Abstract
Future therapeutic intervention that could effectively decelerate the rate of degeneration within the substantia nigra pars compacta (SNc) could add years of mobility and reduce morbidity associated with Parkinson’s disease (PD). Neurodegenerative decline associated with PD is distinguished by extensive damage to SNc dopaminergic (DAergic) neurons and decay of the striatal tract. While genetic mutations or environmental toxins can precipitate pathology, progressive degenerative succession involves a gradual decline in DA neurotransmission/synaptic uptake, impaired oxidative glucose consumption, a rise in striatal lactate and chronic inflammation. Nutraceuticals play a fundamental role in energy metabolism and signaling transduction pathways that control neurotransmission and inflammation. However, the use of nutritional supplements to slow the progression of PD has met with considerable challenge and has thus far proven unsuccessful. This review re-examines precipitating factors and insults involved in PD and how nutraceuticals can affect each of these biological targets. Discussed are disease dynamics (Sections 1 and 2) and natural substances, vitamins and minerals that could impact disease processes (Section 3). Topics include nutritional influences on α-synuclein aggregation, ubiquitin proteasome function, mTOR signaling/lysosomal-autophagy, energy failure, faulty catecholamine trafficking, DA oxidation, synthesis of toxic DA-quinones, o-semiquinones, benzothiazolines, hyperhomocyseinemia, methylation, inflammation and irreversible oxidation of neuromelanin. In summary, it is clear that future research will be required to consider the multi-faceted nature of this disease and re-examine how and why the use of nutritional multi-vitamin-mineral and plant-based combinations could be used to slow the progression of PD, if possible.
1. Introduction
1.1. Pathology
The pathology of Parkinson’s disease (PD) involves chronic degeneration of dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNc). Subsequent decay of the nigrostriatal tract manifests itself clinically by symptomatic rigidity, bradykinesia, postural instability and resting tremor. Prominent pathological manifestations associated with degeneration of SNc DAergic neurons include observations describing mitochondrial abnormalities [1–4], excessive cytosolic dopamine (DA) oxidation, α-synuclein aggregates, autophagolysosome dysfunction, defects in the ubiquitin-proteasome system (UPS), oxidative stress, nitrosative stress, iron released from bound storage and a gradual loss of neuromelanin (NM) [5–7]. These pathological insults are self reinforcing and can advance in cyclical fashion, often intensified by decaying levels of glutathione (GSH), which render greater oxidative damage (via O2, H2O2, OH) [8,9] or lipid/protein nitration (via ONOO−) and accumulation of 3-nitrotyrosine, protein carbonyls, 8-hydroxyguanosine, malondialdehyde and hydroxynonenol in degenerating neurons [10–12]. Neurological degeneration can also be aggravated by chronic central nervous system (CNS) inflammation which can involve recruitment of activated microglial cells, release of cytotoxic molecules, free radicals and glutamate, which can provoke neuritic beading, excitotoxic, apoptotic and necrotic degeneration [13]. While gradual loss of DAergic SNc pigmented cells occur as a natural process of aging—early diagnosis of PD is associated with a 30%/60% reduction of DAergic neurons/striatal DA which is attributable to degeneration of striatal axon terminals [14]. Though the etiology circumscribing the selective loss of SNc DAergic neurons in PD is not fully understood, we do know that reportedly ~5–10% of PD patients display mutations in genes such as DJ-1, PTEN-induced kinase 1 (PINK-I), leucine-rich repeat kinase 2 (LRRK2) G2019S, park-1/Synuclein (SNCA), ubiquitin-carboxy-terminal-hydrolase L1, parkin (Del3-5, T240R, Q311X) [15–18], ATP13A2 (Park 9), β-glucocerebrosidase and mitochondrial proteins such as park 13 Omi/Htra2, Complex I [19–22]. The larger majority of PD cases result from a fusion of natural aging and/or environmental exposures to pesticides, a history of depression, viral/bacterial infections, metals, antipsychotic/antidepressant drugs, rural/farm living or lack of habitual cigarette smoking/tobacco use or consumption of caffeine [23–27]. All of these studies provide partial insight as to the precipitating factors involved with PD onset and progression. Moreover, the greatest commonality appears to be either genetic mutations or environmental triggers that lead to direct/indirect accumulation of malfunctional mitochondria, which precede selective DAergic SNc degeneration.
The extent of DAergic SNc losses in human PD can be imaged using positron emission tomography (PET) or single photon emission computerized tomography (SPECT). Radioactive tracers in PD patients have been used to substantiate (1) compromised integrity of pre-synpatic nigrostriatal projections, i.e., [18F]LDOPA (which monitors DA uptake, metabolism, DOPA decarbarboxylase (DDC), DA storage within intact nerve terminals); (2) faulty DA transporters (DAT) i.e., CFT, C-RTI-32, FP-CIT ligands, [11C]methylphenidate (MP)/99mTc-TRODAT-1 or (3) abnormal type-2-vesicular monoamine transporter (VMAT2) function using tracers such as [11C]-dihydrotetrabenazine (DTBZ) which measures cytoplastic DA uptake into synaptic vesicles [28–30]. Chronic SNc DAergic degeneration parallels a reduction of [18]F-DOPA uptake and DAT binding which are foundational events to faulty circuitry in the basal ganglia that ultimately triggers locomotive disability [31].
1.2. Treatment
In order to counteract the loss of SNc DAergic neurons, medical treatments are aimed at modulating neurotransmitter (NT) function. Prescription medicines allow for fluid voluntary movement, reduction of tremors and a sustained quality of life. Routine adjunct therapies often combine levodopa/dopa-decarboxylase inhibitors Sinemet® and Madopar® with DA receptor agonists, catechol-o-methyltransferase inhibitors, monoamine oxidase (MAO) inhibitors, anti-cholinergics and surgical treatments [32]. While prescription drugs ameliorate the symptoms, they do not necessarily address the central etiology of degeneration and therefore a number of alternative approaches have been considered to slow the progression of this disease.
1.3. Previous Studies on Therapeutic Agents to Slow Progression of PD
Innovative strategies to slow the progression have met with partial success in experimental models, and to a less significant extent in clinical trials. Most neuroprotective strategies seem to fall under the general classes of anti-inflammatory, anti-apoptotic, anti-oxidants, enzyme inhibitors, growth factors, alternative medicine or receptor antagonists/agonists. Experimental trials elucidating efficacy of neuroprotective agents are rapidly expanding, and have thus far included superoxide dismutase (SOD)/catalase/peroxidase mimetics [33], anti-apoptotic MAO inhibitors, rasagiline [34–38], (−)-epigallocatechin-3-gallate, iron chelator/antioxidant/anti-inflammatory combinations [39–41], celastrol, nitric oxide synthase (NOS) inhibitors [42,43], COX, c-jun N-terminal kinase (JNK) inhibitors [44–47], alpha-tocopherol, coenzyme Q10, lipoic acid [48–52], creatine [53,54] melatonin, catalpol from root of Rehmannia glutinosa Libosch [55,56], N-acetyl-l-cysteine (NAC), thiol antioxidants [57], nerve growth factors [58,59], dehydroepiandrosterone [60], estrogen receptor agonists [61], adenosine A2 receptor antagonists [62–68], S-allylcysteine [66], mGlu2/3 metabotropic [67], acupuncture [68] traditional Chinese medicine Zhen-Wu-Tang [69] angiotensin-converting enzyme inhibitors [70], nicotine, ginseng, ginkgo biloba, caffeine and cannabis [71].
Despite the success using a vast range of therapeutic agents in preliminary experiments, there is a general failure of clinical trials to substantiate therapeutic effects that slow disease progression, in particular for antioxidants. This may be attributable to limitations in the current animal or in vitro models that make extrapolation of information for human PD difficult. Further, the pathology is very complex and may not be effectively antagonized with just single therapy antioxidant, ergogenic, anti-inflammatory regimens.
The use of nutritional supplements to slow the progression of PD has also not been fully substantiated by evidenced-based studies. The aim of this review is to re-visit the pathology of PD, and in light of pathological processes further discuss the rationale behind potential use of vitamin/mineral nutraceutical neuroprotective agents. In this review, the details of pathology are presented in Sections 1 and 2, and further discussed relevant to nutrient interactions in Section 3. Discussion includes the role of vitamins and minerals in the established United States recommended daily allowances, as well as macronutrients and plant based constituents that modulate processes with specific relevance to PD. The review is a combination of past literature and proposed theory based on known molecules that affect known biological targets which range from mitochondrial malfunction, inflammation, DA oxidation and defective UPS/lysosomal autophagy processes. Moreover, some of the compounds proposed in this review have not yet been evaluated.
