Eighteen Years of Molecular Genotyping the Hemophilia Inversion Hotspot: From Southern Blot to Inverse Shifting-PCR

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Milestones in Hemophilia A Mutation Characterization
2.1. 1984–1993: Cloning and Characterization of the Human Coagulation Factor VIII
2.2. 1993–2005: F8 Intron 22 Inversion Discovery and Detection
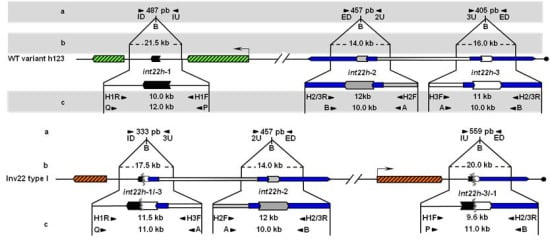
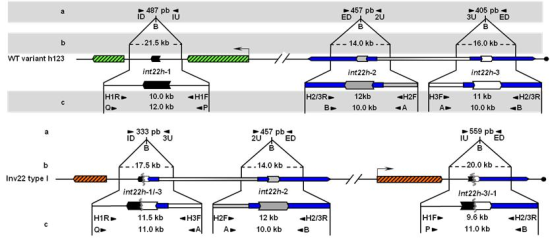
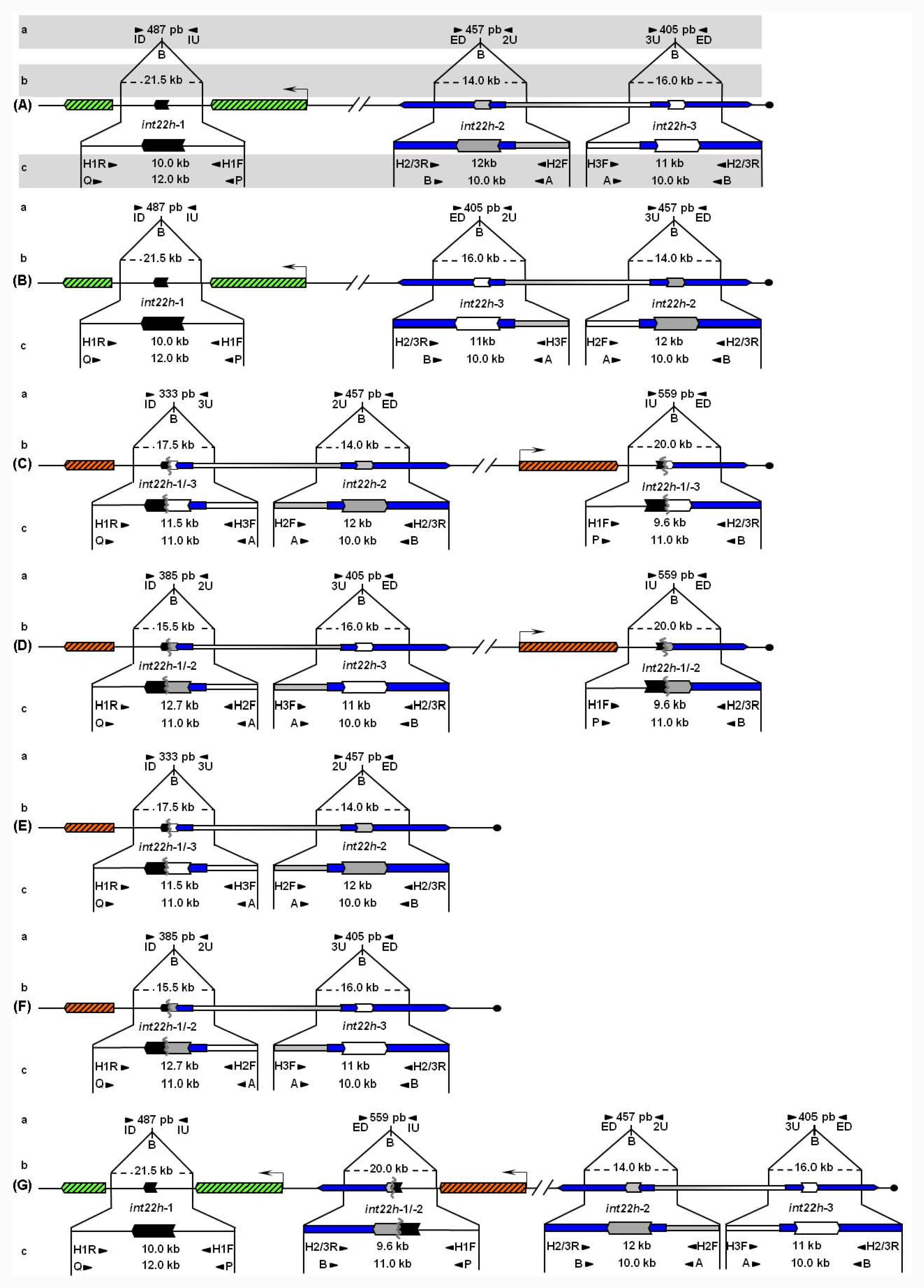
