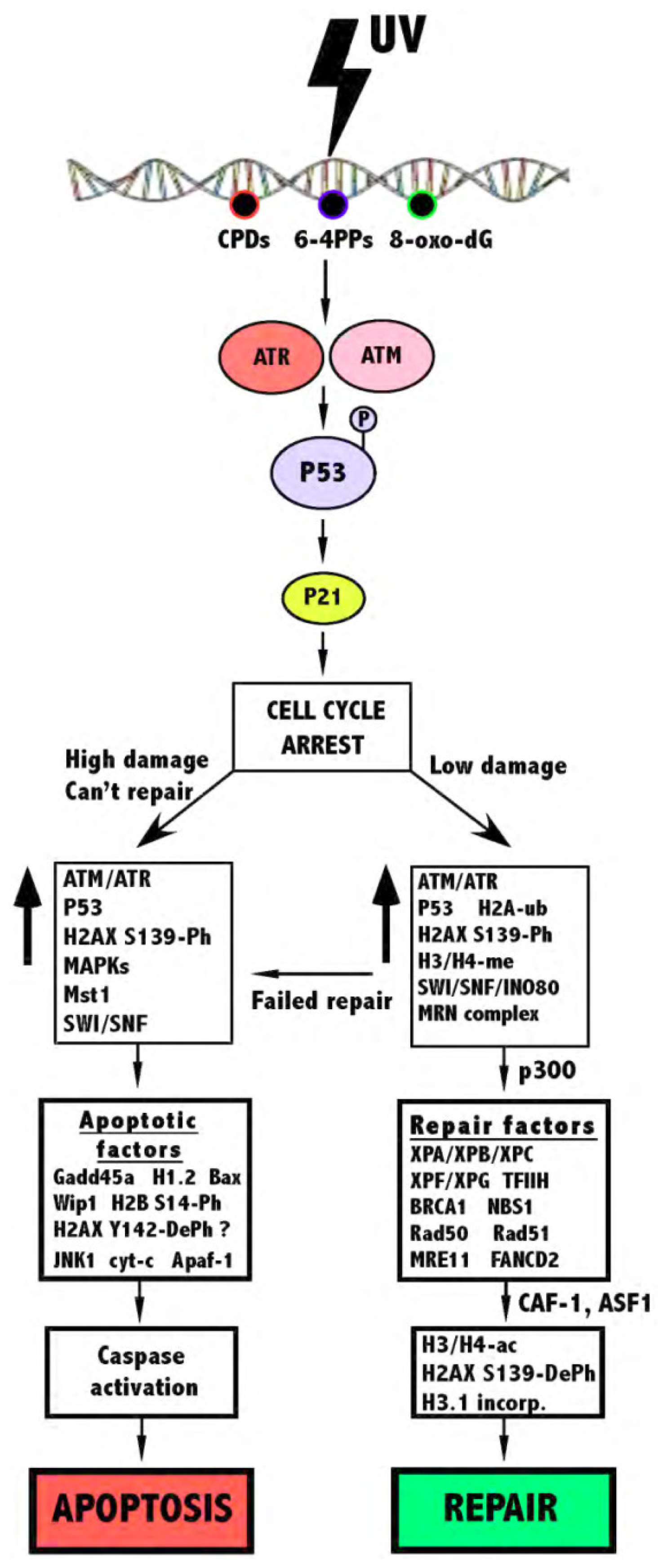
:1. Chromatin
2. UV Damage and the Cellular Response
2.1. DNA Repair
2.2. Apoptosis
2.3. Structural Changes of Chromatin Following UV
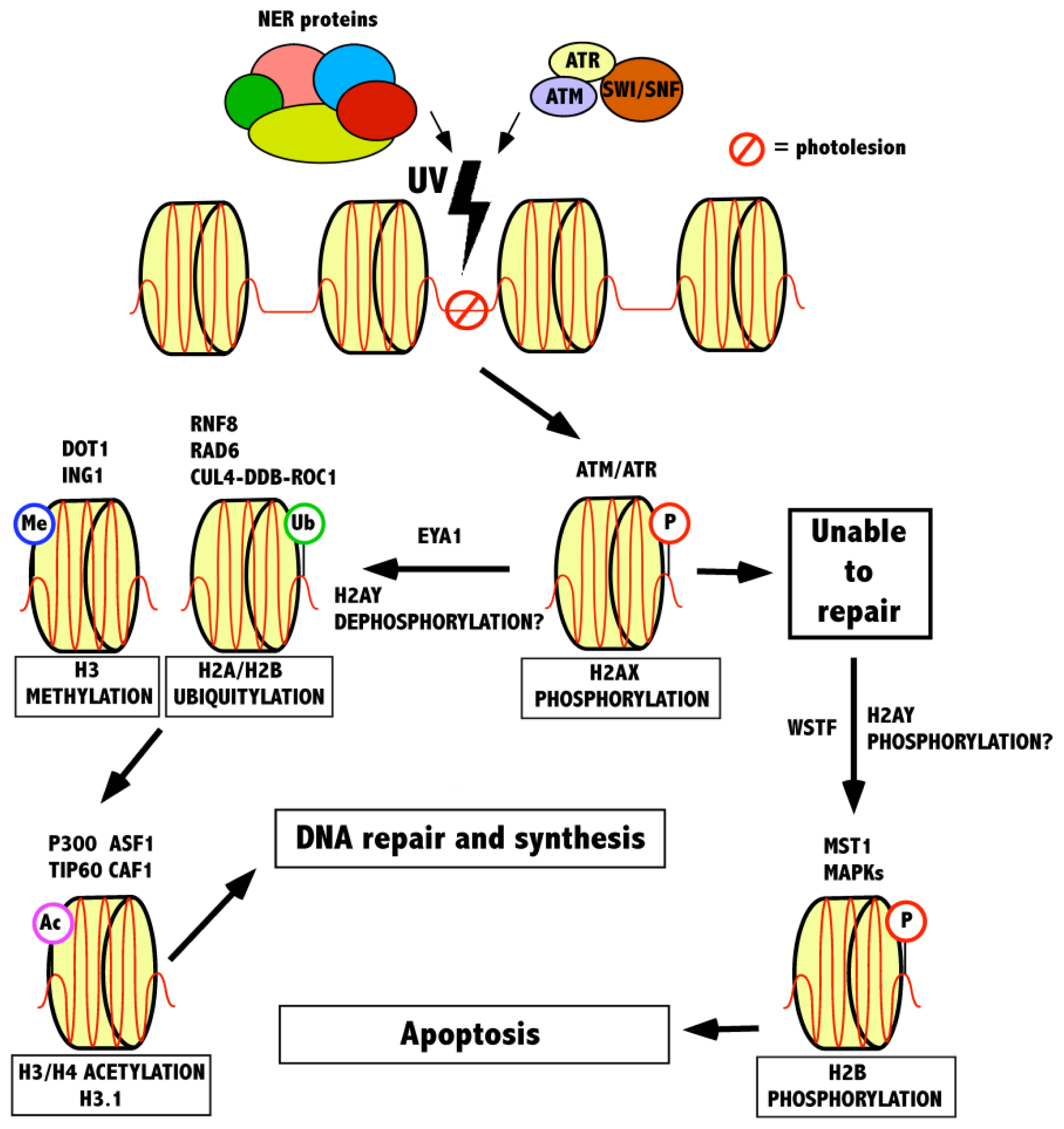
3. The Role of Histone Modifications and Chromatin Remodeling Complexes in Response to UV-Induced DNA Damage
3.1. Recognition of a Lesion by γ-H2AX and the Role of Chromatin Remodeling Complexes
3.2. Modifications Following γ-H2AX in the UV-Induced DNA Damage Response
3.3. Maintenance of Chromatin Following Repair—Histone Acetylation
4. Conclusions
Acknowledgements
- Conflicts of InterestThe authors declare no conflict of interest.