2. Review
2.1. Energy Failure—Loss of OXPHOS, Rise in Anaerobic Glycolysis & Lactate, ATP Depletion
We first review the most prominent issue underlying the loss of DAergic neurons, which is a fundamental failure in glucose metabolism due to aberration of mitochondrial respiration. It is important to note that mitochondrial malfunction could initially occur due to toxic effects of α-synuclein, endogenous neurotoxins or exogenous environmental factors. However, experimental models often employ use of mitochondrial toxins such as 1-methyl-4-phenylpyridinium (MPP+), rotenone or endogenous isoquinolines to selectively target neuropathological damage similar to, but not identical to PD degenerative effects mainly in the SNc and the locus coeruelus (LC) [72–74].
Loss of mitochondrial function leads to immediate failure of DA neurotransmission and acceleration of glycolysis to overcome the loss of oxidative phosphorylation (OXPHOS) through substrate level phosphorylation (SLP) [75–77]. The impact of mitochondrial toxins on these energy processes is almost always observed. In vivo, administration of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP) generates an immediate rise in glucose utilization (detected with [2–14C]deoxyglucose), a drop in ATP, a rise in lactate production, reduction in striatal DAT/DA and loss of tyrosine hydroxylase immunoreactivity, effects which are exacerbated by α-synuclein [78–83]. The drop in ATP suggests that energy deficiency is clearly involved with the process of initiating degenerative decline [84]. Moreover, there is ample information to substantiate that a drop in energy corresponds to a rise in glycolysis to drive SLP, an indicator of metabolic stress. The use of proton magnetic resonance spectroscopy (1H-MRS) has been used to confirm sharp spikes in striatal lactate, which occur within 2 h of MPTP injection in C57BL/6 mice [85]. In primates, a long term study utilizing infusion of MPTP over 14 ± 5 months resulted in loss of DA pre-synaptic re-uptake, parallel to a 23-fold increase in striatal lactate production, which was sustained for up to 10 months post final administration of MPTP [86]. While the CNS is most often studied with respect to biochemical effects induced by systemic injection of MPTP, damage in peripheral tissue such as skeletal muscle also involves heightened anaerobic glycolytic function, elevation of lactic acid dehydrogenase and concomitant decrease of mitochondrial Complex IV, with no changes in mitochondrial Complex I [87]. These shifts toward anaerobic metabolism occur in tissues with capability to uptake MPP+ where similar patterns observed include loss of ATP, loss of OXPHOS, a rise in glycolysis, heightened production in lactate and neurotoxic effects which can be blocked by providing abundant glucose to growth media in order to sustain ATP production through glycolysis [77,88–93]. The reliance of damaged neurons on greater anaerobic glycolytic function is not exclusive to PD, as head trauma, seizure or ischemia can equally provoke a rise in brain/CSF lactate and loss of ATP parallel to neurological damages [94–98]. In Section 3, we discuss nutrients involved with propelling anaerobic function.
In the human brain, in vivo functional imaging strategies to assess glucose metabolism in the brain include PET with a [18F]2-fluoro-2-deoxy-d-glucose ([18F]FDG) tracer. This tracer is used to quantify elevated rates of glycolysis/glucose transport relative to surrounding tissue. Some limitations for this method involve the non-specific manner by which [18F]FDG accumulates in the brain. [18F]FDG enters through the glycolytic cycle prior to conversion of pyruvate, therefore its measurement does not differentiate between aerobic (OXPHOS) and anaerobic (SLP) metabolism. Further, uptake is not selective to cell type and therefore false positives (or heightened metabolic activities) are likely to occur in particular for diseases involving active inflammatory tissue, where metabolic rate of glucose is extremely high [99]. This technique however, has been used in sliced striatal tissue to corroborate that regional exposure to MPP+ can evokes a sharp rise in cerebral glucose metabolic rate (CMRglc) [93]. FDG PET studies could also be beneficial in terms of evaluating patterns in non-diseased, non-inflammatory models. FDG PET imaging techniques clearly show that the process of aging in monkeys, corresponds to loss in both regional cerebral blood flow/[15O] H2O and rCMRglc in many areas of the brain including the cerebellum, hippocampus, striatum, occipital cortex, temporal cortex, and frontal cortex [100]. Age associated hypometabolism in the human brain is also believed to precipitate increased risk for many age associated CNS neurodegenerative diseases [101]. Future research is now considering preventative implementation with nutrients that could assist in minimizing metabolic losses [101].
In brief, the loss of ATP in the SNc is detrimental because this single event can initiate a range of downstream collapse on energy requiring systems that can then lead to (1) catecholamine oxidation and formation of DA neurotoxins/free radicals (2) excitotoxic and programmed cell death (3) mitochondrial transition pore opening, matrix swelling, release of mitochondrial proteins into the cytosol, apoptosis [102–107] and microtubular/cell structure collapse [108].
2.2. Loss of DA Regulation and Trafficking—VMAT2
One of the first events brought about by reduction of ATP is a loss of energy requiring systems that drive DA trafficking. Section 3, also refers to a large number of nutraceuticals that can block these processes. Failure of DA trafficking, not only occurs due to decline in ATP, but can also result from genetic mutations in SNCA (A53T and A30P) [109], mitochondrial insufficiency or oxidative damage by ROS, all which trigger excessive DA release from SNc nerve terminals [110,111]. With regards to energy, a lack of ATP diminishes the capability of intracellular ATPase pumps to sequester DA into synaptic vesicles (where DA is stable due to slightly acidic pH), which is a pivotal factor in initiating a cascade of neurotoxic events [112,113]. Failure of vesicle monoamine transporter 2 (VMAT2) results in immediate leaked DA into the cytosolic compartment (easily subject to oxidative breakdown at neutral pH) where it readily oxidizes to form neurotoxic DA-quinones, o-semiquinones, dopaminergic poisons and related free radicals [114–116] which can then contribute to eventual decay of the striatal tract [117].
Further, functional loss of VMAT2 can occur due to age-associated losses in VMAT2 mRNA expression, which create vulnerability to extensive neurological damage in the presence of mitochondrial toxins such as MPTP in vivo or MPP+ in vitro [118–123]. MPP+ can cause further insult due to its ability to bind directly to VMAT2, gain entrance into synaptic vesicles and initiate extrusion of DA back into the cytoplasmic compartment [124,125].
2.3. Dopamine Oxidation
Inadequate function or expression of VMAT2 mRNA has also been reported in association with PD [126], which could precipitate three main routes by which the oxidation of DA can become pathological. These include (1) the enzymatic oxidation of DA via tyrosinase, phospholipase A2 (PLA2)/prostaglandin H synthase (COX), lipoxygenase and xanthine oxidase to form DA-quinone en route to neuromelanin synthesis (2) non-enzymatic autoxidation of DA by the presence of oxygen, H2O2, or metals and (3) the enzymatic oxidation of DA by MAO which can lead to H2O2 production and synthesis of DA-aldehydes. The heavy oxidation of DA (be it non-enzymatic or enzymatic) seems to initiate neurodegenerative pathogenesis, a depletion of glutathione, oxidation of available ascorbate and subsequent oxidative stress in the SNc area [127]. In Section 3, we provide information on nutraceuticals that may be able to antagonize each of the major routes of DA oxidation.
2.3.1. Enzymatic Oxidation of DA, the Neuromelanin Pathway & DA-Quinones
Understanding the role of target enzymes and how they exacerbate DA oxidation could be beneficial in directing future investigation or design of nutraceutical combinations. First, the enzymatic oxidation of DA occurs through heightened activity of tyrosinase, COX, lipoxygenase and xanthine oxidase which converts DA to DA-quinone en route to neuromelanin (NM) [128]. The neuromelanin pathway if intensified can produce deleterious DA-quinone neurotoxic metabolites such as o-semiquinones or benzothiazolines, which are potent inhibitors of mitochondrial pyruvate dehydrogenase (i.e., complex I/Krebs cycle) and initiators of α-synuclein fibrillization [129–132]. Oxidized DA can further react with thiols producing DA-cysteine adducts such as 5-S-cysteinyldopamine which mediate metal catalyzed oxidation of proteins, which lead to protein misfolding and aggregation [131]. While gradual accumulation of NM in SNc tissue occurs as a natural process of aging [133], an intense heightened dark melanized pigment (hyperpigmentation) appears in the SNc preceding not only neuronal degeneration but also α-synuclein aggregation, inflammation, oxidative stress, apoptosis, Lewy body formation, depletion of GSH, functional loss of DAT and the loss of tyrosine hydroxylase positive neurons [10,128,134,135]. With PD, a biphasic but final loss of NM occurs gradually due to massive oxidation, cell death and release of NM from dying cells [136]. The loss of melanized nigral DAergic neurons is evident in PD brains (Right) when compared to healthy controls (Left) as shown in Figure 3 and is a major part of the pathology [137]. Ultimately, the loss of NM renders failure of its natural protective function, which is to sequester iron, free radicals and toxic quinones [138].