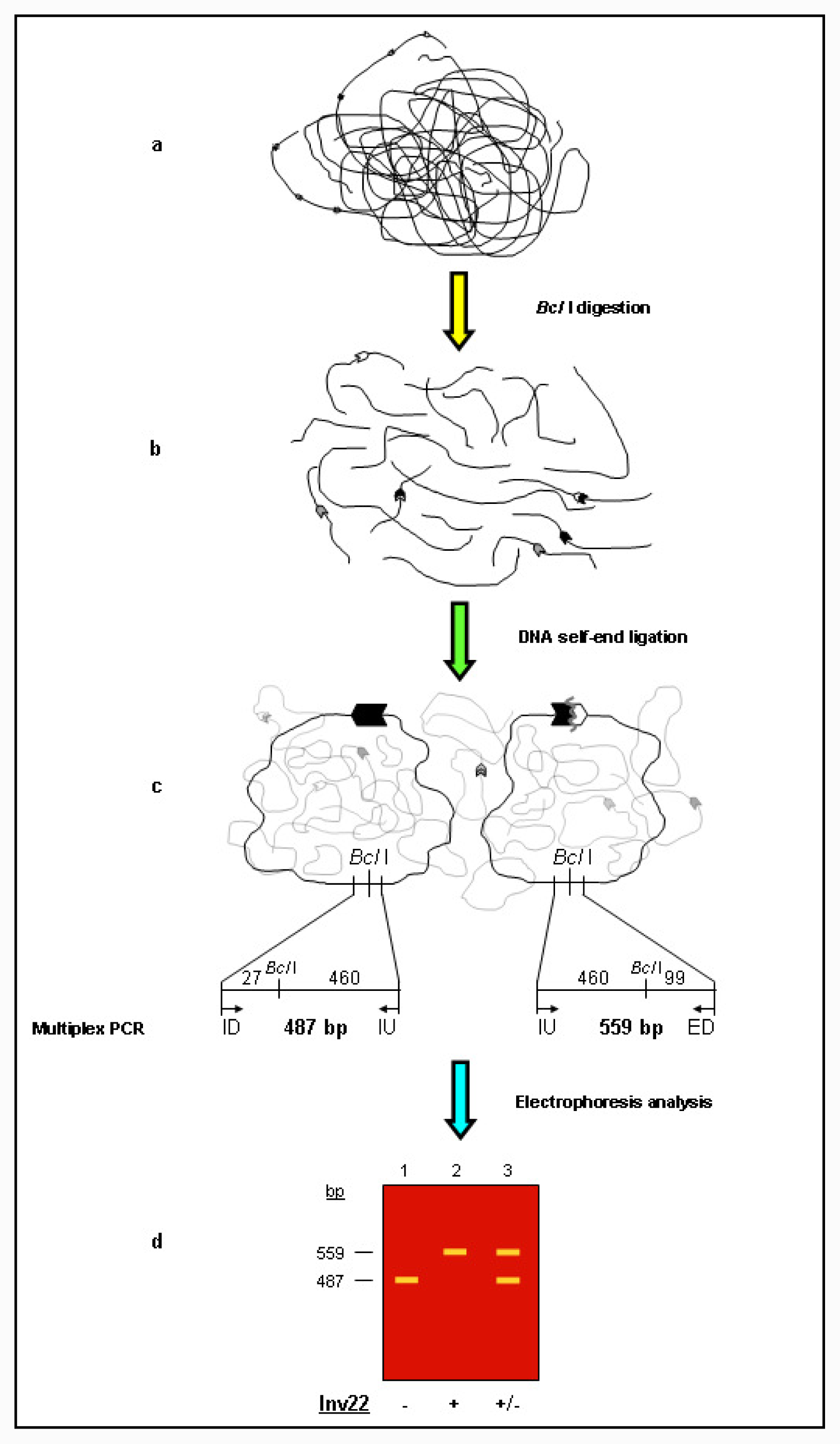
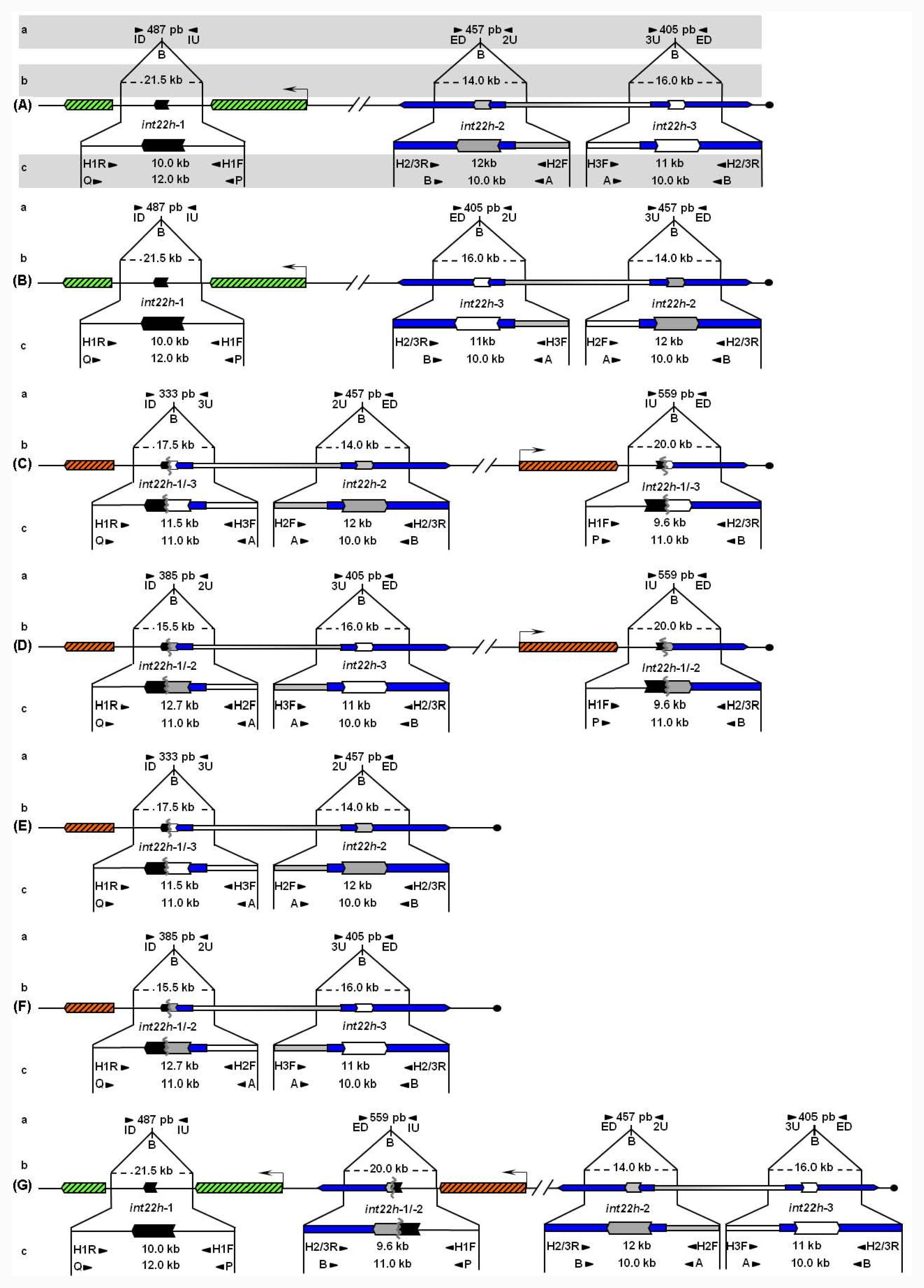
Unbroken F8 gene sequences are shown as green hatched boxes and rearranged F8 sequences as orange hatched boxes; intragenic int22h-1 is shown as a closed chevron; int22h-2 and int22h-3, within the arms of a large imperfect palindrome (blue), are shown as grey and open chevrons, respectively. Chimeric int22h sequences are denoted as [/] e.g., int22h-1/-2 represents the chimera between h-1 and h-2. Each schematic displays: (a) ISPCR based approaches developed by Rossetti et al. [15,16], wherein “B” represents a Bcl I restriction site after self-end ligation; (b) Southern blot analysis as described by Lakich et al. (1993) [11], wherein dashed lines show Bcl I restriction fragment sizes (kb); and (c) LDPCR based approaches of Bagnall et al. (2006) [17] (upper) and Liu et al. (1998) [18] (lower). Please refer to text for further explanation of details, including derivation of primers with orientation marked by arrowheads.