Abbreviations
| 6-4PP | pyrimidine 6-4 pyrimidone photoproduct |
| 8-oxo-dG | 8-oxodeoxyguanine |
| γ-H2AX | phosphorylated histone H2AX |
| APAF-1 | apoptotic protease activating factor-1 |
| ASF1 | anti-silencing function-1 |
| ATM | ataxia-telangiectasia-mutated |
| ATP | adenosine 5′-triphosphate |
| ATR | ataxia-telangiectasia Rad3-related |
| BER | base excision repair |
| CAF1 | chromatin assembly factor-1 |
| CK2 | casein kinase 2 |
| CPD | cyclobutane pyrimidine dimer |
| CSB | Cockayne syndrome B |
| CYT-C | cytochrome-c |
| DNA-PK | DNA-damage-dependent protein kinase |
| DSB | double strand break |
| FADD | Fas-associated death domain protein |
| GADD45a | growth arrest and DNA-damage-inducible alpha |
| GGR | global genome repair |
| HAT | histone acetyltransferase |
| HDAC | histone deacetylase |
| hOGG1 | human 8-oxoguanine DNA glycosylase-1 |
| INO | inositol |
| ISWI | imitation of switch |
| MDC1 | mediator of DNA-damage checkpoint 1 |
| MDM2 | P53 binding protein homolog |
| MST1 | mammalian sterile twenty kinase-1 |
| NER | nucleotide excision repair |
| PARP | poly(ADP ribose) polymerase |
| PcG | Polycomb |
| PCNA | proliferating cell nuclear antigen |
| PIKK | phosphatidylinositol-3 kinase-like kinase |
| RF | replication factor |
| RP | replication protein |
| SWI/SNF | switch/sucrose |
| TCR | transcription coupled repair |
| TFIIH | transcription factor IIH |
| TNF | tumour necrosis factor |
| TRAIL | tumour necrosis factorrelated apoptosis-inducing ligand |
| TSA | Trischostatin A |
| UV | ultraviolet |
| WIP1 | wild-type P53-induced phosphatase-1 |
| WSTF | Williams-Beuren syndrome transcription factor |
| XP | Xeroderma pigmentosum complex. |
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar]
- Horáková, A.H.; Bártova, E.; Kozubek, S. Chromatin structure with respect to histone signature changes during cell differentiation. Cell Struct. Funct 2010, 35, 31–44. [Google Scholar]
- Verzijlbergen, K.F.; Faber, A.W.; Stulemeijer, I.J.; van Leeuwen, F. Multiple histone modifications in euchromatin promote heterochromatin formation by redundant mechanisms in Saccharomyces cerevisiae. BMC Mol. Biol 2009, 10, 76–91. [Google Scholar]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol 2005, 12, 46–53. [Google Scholar]
- Chakravarthy, S.; Park, Y.J.; Chodaparambil, J.; Edaythumangalam, R.S.; Luger, K. Structure and dynamic properties of nucleosome core particles. FEBS Lett 2005, 579, 895–898. [Google Scholar]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar]
- Matsunaga, T.; Hieda, K.; Nikaido, O. Wavelength dependent formation of thymidine dimers and (6-4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm. Photochem. Photobiol 1991, 54, 403–410. [Google Scholar]
- Rochette, P.J.; Therrein, J.P.; Drouin, R.; Perdiz, D.; Bastien, N.; Drobetsky, E.A.; Sage, E. UVA-induced cyclobutane pyrimidine dimers form predominantly at thymidine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells. Nucleic Acids Res 2003, 31, 2786–2794. [Google Scholar]
- Mouret, S.; Baudion, C.; Charvernon, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad Sci. USA 2006, 103, 13765–13770. [Google Scholar]
- Dahle, J.; Kvam, E. Induction of delayed mutations and chromosomal instability in fibroblasts after UVA-, UVB-, and X-radiation. Cancer Res 2003, 63, 1464–1469. [Google Scholar]
- Riedl, T.; Hanaoka, F.; Egly, J.M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003, 22, 5293–5303. [Google Scholar]