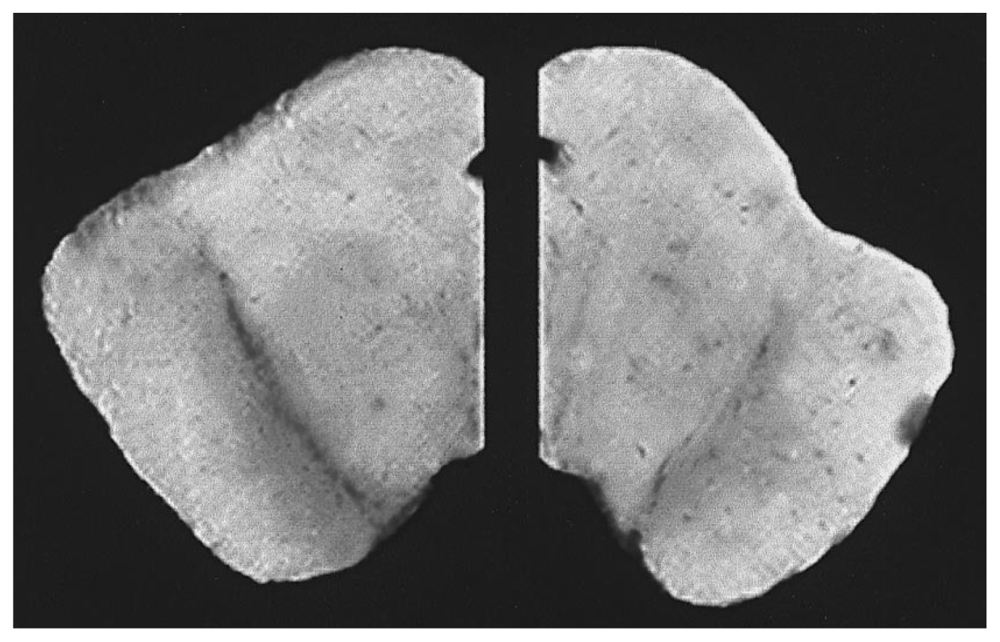
Figure 3.
Melanized dopaminergic neurons of the substantia nigra from post mortem human brain. Brain sections taken through the mid- brain of a normal (left) and a Parkinson’s disease patient (right). The Parkinson’s diseased hemisphere on the right shows a loss of the melanized neurons in the substantia nigra in the ventral midbrain [137].
The generation of DA oxidative toxins also includes enzymatic conversion of dopaminochrome to 5,6,-dihydroxyindole by DT diaphorase, the free radical initiated conversion of o-hydroquinones (protective) to o-semiquinones (toxic) [132] and transglutaminases which incorporate sulfur amino acids into DA-cysteine conjugate toxic precursors to neuromelanin [139]. And, recent studies suggest that transglutaminase inhibitors could be useful to prevent cross-linking reactions that lead to neurodegenerative aggregated proteins [140]. Animal models deficient in enzymes capable of catalytically oxidizing DA to DA-quinone (i.e., absent of PLA2 COX2), show a resistance to DAergic neurotoxicity after administration of MPTP. This is also corroborated where knockout models for SOD/GSH Px show extensive damage with MPTP [140–144], and protective effects are observed with COX/PLA2 inhibitors [145–148].
2.3.2. Non Enzymatic Oxidation of DA, 6-OHDA, Release of Iron & Oxidative Stress
A second route for DA oxidation is non-enzymatically by reactive oxygen species (ROS) and metals (Fe2+, Cu2+, and Mn2+) [149,150]. The autoxidation of DA can render formation of 6-OHDA (a potent neurotoxin) and O2 −. If superoxide reacts with nitric oxide (NO) the formation of ONOO− is evident. Peroxynitrite in turn can cyclically re-oxidize DA, deplete available reduced glutathione/ascorbate (vitamin C), incur a substantial loss of endogenous GSH-peroxidase and destroy the natural ability of GSH to act as an antioxidant [129,151]. While PD patients display depletion of GSH within the SNc [152], the reduction of GSH (i.e., γ-glutamylcysteine synthetase inhibitor) in experimental models also renders the SNc vulnerable to the toxic effects of MPTP and 6-OHDA [8]. For this reason, thiol based dietary antioxidants could be considered for clinical trials, as some have reported they prevent MPTP induced toxicity in mice [57,153], attenuate pathological effects of 6-OHDA, ONOO− and block the formation of DA o-semiquinone neurotoxic radicals [154].
Once 6-OHDA is formed, its presence can trigger neurodegeneration through reduction of striatal zinc and metallothione (otherwise antioxidant/metal detoxification agents) and initiate selective release of free iron from ferritin, where pro-oxidant effects predominate [152,155–160]. This could be perilous given the already high concentrations of iron dispersed throughout the substantia nigra, globus pallidus, red nucleus and locus cerulus [161–163]. Heightened free iron deposits are found in the vicinity of neurodegenerative regions, located in microglia, astrocytes and oligodendrocytes in conjunction with a rise in heme oxygenase (HO-1) (an enzyme which yields free Fe2+ iron from heme) and disappearance of NM (loss of high affinity binding polymer for Fe2+) [164–168]. In PD patients, iron deposition near degenerating neurons [169], could be intensified by hereditary mutations in iron regulatory binding proteins [170,171], iron storage/transport proteins such as ferritin (L/H subunits stabilization: storage/ferroxidase mediated uptake and utilization), caeruloplasmin, iron regulatory protein 2, lactoferrin/melanotransferrin receptors or the divalent metal transporter-1 [172,173]. Further, the accumulation of iron could be heightened by HO-1 which is significantly expressed in SNc dopaminergic neurons, the nigral neuropil, reactive astrocytes and Lewy bodies [174].
To summarize, the dynamics of PD pathogenesis is believed to evolve in part from mitochondrial energy failure, and through a series of events—DA oxidation can perpetuate a cyclical generation of DAergic toxins and precipitate high levels of free iron released throughout the basal ganglia. These events initiate a forward cycle of self-perpetuated DA oxidation, loss of NM and Fe2+ mediated damage which can indirectly fuel this loop by additional damage to the 26S proteasome prompting accelerated α-synuclein protein aggregation [175] or production of OH radicals which can oxidize lipids/proteins and DNA [8]. This degenerative cascade could be worsened by genetic mutations in iron transport proteins such as divalent metal transporter 1 (DMT1/natural resistance associated macrophage protein 2/solute carrier family 11, member 2) as noted in the SN of PD patients [176].
In Section 3, we discuss nutraceuticals that could impact each of these processes, in particular focusing on the important role that iron may play in PD pathology [177–179], the removal of which with iron chelators (i.e., EGCG, VK-28, clioquincol) is protective in a number of experimental models [40,178,179]. We also discuss the importance of using nutrient based combinations that contain chelators as just one module, since equally destructive forces are contributed by energy compromise, DA oxidation, concentration of free Fe2+ and the subsequent down stream metal catalyzed aggregation of insoluble proteins [180].
2.3.3. Enzymatic DA Oxidation by MAO-DA Aldehydes & H2O2
The third major route for oxidation of DA is through routine deamination by MAO A or B. MAO activity increases with the natural process of aging and can yield toxic products such as hydrogen peroxide (H2O2), ammonia, aldehydes, reactive oxygen species [180–182], 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde. The latter two have been reported to condense with H2O2 to form neurotoxic OH radicals [183,184]. And, in catecholamine neurons, DA can directly react with H2O2 leading to formation of 6-OHDA (neurotoxin) or further condense with acetaldehyde to produce toxic endogenous precursors such as 1,2,3,4-tetrahydroisoquinoline, 1,2,3,4-tetrahydro-β-carboline and R-salsolinol which are then subject to methylation [185–190] by either nicotinamide/salsolinol or phenylethanolamine N-methyltransferases forming toxic N-methylated pyridines with structure similar to MPTP/MPP+ [191,186,187,191].
2.4. Excitotoxicity
Mitochondrial energy dysfunction not only leads to collapse of DAergic function, but also instability of neurons to maintain voltage at the plasma membrane. Depolarization can cause over-activation of NMDA receptors throughout the brain, where glycine binds to NR1 and glutamate to the NR2 initiating fast inward Ca2+ currents to the cytoplasm. The general theory of excitotoxicity has remained consistent throughout the years and has been described to involve depolarization of the plasma membrane creating excitability in part due to (1) release of Mg+ as a voltage dependent N-methyl-d-aspartate (NMDA) block at presynaptic receptors (2) greater susceptibility to excitatory postsynaptic inward Ca2+ currents in response to glutamate activation on ionotropic NMDA/AMPA/kainate receptors and (3) a loss of inhibitory GABA metabotropic-inward ion currents upon receptor activation.