2.2.1. First Generation: Southern Blot Analysis as the Gold Standard and Early Findings about Inv22
2.2.2. Second Generation: Long Distance-PCR Based Approaches
2.2.3. Third Generation: Inverse Shifting-PCR Based Approaches
2.3. 2005–2011: Completion of the Human X-Chromosome Sequence and Definition of Hypothetical int22h-Mediated Rearrangements. Unraveling a Complex Picture
2.3.1. More on the Second and Third Generation. New Tests to Allow Comprehensive Detection of int22h-Related Rearrangements
3. Significance of the Human Genome Project for Inv22 Detection and Diagnosis of HA
Acknowledgments
References
- Saiki, R; Gelfand, D; Stoffel, S; Scharf, S; Higuchi, R; Horn, G; Mullis, K; Erlich, H. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar]
- Graw, J; Brackmann, HH; Oldenburg, J; Schneppenheim, R; Spannagl, M; Schwaab, R. Haemophilia A: From mutation analysis to new therapies. Nat. Rev. Genet 2005, 6, 488–501. [Google Scholar]
- Kasper, CK; Buzin, CH. Genetics of Hemophilia A and B. An Introduction for Clinicians, 2009, 1st ed; Southland Publications: Pasadena, CA, USA, 2009; pp. 27–29. [Google Scholar]
- Kemball-Cook, G; Tuddenham, EG; Wacey, AL. The factor VIII structure and mutation resource site: HAMSTeRS version 4. Nucleic Acids Res 1998, 26, 216–219. [Google Scholar]
- Oldenburg, J; Schröder, J; Hermann Brackmann, HH; Müller-Reible, C; Schwaab, R; Tuddenham, E. Environmental and genetic factors influencing inhibitor development. Semin. Hematol 2004, 41, 82–88. [Google Scholar]
- Gitschier, J; Wood, WI; Goralka, TM; Wion, KL; Chen, EY; Eaton, DH; Vehar, GA; Capon, DJ; Lawn, RM. Characterization of the human factor VIII gene. Nature 1984, 312, 326–330. [Google Scholar]
- Fujita, PA; Rhead, B; Zweig, AS; Hinrichs, AS; Karolchik, D; Cline, MS; Goldman, M; Barber, GP; Clawson, H; Coelho, A; et al. The UCSC genome browser DATABASE: Update 2011. Nucleic Acids Res 2010, 39, D876–D882. [Google Scholar]
- Levinson, B; Kenwrick, S; Lakich, D; Hammonds, G; Gitschier, J. A transcribed gene in an intron of the human factor VIII gene. Genomics 1990, 7, 1–11. [Google Scholar]
- Freije, D; Schlessinger, D. A 1.6-Mb contig of yeast artificial chromosomes around the human factor VIII gene reveals three regions homologous to probes for the DXS115 locus and two for the DXYS64 locus. Am. J. Hum. Genet 1992, 51, 66–80. [Google Scholar]
- Levinson, B; Kenwrick, S; Gamel, P; Fisher, K; Gitschier, J. Evidence for a third transcript from the human factor VIII gene. Genomics 1992, 14, 585–589. [Google Scholar]
- Lakich, D; Kazazian, HH, Jr; Antonarakis, SE; Gitschier, J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat. Genet 1993, 5, 236–241. [Google Scholar]
- Peters, MF; Ross, CA. Isolation of a 40 k-Da Huntingtin-associated protein. J. Biol. Chem 2001, 276, 3188–3194. [Google Scholar]
- Valleix, S; Jeanny, JC; Elsevier, S; Joshi, RL; Fayet, P; Bucchini, D; Delpech, M. Expression of human F8B, a gene nested within the coagulation factor VIII gene, produces multiple eye defects and developmental alterations in chimeric and transgenic mice. Hum. Mol. Genet 1999, 8, 1291–1301. [Google Scholar]
- Naylor, J; Brinke, A; Hassock, S; Green, PM; Giannelli, F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum. Mol. Genet 1993, 2, 1773–1778. [Google Scholar]
- Rossetti, LC; Radic, CP; Larripa, IB; De Brasi, CD. Genotyping the hemophilia inversion hotspot by use of inverse PCR. Clin. Chem 2005, 51, 154–158. [Google Scholar]