- Decraene, D.; Agostinis, P.; Pupe, A.; de Heas, P.; Garmyn, M. Acute response of human skin to solar radiation: regulation and function of the p53 protein. J. Photochem. Photobiol. B 2001, 63, 78–83. [Google Scholar]
- Hanawalt, P.C. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002, 21, 8949–8956. [Google Scholar]
- de Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar]
- Palomera-Sanchez, Z.; Zurita, M. Open, repair and close again: Chromatin dynamics and the response to UV-induced DNA damage. DNA Repair (Amst) 2011, 10, 119–125. [Google Scholar]
- Henning, K.A.; Li, L.; Iyer, N.; McDaniel, L.D.; Reagan, M.S.; Legerski, R.; Schultz, R.A.; Stefanini, M.; Lehmann, A.R.; Mayne, L.V.; et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 1995, 82, 555–564. [Google Scholar]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar]
- Schaeffer, L.; Roy, R.; Moncollin, V.; Vermeulen, W.; Hoeijmakers, J.H.; Chambon, P.; Egly, J.M. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science 1993, 260, 58–63. [Google Scholar]
- Evans, E.; Moggs, J.G.; Hwang, J.R.; Egly, J.M.; Wood, R.D. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J 1997, 16, 6559–6573. [Google Scholar]
- O’Donovan, A.; Davies, A.A.; Moggs, J.G.; West, S.C.; Wood, R.D. XPG endonuclease makes the 3′ incision in human nucleotide excision repair. Nature 1994, 371, 432–435. [Google Scholar]
- Matsunaga, T.; Mu, D.; Park, C.H.; Reardon, J.T.; Sancar, A. Human DNA repair excision nuclease. Analysis of the roles of the subunits involved in dual incisions by using anti-XPG and anti-ERCC1 antibodies. J. Biol. Chem 1995, 270, 20862–20869. [Google Scholar]
- Llu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J. Biol. Chem 2007, 282, 13532–13541. [Google Scholar]
- Javeri, A.; Lyons, J.G.; Huang, X.X.; Halliday, G.M. Downregulation of Cockayne syndrome B protein reduces human 8-oxoguanine DNA glycosylase-1 expression and repair of UV radiation-induced 8-oxo-7,8-dihydro-2′-deoxyguanine. Cancer Sci 2011, 102. [Google Scholar] [CrossRef]
- Maher, V.M.; Dorney, D.J.; Mendrala, A.L.; Konze-Thomas, B.; McCormick, J.J. DNA excision repair processes in human cells can eliminate the cytotoxic and mutagenic consequences of ultraviolet irradiation. Mutat. Res 1979, 62, 311–323. [Google Scholar]
- Konze-Thomas, B.; Hazard, R.M.; Maher, V.M.; McCormick, J.J. Extent of excision repair before DNA synthesis determines the mutagenic but not the lethal effect of UV radiation. Mutat. Res 1982, 94, 421–434. [Google Scholar]
- Smith, M.L.; Fornace, A.J.J. P53-mediated protective responses to UV irradiation. Proc. Natl. Acad. Sci. USA 1997, 94, 12255–12257. [Google Scholar]
- Dunkern, T.R.; Fritz, G.; Kaina, B. Ultraviolet light-induced DNA damage triggers apoptosis in nucleotide excision repair-deficient cells via Bcl-2 decline and caspase-3/-8 activation. Oncogene 2001, 20, 6026–6038. [Google Scholar]
- Lehmann, A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85, 1101–1111. [Google Scholar]
- Chigancas, V.; Miyaji, E.N.; Muotri, A.R.; de Fatima Jacysyn, J.; Amarante-Mendes, G.P.; Yasui, A.; Menck, C.F. Photorepair prevents ultraviolet-induced apoptosis in human cells expressing the marsupial photolyase gene. Cancer Res 2000, 60, 2458–2463. [Google Scholar]
- Jans, J.; Schul, W.; Sert, Y.G.; Rijksen, Y.; Rebel, H.; Eker, A.P.; Nakajima, S.; van Steeg, H.; de Gruijl, F.R.; Yasui, A.; et al. Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol 2005, 15, 105–115. [Google Scholar]
- Kaina, B. DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem. Pharmacol 2003, 66, 1547–1554. [Google Scholar]