Mitochondrial toxins such as rotenone can worsen the heightened amplitude of inward ionic currents, effects known to be are reversible by addition of ATP [192]. In terms of circuitry, a deficit of magnesium (Mg) or ATP can lead to failed regulatory control of intracellular Ca2+ systems through changes not only at the NMDA receptor but also intracellularly through influences on inositol 1,4,5-trisphosphate and ryanodine receptors [193,194]. In vivo, studies show that dietary deficiency of Mg lowers NMDA receptor activation threshold and correlates to the overexcitability of glutaminergic neurons [195]. In Section 3, we discuss the importance of dietary Mg, in this and many other processes involved with PD pathology.
In PD, the over-excitability of the NMDA receptor may contribute to neurodegeneration because Ca2+ activation of neuronal nNOS can lead to nitrosative stress—a known primary elemental monomer modification leading to toxic mis-folded and aggregated proteins [196,197]. In reciprocal fashion, the accumulation of α-synuclein can stimulate nNOS, caspase-3 and initiate poly(ADP-ribose) polymerase (PARP-1) cleavage, all events which contribute toward neurotoxicity [198]. The toxic effects of α-synuclein on activation of nNOS are corroborated by studies that demonstrate that effects are blocked in the presence of NMDA receptor antagonists such as MK-801 and APV [199].
In addition, mitochondrial toxicity (i.e., MPTP) also leads to accumulation of glutamate in the SNc to an extent parallel to degenerative lesion [200]. The rise in glutamate stimulates increase influx of Ca2+ calpain activation in the cytosolic compartment, and these toxic effects are reversed by administration of NMDA antagonists, calpain inhibitors or antioxidants [201–203]. While the role of the NMDA receptor as it relates to PD is continually debated, it is noteworthy to mention that there is a very delicate balance between preventing over-activation or under-activation of glutaminergic receptors. The function of glutamate in neurotransmission is required for synaptic plasticity. And, as such, some studies also show that NMDA agonists such as D-cycloserine are protective against MPTP induced DAergic degeneration and microglial activation in the brain [204]. So clearly, this topic is very complex.
2.5. Inflammation
Both dying neurons and aggregated α-synuclein can trigger local gliosis, microglial activation, T cell infiltration and elevated expression/release of immunological participants [205]. These include major histocompatibility antigens, adhesion molecules, COX-2, IL-1b, IL-2, IL-4, IL-6, TNF-alpha, prostaglandins, glutamate, ROS, iNOS, MPO, NO and O2 − the latter two of which can react forming the neurotoxic molecule ONOO− [205–215]. Many of the inflammatory indicators are found in post-mortem tissue obtained form PD patients, particularly in regions of the SNc, striatum, LC and spinal fluid [216]. Major regulators of this response involve tyrosine kinase, phosphatidylinositiol 3-kinase (PI3K)/Akt, and the mitogen activated protein kinase (MAPK) signaling pathways such as c-Jun NH2-terminal Kinase (JNK), extracellular signal-regulated kinases (ERK) ½ p38 MAPK [46,217–222]. MAPK’s are evoked by cytokines or inflammatory stimuli, regulated by protein kinase A/cAMP and ultimately direct gene transcription by phosphorylating nuclear factor-kappa B (NF-κB) [223–226]. PET imaging using PK11195, [1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide] show active microglia occurring around neurodegenerative lesions in idiopathic PD patients vs. controls [227].
The use of anti-inflammatory agents may attenuate DAergic damage and antagonize global effects through targeting a number of signaling routes such as MAPKs, NF-kappaB activation/nuclear translocation or its association with the CREB-binding protein, IkappaB kinase (IKK), activating protein-1 (AP-1) and/or preventing IkappaB degradation or phosphorylation of c-jun N-terminal kinase (JNK) [228–231]. Substances that can inhibit any one of these mechanistic controls should block pro-inflammatory processes and antagonize the formation of iNOS, COX-2, PGE (2) or HO-1, thereby preventing DAergic loss induced by MPTP [232–237]. The co-expression of iNOS and COX-2 could be detrimental because the product formed by NO and O2 − (ONOO−) plays a highly relevant role in the pathological processes involved with PD. Removal of one or both of O2 − and NO/ONOO can prevent the deleterious effects of PD model toxins, as reported in transgenic mice deficient in iNOS, nNOS, NADPH oxidase or with overexpression of Mn/SOD conferring resistance to the toxicological effects of MPTP or intrastriatal injection of 6-OHDA [238–243]. Likewise, administration of specific nNOS/iNOS inhibitors or SOD mimetics can protect against the neurotoxicity of MPTP [33,42,244–246]. In the next section, we review nutraceuticals with multi-capabilities on anti-inflammatory signaling processes.
3. Nutraceuticals
3.1. Energy—Biochemistry and Metabolism
Considering the cascade of degeneration in PD as it involves mutations affecting protein degradation and folding, inadequate energy production in the SNc, DAergic malfunction, degenerative oxidative damage, excitotoxicity and inflammation, the question remains if any nutraceuticals or dietary practices as a lifetime habit can prevent or block one or more of these pathways and as a result slow the progression of PD. The next section provides a review of previous studies and future directives based on mechanisms as discussed above.
3.1.1. Pyruvic Acid
To overcome the loss of ATP, due to single or multiple hits directed at the mitochondria within neurons of the SNc, the first question to arise is to elucidate if it is possible to enhance anaerobic capability of the human brain, when physiological control over glucose concentrations are highly regulated. In order to optimize anaerobic glycolysis within the brain, dietary compounds must pass through the blood brain barrier (BBB) and likely work to promote key glycolytic regulators of substrate level phosphorylation including phosphoglycerate kinase, pyruvate kinase or lactic acid dehydrogenase (LDH), which would propel production of ATP [74,247,248]. Nutrient offsets associated with these enzymes are clearly altered during CNS neurological injury as evidenced by significant elevation in the ratios of lactate/pyruvate, NAD+/NADH and NADP+/NADPH [249]. These nutrient offsets suggest “metabolic stress” and accelerated use of nicotinamide reducing equivalents to drive survival processes to produce ATP through anaerobic shifts, in particular ischemia. The anaerobic process is fueled by the metabolite pyruvate (PY), which is a substrate for the LDH enzyme. Our investigations of the molecule PY, have indicated it to be the most powerful antioxidant of the glycolytic metabolites, also having capability of protect neuroblastoma against MPP+/6-OHDA and H2O2 toxicities in vitro [247–250]. It is possible that oral administration of PY could be capable of entering the brain, serving as a substrate for glycolysis, the Krebs cycle and the GABA shunt [251] and for this reason its use has been effective in preventing neurological damage associated with ischemic stroke [252]. Recently, other attributes of PY include an ability to block NMDA excitotoxicity in hippocampal neurons [253]. Although future research will be required to corroborate this, it would seem logical to combine oral administration of pyruvate, w/Mg (a required cofactor for pyruvate kinase) and niacin (precursor of reducing equivalents) which could help to restore nutrient offsets that occur due to accelerated anaerobic glycolysis in the CNS, when oxygen or mitochondrial function is insufficient.
3.1.2. Niacin
Furthermore, a deficiency of niacin is known to increase the risk for DAergic neurons to degenerate [191]. Likewise, the toxicity of MPTP is associated with a depletion of niacin, likely due to high demand for NAD+ in several biochemical processes including glycolysis and apoptotic over activation of PARP-1 [254–256]. The administration of niacin has shown protective against MPTP induced SNc cell loss and striatal DA depletion in vivo, effects which are possibly due to preventing drop in ATP by fueling glycolysis and preventing PARP-I NAD+ depletion [257–259]. Further upstream, PARP-1 is under regulatory control of tumor suppressor protein p53, a transcription factor that controls programmed cell death and cell cycle arrest. For this reason, it is not surprising that administration of either niacin, PARP-1/p53 inhibitors or PARP-1 knockout mice all show a resistance to MPTP mediated DAergic toxicity [256,257,260–262]. Therefore, the use of niacin as a therapeutic agent could be explored further, given its vast biochemical benefits including additional contribution to the pentose phosphate pathway, which regulates endogenous removal of H2O2 (a major contributing factor to PD pathology) through the GSH-Px system [191]. And, in our previous work, we have also found NADH to be a powerful antioxidant, alone capable of protecting against peroxide induced toxicity in neuroblastoma [247].
With regard to niacin, it is important to make note that a bit of controversy surrounds its use particularly as it relates to PD. Concern has been expressed that its administration could lead to synthesis of endogenous N-methylated nicotinamide, a compound with structural similarity to MPP+ [263]. Nicotinamide N-methyltransferase (NNMT) is the enzyme that can readily convert pyridines to toxic substances very similar to the PD toxic metabolite MPP+ [191]. While future research will be required to investigate these concerns, it may be possible to combine administration of niacin with natural compounds known to down regulate NNMT such as plant derived isoquinoline alkaloids, caffeine ± precursors (i.e., xanthosine—green tea or cocoa tea), which reportedly compete for methyl groups otherwise donated by s-adenosyl-l-methionine to drive NNMT enzyme activity [264–266]. The positive effects of caffeine are consistently reported as both administration of caffeine in animal models is therapeutic against MPTP and most importantly human epidemiological studies show that coffee consumption is associated with a decreased risk for developing PD [267,268]. As a side note, magnesium may also be a downregulator of NNMT [269].