- Rossetti, LC; Radic, CP; Larripa, IB; De Brasi, CD. Developing a new generation of tests for genotyping hemophilia causative rearrangements involving int22h and int1h hotspots in the factor VIII gene. J. Thromb. Haemost 2008, 6, 830–836. [Google Scholar]
- Bagnall, RD; Giannelli, F; Green, PM. Int22h-related Inversions causing hemophilia A: A novel insight into their origin and a new more discriminant PCR test for their detection. J. Thromb. Haemost 2006, 4, 591–598. [Google Scholar]
- Liu, Q; Nozari, G; Sommer, SS. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in haemophilia A. Blood 1998, 92, 1458–1459, Erratum inBlood 1999;93:2141. [Google Scholar]
- Oldenburg, J; Rost, S; El-Maarri, O; Leuer, M; Olek, K; Müller, CR; Schwaab, R. De novo factor VIII gene intron 22 inversion in a female carrier presents as a somatic mosaicism. Blood 2000, 96, 2905–2906. [Google Scholar]
- Rossiter, JP; Young, M; Kimberland, ML; Hutter, P; Ketterling, RP; Gitschier, J; Horst, J; Morris, MA; Schaid, DJ; de Moerloose, P. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum. Mol. Genet 1994, 3, 1035–1039. [Google Scholar]
- Tizzano, EF; Domènech, M; Baiget, M. Inversion of intron 22 in isolated cases of severe hemophilia A. Thromb. Haemost 1995, 73, 6–9. [Google Scholar]
- Naylor, JA; Buck, D; Green, P; Williamson, H; Bentley, D; Giannelli, F. Investigation of the factor VIII intron 22 repeated region (int22h) and the associated inversion junctions. Hum. Mol. Genet 1995, 4, 1217–1224. [Google Scholar]
- Antonarakis, SE; Rossiter, JP; Young, M; Horst, J; de Moerloose, P; Sommer, SS; Ketterling, RP; Kazazian, HH, Jr; Négrier, C; Vinciguerra, C; et al. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood 1995, 86, 2206–2212. [Google Scholar]
- De Brasi, CD; Candela, M; Cermelj, M; Slavutsk, IR; Larripa, IB; Pérez Bianco, R; de Tezanos Pinto, M. Intron 22 factor VIII gene inversions in Argentine families with severe haemophilia A. Haemophilia 2000, 6, 21–22. [Google Scholar]
- Liu, Q; Thorland, EC; Heit, JA; Sommer, SS. Overlapping PCR for bidirectional PCR amplification of specific alleles: A rapid one-tube method for simultaneously differentiating homozygotes and heterozygotes. Genome Res 1997, 7, 389–398. [Google Scholar]
- Barnes, WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc. Natl. Acad. Sci. USA 1994, 91, 2216–2220. [Google Scholar]
- Liu, Q; Sommer, SS. Subcycling-PCR for multiplex long-distance amplification of regions with high and low GC content: Application to the inversion hotspot in the factor VIII gene. Biotechniques 1998, 25, 1022–1028. [Google Scholar]
- Wion, KL; Tuddenham, EGD; Lawn, RM. A new polymorphism in the factor VIII gene for prenatal diagnosis of haemophilia A. Nucleic Acids Res 1986, 14, 4535–4542. [Google Scholar]
- El-Maarri, O; Oldenburg, J; Cağlayan, SH. Intron 22-specific long PCR for the Xba I polymorphism in the factor VIII gene. Br. J. Haematol 1999, 105, 1120–1122. [Google Scholar]
- De Brasi, CD; Bowen, DJ; Collins, PW; Larripa, IB. Specific analysis of the intron 22 XbaI polymorphism of the human factor VIII gene using long-distance PCR. Br. J. Haematol 1999, 107, 566–568. [Google Scholar]
- Bowen, DJ; De Brasi, CD; Larripa, IB; Collins, PW. A new polymorphism in the human factor VIII gene: implications for linkage analysis in haemophilia A and for the evolution of int22h sequences. Br. J. Haematol 2000, 111, 544–548. [Google Scholar]