- Yuan, J. Evolutionary conservation of a genetic pathway of programmed cell death. J. Cell Biochem 1996, 60, 4–11. [Google Scholar]
- Vermuelen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: mechanisms and relevance in cancer. Ann. Hematol 2005, 84, 627–639. [Google Scholar]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther 2005, 4, 139–163. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Kulms, D.; Schwarz, T. Molecular mechanisms of UV-induced apoptosis. Photodermatol. Photoimmunol. Photomed 2000, 16, 195–201. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffel, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar]
- Chaturvedi, V.; Sitailo, L.A.; Qin, J.Z. Knockdown of p53 levels in human keratinocytes accelerates Mcl-1 and Bcl-x(L) reduction thereby enhancing UV-light induced apoptosis. Oncogene 2005, 24, 5299–5312. [Google Scholar]
- Hildesheim, J.; Bulavin, D.V.; Anver, M.R.; Alvord, W.G.; Hollander, M.C.; Vardanian, L.; Fornace, A.J. Gadd45a protects against UV irradiation-induced skin tumors, and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 2002, 62, 7305–7315. [Google Scholar]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma—a role for antiapoptotic signalling pathways. Br. J. Dermatol 2009, 161, 107–115. [Google Scholar]
- Kulms, D.; Pöppelmann, B.; Yarosh, D.; Luger, T.A.; Krutmann, J.; Schwarz, T. Nuclear and cell membrane effects contribute independently to the induction of apoptosis in human cells exposed to UVB radiation. Proc. Natl. Acad. Sci. USA 1999, 96, 7974–7979. [Google Scholar]
- Higuchi, Y.; Mizukami, Y.; Yoshimoto, T. Ultraviolet ray induces chromosomal giant DNA fragmentation followed by internucleosomal DNA fragmentation associated with apoptosis in rat glioma cells. Ann. NY Acad. Sci 2003, 1010, 326–330. [Google Scholar]
- Ujvarosi, K.; Hunyadi, J.; Nagy, G.; Pocsi, I.; Banfalvi, G. Preapoptotic chromatin changes induced by ultraviolet B irradiation in human erythroleukemia K562 cells. Apoptosis 2007, 12, 2089–2099. [Google Scholar]
- Nagy, G.; Gacsi, M.; Rehak, M.; Basnakian, A.G.; Klaisz, M.; Banfalvi, G. Gamma irradiation-induced apoptosis in murine pre-B cells prevents the condensation of fibrillar chromatin in early S phase. Apoptosis 2004, 9, 765–776. [Google Scholar]
- Zierler, S.; Klein, B.; Furtner, T.; Bresgen, N.; Lütz-Meindl, U.; Kerschbaum, H.H. Ultraviolet irradiation-induced apoptosis does not trigger nuclear fragmentation but translocation of chromatin from nucleus into cytoplasm in the microglial cell-line, BV-2. Brain Res 2006, 1121, 12–21. [Google Scholar]
- Saitoh, Y.; Miyanishi, A.; Mizuno, H.; Kato, S.; Aoshima, H.; Kokubo, K.; Miwa, N. Super-highly hydroxylated fullerene derivative protects human keratinocytes from UV-induced cell injuries together with the decreases in intracellular ROS generation and DNA damages. J. Photochem. Photobiol 2011, 102, 69–76. [Google Scholar]
- Vaquero, A.; Loyola, A.; Reinberg, D. The constantly changing face of chromatin. Sci. Aging Knowledge Environ 2003, 2003, RE4. [Google Scholar]
- Martelli, A.M.; Zweyer, M.; Ochs, R.L.; Tazzari, P.L.; Tabellini, G.; Narducci, P.; Bortul, R. Nuclear apoptotic changes: an overview. J. Cell Biochem 2001, 82, 634–646. [Google Scholar]
- Imbalzano, A.N.; Xiao, H. Functional properties of ATP-dependent chromatin remodeling enzymes. Adv. Protein Chem 2004, 67, 157–179. [Google Scholar]
- Altaf, M.; Saksouk, N.; Côté, J. Histone modifications in response to DNA damage. Mutat. Res 2007, 618, 81–90. [Google Scholar]