3.1.3. Magnesium
Dietary magnesium (Mg) has a vast role in integrated human metabolism and is critically involved with production/utilization of ATP in the human brain. A number of studies suggest that a dietary deficiency of Mg is associated with greater loss of DAergic neurons [270]. And. low Mg brain tissue concentrations are evident in human PD patients [271]. With regards to the review on the pathology of PD above, Mg plays an indispensable role in proper DA uptake and vesicular storage and transport [272]. Heightened levels of Mg can attenuate effects of Ca2+ overload [273,274], augment the function of VMAT2 for sequestration of DA and provide a voltage dependent non-competitive block of the NMDA receptor otherwise responsible for excitability of neurons [199,275,276]. Ample Mg+ in the diet could be critical for PD patients, because of its diversity in energy related functions, energy storage processes (phosphocreatine), and its ability to thwart Ca2+ mediated neurotoxicity [276,277]. Mg also plays a critical functional role in activation of CuZn-SOD, and could thereby attenuate formation of ONOO−, involved with α-synuclein aggregation [278]. As a note, oral administration of Mg could benefit when co-administered with vitamin B6 and vitamin D. both which assist to maximize its adsorption and utilization.
3.1.4. B Vitamins and Regulation of Physiological Homocysteine
Another role for vitamin B6 pertaining to the pathology of PD is its conjunction with vitamin B12 and folate which regulate homocysteine by aiding its breakdown to methionine and tetrahydrofolate. These effects may attenuate neurotoxicity associated with hyperhomocysteinemia—a condition that is not only associated with PD pathology, but also the toxicity of MPTP in experimental models and as a side-effect of L-DOPA [279–281]. High levels of homocysteine could greater the severity of PD because it mediates toxicity by acting on NMDA receptors to precipitate oxidative stress, Ca2+ overload and apoptosis [282]. Vitamin B6 could also help to antagonize the hyperhomocysteinemic effects of nicotinamide via enhanced methylation [279]. Folate is another critical nutrient, where deficiencies in human PD patients have been observed in association with greater levels of plasma homocysteine [283,284]. In experimental models, the effects of hyperhomocysteinemia are known to potentiate the neurotoxic effects of MPTP [285]. For this reason, folate, B12 and vitamin B6 could be combined with a nutraceutical such as betaine and/or serine, which reduce homocysteine levels through aiding in its regulatory conversion to methionine or cysteine, respectively [286,287]. Garlic is another natural compound which can prevent the build up of homocysteine given its ability to stimulate cystathionine β-synthase and inhibit N5,N10-methylenehydrofolate reductase [288]. Further research will be required to explore the combined use of these particular B-vitamins with some of these nutraceuticals as it relates to homocysteine accumulation and neurotoxicity in human PD.
3.1.5. B Complex Vitamins, Riboflavin and Mitochondrial Disorders
There is also rationale to substantiate use of B-Complex vitamins for PD patients, due to the critical role these nutrients play in glucose metabolism and mitochondrial respiration. Vitamin B2 derivatives such as flavin adenine dinucleotide (FAD)/flavin mononucleotide (FMN) regulate aerobic mitochondrial metabolism by mediating redox reactions through the electron transport chain [74,289]. Interestingly, the use of oral riboflavin supplements in humans can reverse clinical symptoms associated with mitochondrial myopathy/pathologies (involving complex I–II), where reduction of lactate and restored mitochondrial function are associated with clinical improvements [290–294]. And, use of coenzyme Q10 (which plays a role in complex I–II function) for treatment of PD has been of considerable interest, although clinical trials have not yet confirmed therapeutic effects [295,258]. The B-Complex vitamins such as thiamin (vitamin B1), lipoic acid, biotin, vitamin B6, B12 folate, and pantothenate work together symbiotically to drive pyruvate dehydrogenase complex, gluconeogenesis and blood, glucose, oxygen delivery to the brain. The B-complex vitamins each play a unique role of equal importance but work collectively to optimize mitochondrial function, in particular when challenged with toxins such as rotenone [296]. Clinical trials for multi-vitamin supplements therefore could be considered.
3.1.6. Creatine, Chromium
Nutraceuticals that optimize ATP storage reserves may further strengthen the capacity of energy requiring systems. Known disturbances in choline/creatine have been observed in PD patients [297], and creatine supplements have been shown to protect against MPP+/MPTP, 6-OHDA and glucose deprivation [53,54,298]. However, preliminary studies in our lab have failed to show protective effects by creatine against MPTP induced DA degeneration in the mouse model, a topic under current investigation (unpublished). While creatine could be beneficial in augmenting ATP storage, chromium salts would be equally important in maintaining physiological glucose, glucose tolerance, insulin sensitivity [299] and glycemic functions [300]. Adequate chromium in the diet seems fitting given its role in optimizing systemic glucose metabolism, despite a lack of evidence to suggest chromium aberrations in cerebral spinal fluid of PD patients [301].
3.2. Plant Polyphenols—Attenuation of DA Oxidation
The use of vitamins to support energy function could further be combined with plant derived polyphenolic compounds (PDPC) that specifically target downstream toxic effects as a direct result to the loss of ATP. These include collapse of DA trafficking, DA oxidation, generation of ROS, fenton reactions, DAergic neurotoxins, loss of NM and CNS glial inflammation. A number of food-based molecules have previously been reported in the literature as being effective in antagonizing specific events within these processes.
3.2.1. Tyrosinase Inhibitors
As stated previously, the initial oxidation of DA to DA-quinone, or from DA quinone to its toxic metabolites are believed to contribute toward DAergic degeneration. Although future research will be required to substantiate this, these processes could be blocked by nutraceuticals such as polyphenolic inhibitors of tyrosinase, COX, lipoxygenase, PLA2, xanthine oxidase or antioxidants/metal chelators.
The first to review is tyrosinase/polyphenol oxidase (PPO) which is a copper requiring metalloenzyme that catalyzes formation of o-quinones. A heightened enzyme activity of tyrosinase could be associated with elevated risk for PD [302] and skin hyperpigmentation disorders [303] both which involve heightened oxidation of L-DOPA to form dopachrome [304,305]. These same processes are often researched in the field of food chemistry, due to food browning reactions occurring through PPO enzymes in vegetables such as potato or mushroom. Creatively, it has been proposed that such as model could serve practical for the investigation or screening of nutraceuticals against DA oxidation processes as it relates to PD [306]. Future research could be done to consider analysis of established nutraceuticals known to inhibit tyrosinase, some of which include the following:
3.2.2. COX Inhibitors
Natural inhibitors of COX could also block the initial step of enzymatic DA oxidation to DA quinone through PGH2 synthase. A review of the literature shows a number of promising plant derived polyphenolic compounds (PDPCs) as effective COX inhibitors such as:
3.2.3. Lipoxygenase Inhibitors
PDPC’s that may be able to block the initial step of enzymatic DA oxidation to DA-quinone through inhibition of lipoxygenase (5-LOX, 12-LOX) and include the following:
3.2.4. Phospholipase A2 Inhibitors
While PLA2 inhibitors attenuate DA oxidation reactions, they may serve dual function in PD pathology because they also block formation of arachidonic acid as a substrate for prostaglandins. PLA2 inhibitors could be combined with administration of omega-3 fatty acids (i.e., canola/fish oil), thereby reducing PGE2 (a pro-inflammatory prostaglandin specifically associated with PD pathology) [377]. Co-administration of vitamin E may enhance absorption of omega-3 fatty acids and prevent fatty acid oxidation. Future research could consider analysis of plant-derived compounds that are known to inhibit PLA2 in experimental models of SNc DAergic damage, some of which are known to include:
3.2.5. Xanthine Oxidase Inhibitors
The initial step of enzymatic DA oxidation to DA quinone could be attenuated by xanthine oxidase inhibitors, some of which are known to include the following:
3.2.6. Xanthine Oxidase and Superoxide Scavengers
Combined xanthine oxidase/superoxide scavengers may reduce oxidative stress, prevent formation of ONOO and attenuate the degenerative process, some of which are known to include:
3.3. Histidine, Quercetin and Zinc
Other polyphenolic compounds that may block the initial step of enzymatic DA oxidation include substances which down regulate DT diaphorase or mono-oxygenases such as EGCG [432], flavones [433] baicalin, oroxylin-A glucoronides [434], quercetin [435] or histidine [436]. While we mention the protective properties of EGCG and quercetin on PD related processes throughout this review, noted effects of histidine may also include its ability to augment the uptake and transport of zinc into the brain, where zinc can counteract the pro-oxidant effects of iron [437], ischemia-reperfusion [438,439] or mitochondrial toxins such as MPP+ [440]. See Section 3.8.