- De Brasi, CD; Rossetti, LC; Larripa, IB. Rapid genotyping of XbaI and MspI DNA polymorphisms of the human factor VIII gene: Estimation of their combined heterozygosity in the Argentinean population. Haematologica 2003, 88, 232–234. [Google Scholar]
- Bowen, DJ; Keeney, S. Unleashing the long-distance PCR for detection of the intron 22 inversion of the factor VIII gene in severe haemophilia A. Thromb. Haemost 2003, 89, 201–202. [Google Scholar]
- Ochman, H; Gerber, AS; Hartl, DL. Genetic applications of an inverse polymerase chain reaction. Genetics 1988, 120, 621–623. [Google Scholar]
- Ross, MT; Grafham, DV; Coffey, AJ; Scherer, S; McLay, K; Muzny, D; Platzer, M; Howell, GR; Burrows, C; Bird, CP; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar]
- Bagnall, RD; Giannelli, F; Green, PM. Polymorphism and hemophilia A causing inversions in distal Xq28: A complex picture. J. Thromb. Haemost 2005, 3, 2598–2599. [Google Scholar]
- Bagnall, RD; Giannelli, F; Green, PM. Int22h-related inversions causing hemophilia A: A novel insight into their origin and a new more discriminant PCR test for their detection. J. Thromb. Haemost 2006, 4, 591–598. [Google Scholar]
- Radic, CP; Rossetti, LC; Zuccoli, JR; Abelleyro, MM; Larripa, IB; De Brasi, CD. Inverse shifting PCR based prenatal diagnosis of hemophilia-causative inversions involving int22h and int1h hotspots from chorionic villus samples. Prenat. Diagn 2009, 29, 1183–1185. [Google Scholar]
- Abou-Elew, H; Ahemed, H; Raslan, H; Abdelwahab, M; Hammoud Mokhtar, D; Arnaout, H. Genotyping of intron 22-related rearrangements of F8 by inverse-shifting PCR in Egyptian hemophilia A patients. Ann. Hematol 2011, 90, 579–584. [Google Scholar]
- Abelleyro, MM; Rossetti, LC; Radic, CP; Candela, M; Larripa, IB; De Brasi, CD. Are int22h-mediated deletions a common cause of hemophilia. Ann Hematol 2011, in press. [Google Scholar]
- De Brasi, CD; Bowen, DJ. Molecular characteristics of the intron 22 homologs of the coagulation factor VIII gene: an update. J. Thromb. Haemost 2008, 6, 1822–1824. [Google Scholar]
- Altschul, SF; Madden, TL; Schäffer, AA; Zhang, J; Zhang, Z; Miller, W; Lipman, DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Smith, TF; Waterman, MS. Identification of common molecular subsequences. J. Mol. Biol 1981, 147, 195–197. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rossetti, L.C.; Radic, C.P.; Abelleyro, M.M.; Larripa, I.B.; De Brasi, C.D. Eighteen Years of Molecular Genotyping the Hemophilia Inversion Hotspot: From Southern Blot to Inverse Shifting-PCR. Int. J. Mol. Sci. 2011, 12, 7271-7285. https://doi.org/10.3390/ijms12107271
Rossetti LC, Radic CP, Abelleyro MM, Larripa IB, De Brasi CD. Eighteen Years of Molecular Genotyping the Hemophilia Inversion Hotspot: From Southern Blot to Inverse Shifting-PCR. International Journal of Molecular Sciences. 2011; 12(10):7271-7285. https://doi.org/10.3390/ijms12107271
Chicago/Turabian StyleRossetti, Liliana C., Claudia P. Radic, Miguel M. Abelleyro, Irene B. Larripa, and Carlos D. De Brasi. 2011. "Eighteen Years of Molecular Genotyping the Hemophilia Inversion Hotspot: From Southern Blot to Inverse Shifting-PCR" International Journal of Molecular Sciences 12, no. 10: 7271-7285. https://doi.org/10.3390/ijms12107271