- Ward, I.M.; Chen, J. Histone H2AX is phoshorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem 2001, 276, 47759–47762. [Google Scholar]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 2004, 64, 2390–2396. [Google Scholar]
- Solier, S.; Sordet, O.; Kohn, K.W.; Pommier, Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell Biol 2009, 29, 68–82. [Google Scholar]
- Garinis, G.A.; Mitchell, J.R.; Moorhouse, M.J.; Hanada, K.; de Waard, H.; Vandeputte, D.; Jans, J.; Brand, K.; Smid, M.; van der Spek, P.J.; et al. Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J 2005, 24, 3952–3962. [Google Scholar]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar]
- Halicka, H.D.; Huang, X.; Traganos, F.; King, M.A.; Dai, W.; Darzynkiewicz, Z. Histone H2AX phosphorylation after cell irradiation with UV-B: relationship to cell cycle phase and induction of apoptosis. Cell Cycle 2005, 4, 339–345. [Google Scholar]
- Han, W.; Ming, M.; He, Y.Y. Caffeine Promotes Ultraviolet B-induced Apoptosis in Human Keratinocytes without Complete DNA Repair. J. Biol. Chem 2011, 286, 22825–22832. [Google Scholar]
- Kim, J.E.; Minter-Dykhouse, K.; Chen, J. Signaling networks controlled by the MRN complex and MDC1 during early DNA damage responses. Mol. Carcinog 2006, 45, 403–408. [Google Scholar]
- Oh, K.S.; Bustin, M.; Mazur, S.J.; Appella, E.; Kraemer, K.H. UV-induced histone H2AX phosphorylation and DNA damage related proteins accumulate and persist in nucleotide excision repair-deficient XP-B cells. DNA Repair (Amst) 2011, 10, 5–15. [Google Scholar]
- Kim, H.; Chen, J. New players in the BRCA1 mediated DNA damage responsive pathway. Mol. Cells 2008, 25, 457–461. [Google Scholar]
- Chai, B.; Huang, J.; Cairns, B.R.; Laurent, B.C. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 2005, 19, 1656–1661. [Google Scholar]
- Gong, F.; Fahy, D.; Liu, H.; Wang, W.; Smerdon, M.J. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 2008, 7, 1067–1074. [Google Scholar]
- Park, J.H.; Park, E.J.; Lee, H.S.; Kim, S.J.; Hur, S.K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 2006, 25, 3986–3997. [Google Scholar]
- Lee, H.S.; Park, J.H.; Kim, S.J.; Kwon, S.J.; Kwon, J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J 2010, 29, 1434–1445. [Google Scholar]
- Cheung, W.L.; Ajiro, K.; Samejima, K.; Kloc, M.; Cheung, P.; Mizzen, C.A.; Beeser, A.; Etkin, L.D.; Chernoff, J.; Earnshaw, W.C.; et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003, 113, 507–517. [Google Scholar]
- Hu, Y.; Liu, Z.; Yang, S.J.; Ye, K. Acinus-provoked protein kinase C delta isoform activation is essential for apoptotic chromatin condensation. Cell Death Differ 2007, 14, 2035–2046. [Google Scholar]
- Lu, C.; Shi, Y.; Luo, Y.; Duan, L.; Hou, Y.; Hu, H.; Wang, Z.; Xiang, P. MAPKs and Mst1/Caspase-3 pathways contribute to H2B phosphorylation during UVB-induced apoptosis. Sci. China Life Sci 2010, 53, 663–668. [Google Scholar]
- Recht, J.; Osley, M.A. Mutations in both the structured domain and N-terminus of histone H2B bypass the requirement for SWI/SNF in yeast. EMBO J 1999, 18, 229–240. [Google Scholar]
- Parra, M.A.; Kerr, D.; Fahy, D.; Pouchnik, D.J.; Wyrick, J.J. Deciphering the roles of histone H2B N-terminal domain in genome-wide transcription. Mol. Cell Biol 2006, 26, 3842–3852. [Google Scholar]
- Nag, R.; Kyriss, M.; Smerdon, J.W.; Wyrick, J.J.; Smerdon, M.J. A cassette of N-terminal amino acids of histone H2B are required for efficient cell survival, DNA repair and SWI/SNF binding in UV irradiated yeast. Nucleic Acids Res 2009, 38, 1450–1460. [Google Scholar]