3.4. N Acetyl Cysteine
Thiol based compounds are believed to help slow non-enzymatic autoxidation of DA in the presence of ROS and metals (Fe2+, Cu2+, and Mn2+) [149,150]. Autoxidation of DA to 6-OHDA (a potent neurotoxin) and O2 − can be lethal in the presence of NO, forming ONOO−. Peroxynitrite can then re-oxidize DA and deplete available reduced glutathione and ascorbate [129,151]. Possible dietary counter intervention could include thiol antioxidants such as NAC which in experimental models blocks the autoxidation of DA, prevents MPTP induced toxicity in mice [57,153] attenuates pathological effects of 6-OHDA, ONOO− and blocks the formation of DA o-semiquinone neurotoxic radicals [154].
3.5. Hydrogen Peroxide Scavengers
The third route of DA oxidation is through deamination by MAO A or B which yields H2O2, ammonia [180–182], 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde. The latter two condense with H2O2 to form OH radicals [183,184] and DA reacts with H2O2 leading to form 6-OHDA or condenses with acetaldehyde to produce toxic precursors subject to methylation [185–190]. Due to the importance of MAO activity and the initial condensation reaction between catecholamines and aldehydes that create precursors subject to methylation, future research could investigate therapeutic food based compounds that work as (1) MAO inhibitors (2) compounds that potentiate aldehyde dehydrogenase such as GSH, NAD+ (3) down regulate nicotinate/phenylethanolamine N-methyltransferases such as caffeine or 4) scavenge H2O2.
Removing hydrogen peroxide generated by MAO or DA autoxidation could be very beneficial in slowing the rate of progression in PD. Hydrogen peroxide, if present in high quantities can oxidize DA to 6-OHDA, which in turn can then react with 6-OHDA to propagate OH radicals, contributing to the formation of α-synuclein-Fe aggregates and insoluble filaments [35,441]. The generation of H2O2 in DAergic neurons initiates multiple degenerative processes such as improper degradation of oxidized proteins through the ubiquitin proteasome pathway, formation of dopachrome and toxic DA quinones [132,442,443]. The role for peroxide in PD pathogenesis is evidenced by the fact that its removal via potentiation of catalase/SOD prevents injury in MPTP models of injury. Transgenic mice that over express cytosolic CuZn-SO/GSH-Px or applied administration of SOD/catalase mimetics (which both dismutase O2 −, and convert subsequent H2O2 to water) provide protection against MPTP, paraquat and 6-OHDA in vivo models of injury [33,243,444–446]. In contrast, reduction in GSH-Px/CuZn SOD (i.e., knockout mice) leaves the SNc area vulnerable to oxidative stress and MPTP injury [143,447]. For these reasons, beneficial nutritional substances could include those that upregulate endogenous glutathione peroxidase and/or catalase, such as NAC, GSH, selenium, vitamin E, NADPH and curcumin [448]. Co-administration of niacin (which provides NADPH to drive GSH-Px) along with substances that augment function of GSH-PX could provide synergy in protecting SNc neurons from oxidative stress [57,153]. Other useful nutritional substances could include those that aid in SOD such as methionine, manganese, copper, zinc and propolis [449] and H2O2 scavengers which are known to include the following:
3.6. Iron Chelators
6-OHDA generated during DA oxidation, reduces metallothione and causes release of free iron from ferritin [152,155,160]. Natural substances that antagonize 6-OHDA toxicity such as NAC, GSH, cysteine, pyruvic acid, [455] and zingerone [456] or are integral constituents of metallothioneine such as serine, lysine and cysteine [155] could be further researched. The accumulation of free iron is deleterious because it is associated with degenerating SNc neurons, surrounding glial cells and found after administration of MPTP/6-OHDA in animals [164–169]. Faulty iron homeostasis in the basal ganglia could lead to a number of oxidative reactions, the acceleration of α-synuclein protein aggregation [175] and formation of OH radicals which can damage neuronal lipid/protein and DNA [8]. It is reported that the use of iron chelators protect against MPTP and 6-OHDA models of PD toxicity [40,178,179]. A number of natural substances are capable of reducing/chelating complex iron including the following:
3.7. Heme Oxygenase Inhibitors
The accumulation of iron can also occur due to overactivity of the HO-1 enzyme, which can convert heme to free Fe2+, and carbon monoxide, this also being significantly expressed in SNc dopaminergic neurons, the nigral neuropil, surrounding reactive astrocytes and Lewy bodies [174]. Up regulation of HO-1 occurs as a natural response to oxidative stress and correlates to iron deposition in the nigral area with degenerative SNc lesions. For this reason, potentially helpful nutritional substances may include those that can inhibit HO-1 directly such as cysteine, resevatrol, vitamin C, sulfur compounds (i.e., NAC, GSH) [471], apigenin [472], quercetin and kaempferol [473].
3.8. Zinc and Selenium
While reactive iron contributes to the degeneration in SNc, the administration of zinc (Zn) and selenium (Se) could strengthen combination nutraceutical strategies [474–476]. Dietary intake of Se, Zn are required for the function/expression of endogenous antioxidant enzymes and ample amounts can attenuate iron-induced, MPTP and 6-OHDA induced DAergic degeneration [150,475,477]. Furthermore, chronic inflammation can bring about a Zn deficiency due to the use of Zn-dependent transcription factors that regulate DNA/nucleic acid synthesis in response to cytokine activation in immunocompetant cells (i.e., hypozincemia) [478,479]. A Zn deficiency can also evoke a shift in the ratio of Cu/Zn rending less than normal function of the CuZn SOD, turning it from an antioxidant to a pro-oxidant enzyme [479]. A requirement for zinc in the body could be justified with PD patients, since Zn mediates (a) downregulation of glutamate release, inhibition of NMDA/mGlu-R receptors, protection against NMDA neurotoxicity (b) renders a positive modulation on GABA release (c) stimulates endogenous antioxidant enzymes and nerve growth factors (d) inhibits nNOS, endonucleases, pro-apoptotic cascades (e) augments synaptic plasticity and (f) is known to prevent age related deterioration of learning and memory [437].
Both zinc and selenium contribute to anti-inflammatory effects through downregulation of MAPK p38, JNK and NF-κB DNA binding/AP-1 c Jun activation, where the therapeutic effects of Se also involve a rise in glutathione peroxidase/reduction of lipid peroxidation, increased glucose uptake, ATP production through glycolysis and an anti-apoptotic effects [474,480].
3.9. Anti-Inflammatory Nutraceuticals
The CNS inflammatory response is under the ultimate control of kinases such as tyrosine kinase [217], PI3K/Akt, and mitogen activated protein kinase signaling pathways such as JNK, ERK ½ p38 MAPK [46,218–222]. The topic of inflammatory is far too large for this review and therefore is summarized as follows. Briefly, MAPK’s are evoked by cytokines or inflammatory stimuli, regulated by protein kinase A/cAMP and ultimately control gene transcription by phosphorylating NF-κB which then binds to the promoter region of genes to initiate transcription for a range of pro-inflammatory proteins [223–226]. Anti-inflammatory agents can antagonize global effects through targeting a number of these signaling routes such as MAPKs, NF-κB activation/nuclear translocation or its association with the CREB-binding protein, IkappaB kinase (IKK), activating protein-1 (AP-1) and/or preventing IkappaB degradation or phosphorylation of JNK [228–231]. In brief summary, natural substances that may provide protection include those that can inactivate phosphorylated MAPK’s such as ERK ½ kinase, p38 MAPK, JNK, inhibit IkappaB kinase, IkappaB degradation, NF-κB, AP-1 activation, antagonize COX-2/PGE2/iNOS and reduce expression of TNF-alpha and other pro-inflammatory proteins in immuno-competent cells some of which are listed as follows:
Additionally, phosphodiesterase (PDE) inhibitors, in particular PDE 1 and IV through altering cAMP can downregulate iNOS [511] and protect against MPTP toxicity [512]. Food based compounds known to inhibit PDE include the following:
3.10. Toxic Protein Aggregates
In this section we briefly discuss a potential for targeted nutraceutical therapies which would prevent accumulation of α-synuclein, augment the ubiquinone-proteasome system (UPS) or inhibit mammalian target of rapamycin (mTOR) signaling to upregulate autophagy, which may in the long term slow the progression of this disease.