- Kratzmeier, M.; Albig, W.; Hanecke, K.; Doenecke, D. Rapid dephosphorylation of H1 histones after apoptosis induction. J. Biol. Chem 2000, 275, 30478–30486. [Google Scholar]
- Goebel, W.; Obermeyer, N.; Bleicher, N.; Kratzmeier, M.; Eibl, H.J.; Doenecke, D.; Albig, W. Apoptotic DNA fragmentation is not related to the phosphorylation state of histone H1. Biol. Chem 2007, 388, 197–206. [Google Scholar]
- Ruiz-Vela, A.; Korsmeyer, S.J. Proapoptotic histone H1.2 induces CASP-3 and -7 activation by forming a protein complex with CYT c, APAF-1 and CASP-9. FEBS Lett 2007, 581, 3422–3428. [Google Scholar]
- Konishi, A.; Shimizu, S.; Hirota, J.; Takao, T.; Fan, Y.; Matsuoka, Y.; Zhang, L.; Yoneda, Y.; Fujii, Y.; Skoultchi, A.I.; et al. Involvement of histone 1.2 in apoptosis induced by DNA double-strand breaks. Cell 2003, 114, 673–688. [Google Scholar]
- Cheung, W.L.; Turner, F.B.; Krishnamoorthy, T.; Wolner, B.; Ahn, S.H.; Foley, M.; Dorsey, J.A.; Peterson, C.L.; Berger, S.L.; Allis, C.D. Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr. Biol 2005, 15, 656–660. [Google Scholar]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 2002, 12, 1052–1058. [Google Scholar]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar]
- Toh, G.W.; O’Shaughnessy, A.M.; Jimeno, S.; Dobbie, I.M.; Grenon, M.; Maffini, S.; O’Rorke, A.; Lowndes, N.F. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 2006, 5, 693–703. [Google Scholar]
- Bostelman, L.J.; Keller, A.M.; Albrecht, A.M.; Arat, A.; Thompson, J.S. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair (Amst) 2007, 6, 383–395. [Google Scholar]
- Evans, M.L.; Bostelman, L.J.; Albrecht, A.M.; Keller, A.M.; Strande, N.T.; Thompson, J.S. UV sensitive mutations in histone H3 in Saccharomyces cerevisiae that alter specific K79 methylation states genetically act through distinct DNA repair pathways. Curr. Genet 2008, 53, 259–274. [Google Scholar]
- Chaudhuri, S.; Wyrick, J.J.; Smerdon, J.W. Histone H3 Lys79 methylation is required for efficient nucleotide excision repair in a silenced locus of Saccharomyces cerevisiae. Nucleic Acids Res 2009, 37, 1690–1700. [Google Scholar]
- Peña, P.V.; Hom, R.A.; Hung, T.; Lin, H.; Kuo, A.J.; Wong, R.P.; Subach, O.M.; Champagne, K.S.; Zhao, R.; Verkhusha, V.V.; et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J. Mol. Biol 2008, 380, 303–312. [Google Scholar]
- Palomera-Sanchez, Z.; Bucio-Mendez, A.; Valadez-Graham, A.V.; Reynaud, E.; Zurita, M. Drosophila p53 is required to increase levels of the Dkdm4B demethylase after UV induced DNA damage to demethylate histone H3 lysine 9. J. Biol. Chem 2010, 285, 31370–31379. [Google Scholar]
- Magnaghi-Jaulin, L.; Groisman, R.; Naguibneva, I.; Robin, P.; Lorain, S.; Le Villain, J.P.; Troalen, F.; Trouche, D.; Harel-Bellan, A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 1998, 391, 601–605. [Google Scholar]
- Kim, M.; Shin, J.; Eun, H.C.; Chung, J.H. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS One 2009, 4, e4864. [Google Scholar]
- Rebollar, E.; Valadez-Graham, V.; Vászquez, M.; Reynaud, E.; Zurita, M. Role of the p53 homologue from Drosophila melanogaster in the maintenance of hsitone H3 acetylation and response to UV-light irradiation. FEBS Lett 2006, 580, 642–648. [Google Scholar]
- Recht, J.; Tsubota, T.; Tanny, J.C.; Diaz, R.L.; Berger, J.M.; Zhang, X.; Garcia, B.A.; Shabanowitz, J.; Burlingame, A.L.; Hunt, D.F.; et al. Histone acetylation Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. USA 2006, 103, 6988–6993. [Google Scholar]
- Battu, A.; Ray, A.; Wani, A.A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 2011, 39. [Google Scholar] [CrossRef]
- Allison, S.J.; Milner, J. Loss of p53 has site-specific effects on histone H3 modification, including serine 10 phosphorylation important for the maintenance of ploidy. Cancer Res 2003, 63, 6674–6679. [Google Scholar]
- Koprinarova, M.; Russev, G. Histone H4 acetylation during UV light induced G1 and S phase arrest of the cell cycle. Cell Cycle 2008, 7, 1–3. [Google Scholar]
- Bolx-Chornet, M.; Fraga, M.F.; Villar-Garea, A.; Caballero, R.; Espada, J.; Nuñez, A.; Casado, J.; Largo, C.; Casal, J.I.; Cigudosa, J.C.; et al. Release of hypoacetylated and trimethylated histone H4 is an epigenetic marker of early apoptosis. J. Biol. Chem 2006, 281, 13540–13547. [Google Scholar]
- Bergink, S.; Salomons, F.A.; Hoogstraten, D.; Groothuis, T.A.; de Waard, H.; Wu, J.; Yuan, L.; Citterio, E.; Houtsmuller, A.B.; Neefjes, J.; et al. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev 2006, 20, 1343–1352. [Google Scholar]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermuelen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol 2009, 186, 835–847. [Google Scholar]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol 2010, 191, 45–60. [Google Scholar]
- Robzyk, K.; Recht, J.; Osley, M.A. Rad6-dependent ubiquitination of histone H2B in yeast. Science 2000, 287, 501–504. [Google Scholar]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar]
- Polo, S.E.; Roche, D.; Almouzni, G. New histone incorporation marks site of UV repair in human cells. Cell 2006, 127, 481–493. [Google Scholar]
- Park, J.H.; Park, E.J.; Hur, S.K.; Kim, S.J.; Kwon, J. Mammalian SWI/SNF chromatin remodeling complexes are required to prevent apoptosis after DNA damage. DNA Repair (Amst) 2009, 8, 29–39. [Google Scholar]
- Bock, V.L.; Lyons, J.G.; Huang, X.X.; Jones, A.M.; McDonald, L.A.; Scolyer, R.A.; Moloney, F.J.; Barnetson, R.S.; Halliday, G.M. BRM and BRG1 subunits of the SWI/SNF chromatin remodelling complex are downregulated upon progression of benign skin lesions into invasive tumours. Br. J. Dermatol 2011, 164. [Google Scholar] [CrossRef]
- Moloney, F.J.; Lyons, J.G.; Bock, V.L.; Huang, X.X.; Bugeja, M.J.; Halliday, G.M. Hotspot mutation of brahma in non-melanoma skin cancer. J. Invest. Dermatol 2009, 129, 1012–1015. [Google Scholar]
- Reyes, J.C.; Barra, J.; Muchardt, C.; Camus, A.; Babinet, C.; Yaniv, M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α). EMBO J 1998, 17, 6979–6991. [Google Scholar]
- Biggs, J.R.; Yang, J.; Gullberg, U.; Muchardt, C.; Yaniv, M.; Kraft, A.S. The human brm protein is cleaved during apoptosis: The role of cathepsin G. Proc. Natl. Acad. Sci. USA 2001, 98, 3814–3819. [Google Scholar]
- Klochendler-Yeivin, A.; Picarsky, E.; Yaniv, M. Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini5 subunit of the SWI/SNF chromatin remodeling complex. Mol. Cell Biol 2006, 26, 2661–2674. [Google Scholar]
- Isakoff, M.S.; Sansam, C.G.; Tamayo, P.; Subramanian, A.; Evans, J.A.; Filmore, C.M.; Wang, X.; Biegel, J.A.; Pomeroy, S.L.; Mesirov, J.P.; et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl. Acad. Sci. USA 2005, 102, 17745–17750. [Google Scholar]
- Ray, A.; Mir, S.N.; Wani, G.; Zhao, Q.; Battu, A.; Zhu, Q.; Wang, Q.E.; Wani, A.A. Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol. Cell Biol 2009, 29, 6206–6219. [Google Scholar]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar]
- van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar]