3.10.1. Nutraceuticals—Reduction of aggregated α-SYNUCLEIN (PARK1)
In brief, the kinetics of α-synuclein aggregation involves a number of progressive stages some of which could be altered by nutraceuticals. A higher propensity for α-synuclein aggregation can occur due to missense mutations (A30P, A53T, E46K) in human PD [524]. The general kinetics of aggregation involves three stages: (1) a protein monomer must undergo a modification; (2) modified monomers can then readily interact with each other to form small aggregates and (3) aggregates after reaching a certain size, referred to as a “nucleus”, can undergo irreversible rapid volume expansion called elongation which result in the formation of fibrils, then taking up residence as toxic entities in neurons and Lewy bodies. The initial protein modifications can occur due to phosphorylation of α-Synuclein at Ser 129 (p-Ser 129), nitration at tyrosine residues and C-terminal truncation- all of which can lead to nucleation where aggregation becomes probable [525,526]. The initial protein modification stage can also occur due to neurotoxic insults including but not limited to DA oxidative products, NO, ROS and high concentration of metals [527–529]. In turn, α-synuclein can directly initiate increased membrane ion permeability, vesicle leakage of DA and decreased mitochondrial respiration [530,531], which in turn can generate compounds that lead to α-synuclein modification. In essence, α-synuclein can lead to toxicity, and neurotoxicity can lead to α-synuclein aggregation. In this review, we have covered information on nutraceuticals that indirectly attenuate events known to evoke the initial stages of propagative α-synuclein misfolding, such as iNOS, nNOS, DA oxidative products and the enzyme pathways by which DA quinones are produced (Sections 3.1–3.9). In addition, nutraceuticals that inhibit enzymes that otherwise phosphorylate α-synuclein such as polo-like kinases (i.e., thymoquinone–black cumin) [532], casein kinase II (i.e., ellagic acid) [533,534], Gprk2GRK2/5 [535] or proteases such as calpains, calcium-dependent non-lysosomal cysteine proteases may prevent a tendency for α-synuclein to aggregate or result in truncated toxins of α-synuclein. The use of any nutri-therapy which can prevent likelihood of aggregation, should lessen cell burden of accumulated insoluble proteins which otherwise has affinity for lipids, presynaptic vesicles, membranes and can cause considerable damage to organelles including mitochondria [536].
3.10.2. (Parkin) E3 ubiquitin ligase and Proteosomal Dysfunction
Once α-synuclein aggregates are formed, a second vulnerability for continued accumulation would be improper recognition and ubiquitination of specific target proteins for degradation by the proteasome. This can occur in part due to genetic defects in parkin-E3 ubiquitin ligase or its associate SCF complex (Skp1-Cullin-F-box protein complex) [537,538]. While nutritional constituents may not be able to halt faulty processes in ubiquitination, it may be possible to optimize the function of the proteasomal complex with dietary agents.
The proteasomal complex consists of a 20S proteolytic core with two 19S regulatory caps, responsible for recognition, proteolysis, unfolding and transport of proteins into the core lumen for processing. Inhibiting the function of the proteasome with lactacystin, PSI or MG-132 can effectively mimic PD pathology including selective SNc degeneration, α-synuclein positive inclusion like granules and activation of glial cells [539,540]. Nutraceutical substances such as iron chelators can protect against the adverse effects of such proteasomal inhibitors with capability to prevent lactacystin-induced DA neurodegeneration in vivo [541]. These protective effects are likely because the proteasome can also be adversely affected or inhibited by DA oxidative metabolites, DA quinones or ROS, effects that are also blocked by antioxidants such as GSH, ascorbic acid, vitamin E, SOD or catalase [542]. In this aspect nutraceuticals could serve useful to protect against further insult to an already vulnerable proteosomal complex, not only due to mutations in parkin, but also due to lack of endogenous proteasome activator PA28 expression in the SNc, concomitant to reduced function of α-subunit of the 20S proteasome in the SNc of sporadic PD patients [543,544].
3.10.3. Nutraceuticals, Autophagy and mTOR signaling
A second degradation route for eliminating α-synuclein aggregates and malfunctional mitochondria is through the process of autophagy. The removal of depolarized damaged mitochondria is mediated through a process called mitochondrial fission which is regulated by membrane constriction through dynamin-related protein (Drp1) mitochondrial fission 1 and GTP hydrolysis [545] in preparation for clearance through autolysosomes. Notable mutations in PINK1 can adversely affect this process, by preventing both Drp1-dependent fragmentation and phosphorylation/relocation of Parkin to mitochondria where it then fails to catalyze mitochondrial ubiquitination, recruitment of ubiquitin-binding autophagic components, HDAC6 and p62, and subsequent mitochondrial clearance [546]. Together, genetic mutations in both PINK1 and Parkin lead not only to failure of mitochondria, but also a lack of mitochondrial quality control for proper degradation of mitochondria that are no longer functional. While nutritional constituents may not be able to reverse protein defects associated with function of Park and PINK1 mutations, dietary factors can largely influence and activate autophagy-lysosomal function.
Autophagy is described as the means by which cells degrade oxidized and damaged membranes, organelles and mis-folded proteins. This process is initiated by formation of a phagophore which expands and engulfs portions of the cytoplasm then forming a autophagosome [547]. The initiation stages of autophagosome formation is under control of signaling by class III phosphoinositide 3-kinase and Atg 6 (Beclin-1), which regulates phosphorylation of microtubule-associated protein 1 light chain 3LC3 [548]. These phosphorylated LC3 marked vesicles are then trafficked along microtubules in a dynein reliant fashion and eventually fuse with lysosomes (autolysosomes), where contents are degraded by acidic lysosomal hydrolases. Lysosomes can also reach out on their own in a process called microautophagy where they directly engulf cytoplasm by invagination or septation. And, once inside the lysosome, cathepsin D becomes the main lysosomal enzyme involved in the degradation of α-synuclein [549].
The process of UPS and the autophagy-lysosomal systems are under direct control of mammalian target of rapamycin (mTOR) signaling. Stimuli that lead to upregulation of mTOR serve to block autophagy-lysosomal function and its contribution toward accumulated oxidized and damaged organelles/proteins. Signals that upregulate mTOR include those registering as high nutrient energy status signals, such as glucose, insulin, a high ratio of ATP/AMP ratio, leucine, oxidative stress [550], arginine [551] and high levels of amino acids [552]. The rise in mTOR and reduction in the autophagy-lysosome pathway can be chemically induced by 3-methyladenine or chloroquine, effects which lead to accumulation of Ser-129-phosphorylated α-synuclein [553]. This is opposite to the effects of rapamycin which through inhibition of mTOR activate clearance of aggregate-prone proteins, including α-synuclein as well as faulty mitochondria and prevent the toxic effects of proteosomal inhibitors on DAergic systems [554]. A number of substances in the diet are known to upregulate autophagy lysosomal function by downregulation of mTOR, some of which include resveratrol, spermidine, curcumin, piperine, caffeine, epigallocatechin gallate, garlic, S-allylcysteine [555–557], anthocyanins [558], selenium [559], eicosapentaenoic acid and lycopene [560]. Also, it is likely that nutraceuticals that could selectively inhibit IMPase, IP3, adenylate cyclase [561] or Akt signaling may downregulate mTOR and induce autophagosomal clearance [562].
4. Conclusion
In conclusion, this review provides information on nutritional biochemistry as it relates to pathological processes inherent to PD. PD pathology involves both regional and systemic nutrient offsets that are largely related to heightened anaerobic glycolysis, homocysteine metabolism, faulty aerobic energy metabolism, metabolic stress, iron deposition and catecholamine mediated oxidative stress. These offsets could be aggravated by blood -tissue nutrient deficiencies as commonly reported in human PD patients or the process of aging itself, both which could exacerbate protein mis-folding/aggregation, disruption of proteasomal processes or losses in DAergic neurotransmission. Future research will be needed to investigate a strategic means to employ combined nutraceuticals that work effectively and collectively to alter metabolism or pathological processes in such a way as to slow the progression of PD in humans. Any therapeutic strategy that can effectively do so, will afford extended quality of life to human PD patients.
Acknowledgment
This research was supported by a grant from NIH NCRR RCMI program (G12RR 03020).