- van Attikum, H.; Fritsch, O.; Gasser, S.M. Distinct roles for the SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 2007, 26, 4113–4125. [Google Scholar]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of double-strand breaks. Nat. Cell Biol 2006, 8, 91–99. [Google Scholar]
- Cha, H.; Lowe, J.M.; Li, H.; Lee, J.S.; Belova, G.I.; Bulavin, D.V.; Fornace, A.J. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res 2010, 70, 4112–4122. [Google Scholar]
- Moon, S.; Lin, L.; Zhang, X.; Nguyen, T.; Darlington, Y.; Waldman, A.S.; Lu, X.; Donehower, L.A. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant γ-H2AX and suppresses DNA double strand break repair. J. Biol. Chem 2010, 285, 12935–12947. [Google Scholar]
- Füllgrabe, J.; Hajji, N.; Joseph, B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ 2010, 17, 1238–1243. [Google Scholar]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci 2010, 35, 323–332. [Google Scholar]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar]
- Garkavtsev, I.; Grigorian, I.A.; Ossovskaya, V.S.; Chernov, M.V.; Chumakov, P.M.; Gudkov, A.V. The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control. Nature 1998, 391, 295–298. [Google Scholar]
- Zeremski, M.; Hill, J.E.; Kwek, S.S.; Grigorian, I.A.; Gurova, K.V.; Garkavtsev, I.; Diatchenko, L.; Koonin, E.V.; Gudkov, A.V. Structure and regulation of the mouse ing1 gene. Three alternative transcripts encode two phd finger proteins that have opposite effects on p53 function. J. Biol. Chem 1999, 274, 4890–4893. [Google Scholar]
- Kim, M.S.; Baek, J.H.; Chakravarty, D.; Sidransky, D.; Carrier, F. Sensitization to UV-induced apoptosis by the histone deacetylase inhibitor Trichostatin A (TSA). Exp. Cell Res 2005, 306, 94–102. [Google Scholar]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol 2009, 27, 5459–5468. [Google Scholar]
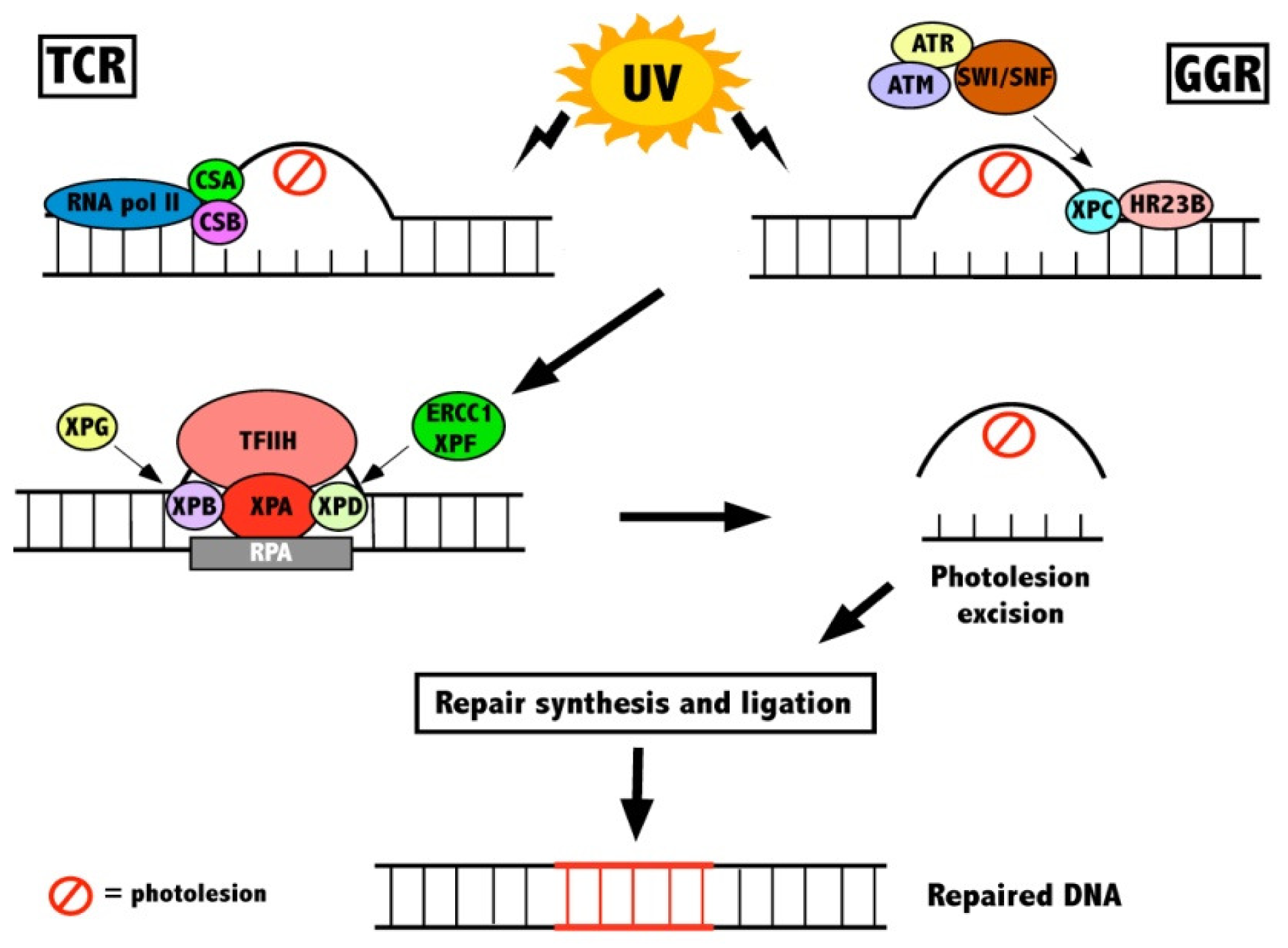


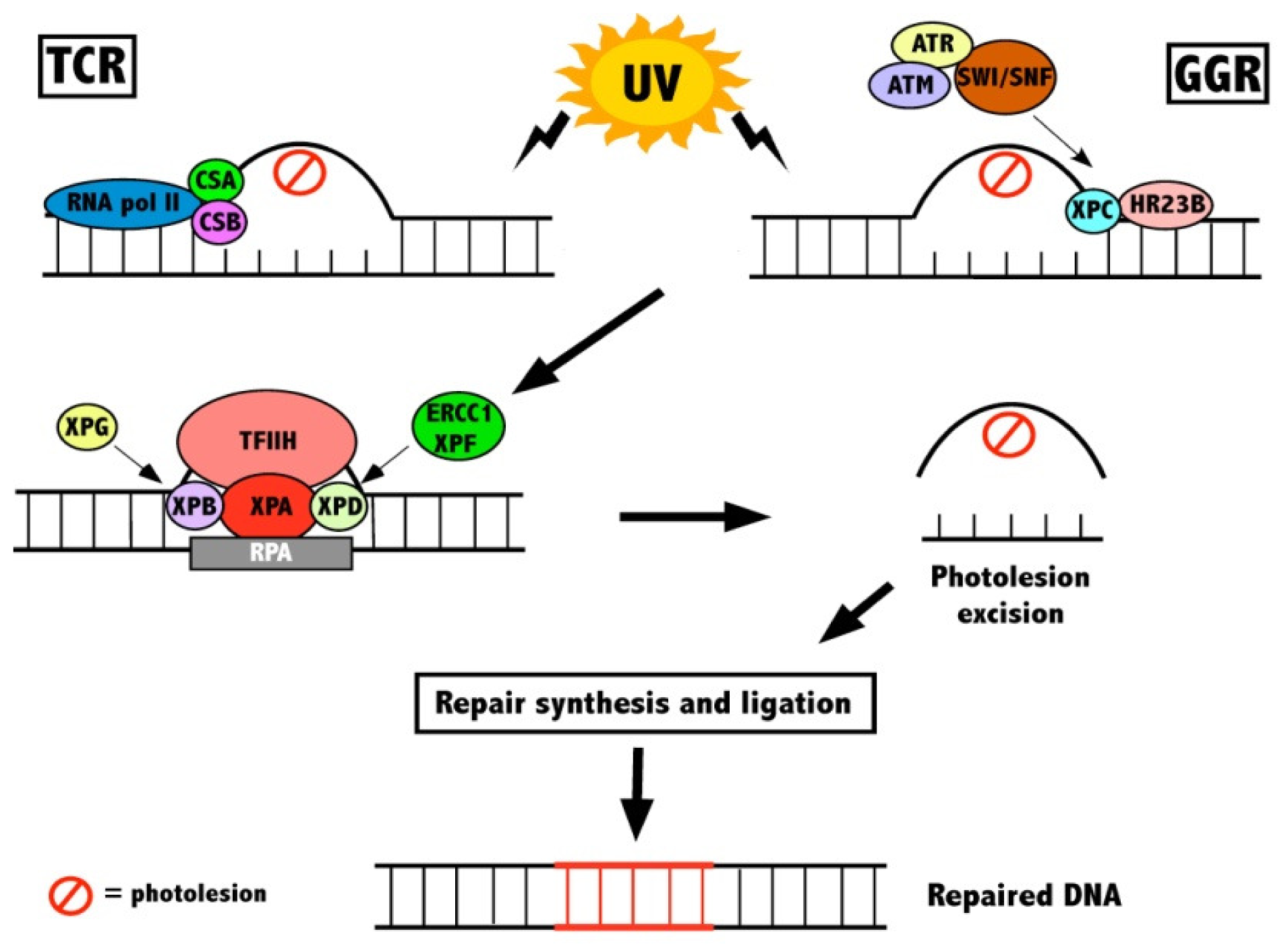{kind=link}
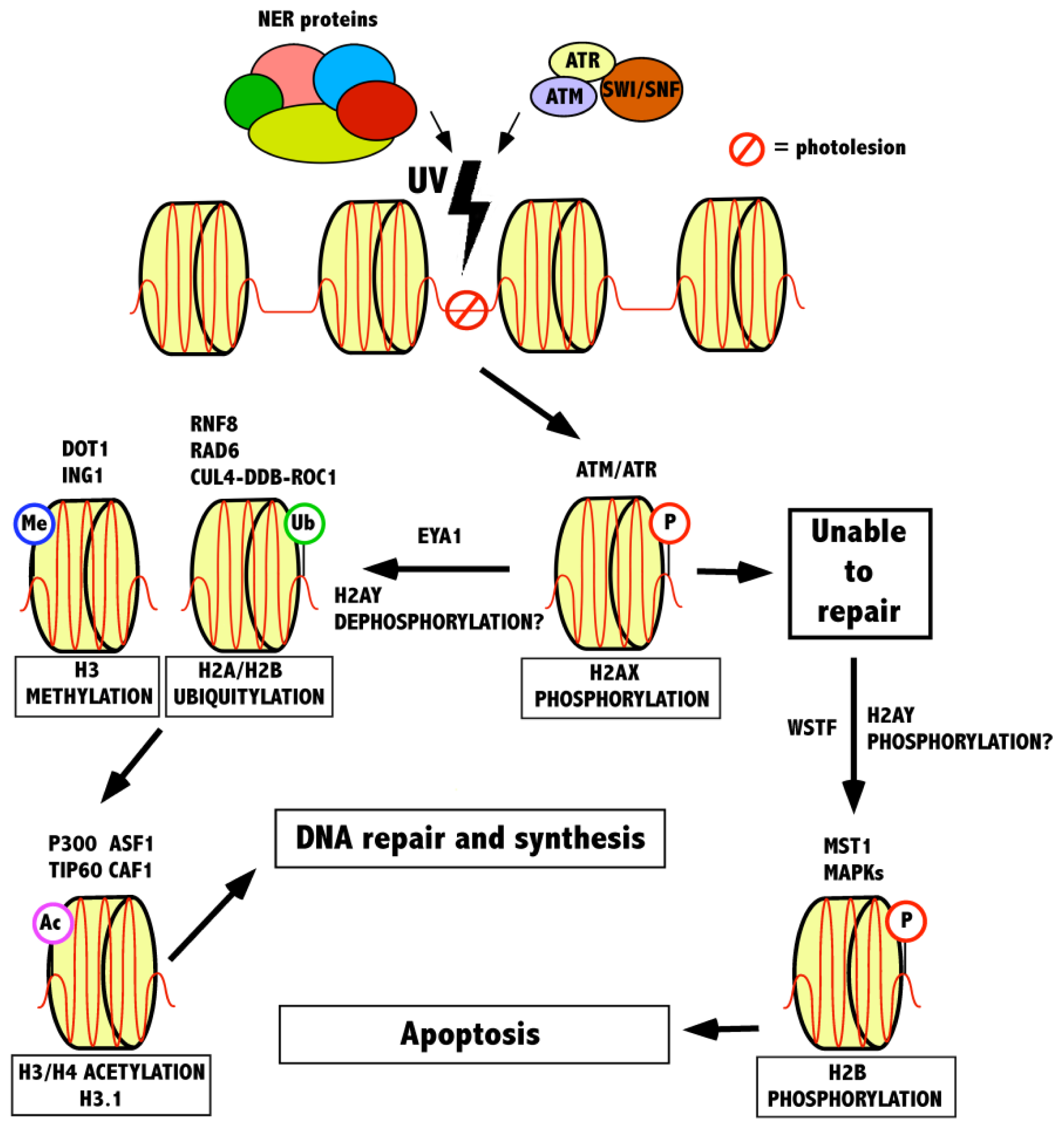{kind=link}
{kind=link}
| Histone residue | Modification type | Enzymes involved | Associated with? | References |
|---|---|---|---|---|
| H2AX S139 | Phosphorylation | ATM/ATR, DNA-PK, P53, MDC1, SWI/SNF | Repair and apoptosis | [50–65] |
| H2B S14 | Phosphorylation | MST1, MAPKs | Apoptosis | [66–71] |
| H1 H1.2 | Phosphorylation/Dephosphorylation | ND | Apoptosis | [72–76] |
| H2AX Y142 (?) | Phosphorylation | WSTF | Apoptosis | [77] |
| H2AX Y142 (?) | Dephosphorylation | EYA1 | Repair | [78] |
| H3 K79 H3 K4 H3 K9 | Methylation | DOT1, SIR ING1 P53 | Repair and apoptosis | [79–84] [85] [16,86] |
| H3 | Acetylation | P53, P300, ASF1, TIP60 | Repair | [87–92] |
| H4 H2A | Acetylation | ND RNF8, PcGs | Repair | [93,94] [95–97] |
| H2B H3/H4 | Ubiquitylation | RAD6 CUL4-DDB-ROC1 | Repair | [98] [99] |
| H3.1 | Incorporation | CAF-1 | Repair | [100] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Farrell, A.W.; Halliday, G.M.; Lyons, J.G. Chromatin Structure Following UV-Induced DNA Damage—Repair or Death? Int. J. Mol. Sci. 2011, 12, 8063-8085. https://doi.org/10.3390/ijms12118063
Farrell AW, Halliday GM, Lyons JG. Chromatin Structure Following UV-Induced DNA Damage—Repair or Death? International Journal of Molecular Sciences. 2011; 12(11):8063-8085. https://doi.org/10.3390/ijms12118063
Chicago/Turabian StyleFarrell, Andrew W., Gary M. Halliday, and James Guy Lyons. 2011. "Chromatin Structure Following UV-Induced DNA Damage—Repair or Death?" International Journal of Molecular Sciences 12, no. 11: 8063-8085. https://doi.org/10.3390/ijms12118063