Abbreviations
| 6-OHDA | 6 Hydroxydopamine |
| AADC | Aromatic amino acid decarboxylase |
| AMPA | α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate |
| AP-1 | Activator protein 1 |
| ATP | Adenosine triphosphate |
| BBB | Blood brain barrier |
| CBF | Cerebral blood flow |
| CMRG | Cerebral metabolic rate of glucose |
| CMRG | Cerebral metabolic rate of glucose |
| COMT | Catechol-O-methyltransferase |
| COX | Cyclooxygenase |
| CSF | Cerebral spinal fluid |
| DA | Dopamine |
| DAergic | Dopaminergic |
| DAT | Dopamine transporter |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| DTBZ | [11C]-dihydrotetrabenazine |
| ERK | Extracellular signal-regulated kinases |
| FAD | Flavin adenine dinucleotide |
| FD | [18F]-fluoro-l dopa |
| FDG | [18F]-Fluoro-deoxyglucose |
| FMN | Flavin mononucleotide |
| GABA | Γ-Aminobutyric acid |
| GSHPx | Glutathione peroxidase |
| GSH | Reduced Glutathione |
| H2O2 | Hydrogen Peroxide |
| HO-1 | Heme oxygenase 1 |
| HVA | Homovanillic acid |
| IKK | I kappaB kinase |
| IL | Interleukin |
| INOS | Inducible NOS |
| mRNA | Messenger ribonucleic acid |
| mTOR | Mammalian target of rapamycin |
| NA | Nucleus accumbens, |
| NAC | N acetyl L cysteine |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Nicotinamide adenine dinucleotide reduced |
| NADPH | Nicotinamide adenine dinucleotide phosphate reduced |
| NF-κB | Nuclear factor-kappa B |
| NM | Neuromelanin |
| NMDA | N-methyl-D-aspartate |
| NNMT | Nicotinamide N-methyl transferase |
| NOS | Nitric oxide synthase |
| NT | Neurotransmitter |
| O2− | Superoxide |
| OXPHOS | Oxidative Phosphorylation |
| PARP-1 | Poly [ADP-ribose] polymerase 1 |
| PD | Parkinson’s disease |
| PDE | Phosphodiesterase |
| PDPC | Plant derived polyphenolic compounds |
| PET | Positron emission tomography |
| PGE2 | Prostaglandin E2 |
| PGH2 | Prostaglandin H2 synthase |
| PI3K | Phosphoinositide 3 kinase |
| PINK-1 | PTEN-induced putative kinase 1 |
| PK | Pyruvate kinase |
| PLA2 | Phospholipase A2 |
| PPO | Polyphenol Oxidase |
| PY | Pyruvate |
| ROS | Reactive Oxygen Species |
| Se | Selenium |
| SLP | Substrate level phosphorylation |
| JNK | c-jun N-terminal kinase |
| LC | Locus coeruleus |
| LDH | Lactate Dehydrogenase |
| l-DOPA | l-3,4-dihydroxyphenylalanine |
| LOX | Lipoxygenase |
| MAO | Monoamine oxidase |
| MAPK | Mitogen-activated protein kinase |
| MP | 11C-methylphenidate |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| SN | Substantia nigra |
| SNc | Substantia nigra pars compacta |
| SNCA | α-synuclein |
| SOD | Superoxide dismutase |
| SPECT | Single photon emission computed tomography |
| TH | Tyrosine hydroxylase |
| TNF | Tumor necrosis factor |
| UPS | Ubiquitin-proteasome system |
| VMAT2 | Type-2-vesicular monoamine transporter |
| Zn | Zinc |
References
- Burch, D; Sheerin, F. Parkinson’s disease. Lancet 2005, 365, 622–627. [Google Scholar]
- Lin, TK; Liou, CW; Chen, SD; Chuang, YC; Tiao, MM; Wang, PW; Chen, JB; Chuang, JH. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung Med. J 2009, 32, 589–599. [Google Scholar]
- Dagda, RK; Chu, CT. Mitochondrial quality control: insights on how Parkinson’s disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr 2009, 41, 473–479. [Google Scholar]
- Nishioka, K; Vilariño-Güell, C; Cobb, SA; Kachergus, JM; Ross, OA; Hentati, E; Hentati, F; Farrer, MJ. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism Relat. Disord 2010, 10, 686–687. [Google Scholar]
- Levy, OA; Malagelada, C; Greene, LA. Cell death pathways in Parkinson’s disease: Proximal triggers, distal effectors, and final steps. Apoptosis 2009, 14, 478–500. [Google Scholar]
- Nagatsu, T. Parkinson’s disease: changes in apoptosis-related factors suggesting possible gene therapy. J. Neural. Transm 2002, 109, 731–745. [Google Scholar]
- Tofaris, GK; Spillantini, MG. Alpha-synuclein dysfunction in Lewy body diseases. Mov. Disord 2005, 20, S37–S44. [Google Scholar]
- Bharath, S; Hsu, M; Kaur, D; Rajagopalan, S; Andersen, JK. Glutathione, iron and Parkinson’s disease. Biochem. Pharmacol 2002, 64, 1037–1048. [Google Scholar]
- Johnson, MD; Yu, LR; Conrads, TP; Kinoshita, Y; Uo, T; McBee, JK; Veenstra, TD; Morrison, RS. The proteomics of neurodegeneration. Am. J Pharmacogenomics 2005, 5, 259–270. [Google Scholar]
- Hald, A; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp. Neurol 2005, 193, 279–290. [Google Scholar]
- Sato, S; Mizuno, Y; Hattori, N. Urinary 8-hydroxydeoxyguanosine levels as a biomarker for progression of Parkinson disease. Neurology 2005, 64, 1081–1083. [Google Scholar]
- Pennathur, S; Jackson-Lewis, V; Przedborski, S; Heinecke, JW. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o′-dityrosine in brain tissue of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson’s disease. J. Biol. Chem 1999, 274, 34621–34628. [Google Scholar]
- Takeuchi, H; Mizuno, T; Zhang, G; Wang, J; Kawanokuchi, J; Kuno, R; Suzumura, A. Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J. Biol. Chem 2005, 280, 10444–10454. [Google Scholar]
- Cheng, HC; Ulane, CM; Burke, RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol 2010, 67, 715–725. [Google Scholar]
- Fahn, S; Sulzer, D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx 2004, 1, 139–154. [Google Scholar]
- Bertram, L; Tanzi, RE. The genetic epidemiology of neurodegenerative disease. J. Clin. Invest 2005, 115, 1449–1457. [Google Scholar]
- Hyun, DH; Lee, M; Halliwell, B; Jenner, P. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J. Neurosci. Res 2005, 82, 232–244. [Google Scholar]
- Mortiboys, H; Johansen, KK; Aasly, JO; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar]
- Henchcliffe, C; Beal, MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol 2008, 4, 600–609. [Google Scholar]
- Arvanitakis, Z; Wilson, RS; Schneider, JA; Bienias, JL; Evans, DA; Bennett, DA. Diabetes mellitus and progression of rigidity and gait disturbance in older persons. Neurology 2004, 63, 996–1001. [Google Scholar]
- Klein, RC; de Jong, BM; de Vries, JJ; Leenders, KL. Direct comparison between regional cerebral metabolism in progressive supranuclear palsy and Parkinson’s disease. Mov. Disord 2005, 20, 1021–1030. [Google Scholar]
- Lesage, S; Brice, A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet 2009, 18, R48–R59. [Google Scholar]
- Tanner, CM; Ross, GW; Jewell, SA; Hauser, RA; Jankovic, J; Factor, SA; Bressman, S; Deligtisch, A; Marras, C; Lyons, KE; Bhudhikanok, GS; Roucoux, DF; Meng, C; Abbott, RD; Langston, JW. Occupation and risk of parkinsonism: a multicenter case-control study. Arch. Neurol 2009, 66, 1106–1113. [Google Scholar]
- Nguyen, N; Pradel, V; Micallef, J; Montastruc, JL; Blin, O. Drug-induced Parkinson syndromes. Therapie 2004, 59, 105–112. [Google Scholar]
- Sanyal, J; Chakraborty, DP; Sarkar, B; Banerjee, TK; Mukherjee, SC; Ray, BC; Rao, VR. Environmental and familial risk factors of Parkinsons disease: case-control study. Can. J. Neurol. Sci 2010, 37, 637–642. [Google Scholar]
- Allam, MF; Del Castillo, AS; Navajas, RF. Parkinson’s disease risk factors: genetic, environmental, or both? Neurol. Res 2005, 27, 206–208. [Google Scholar]
- Logroscino, G. The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environ. Health Perspect 2005, 113, 1234–1238. [Google Scholar]
- Pavese, N; Brooks, DJ. Imaging neurodegeneration in Parkinson’s disease. Biochim. Biophys Acta 2009, 1792, 722–729. [Google Scholar]
- Thobois, S; Guillouet, S; Broussolle, E. Contributions of PET and SPECT to the understanding of the pathophysiology of Parkinson’s disease. Neurophysiol. Clin 2001, 31, 321–340. [Google Scholar]
- Au, WL; Adams, JR; Troiano, AR; Stoessl, AJ. Parkinson’s disease: in vivo assessment of disease progression using positron emission tomography. Brain Res. Mol. Brain Res 2005, 134, 24–33. [Google Scholar]
- Galvan, A; Wichmann, T. Pathophysiology of parkinsonism. Clin. Neurophysiol 2008, 119, 1459–1474. [Google Scholar]
- Pal, PK; Netravathi, M. Management of neurodegenerative disorders: Parkinson’s disease and Alzheimer’s disease. J. Indian Med. Assoc 2005, 103, 168–170. [Google Scholar]
- Samai, M; Sharpe, MA; Gard, PR; Chatterjee, PK. Comparison of the effects of the superoxide dismutase mimetics EUK-134 and tempol on paraquat-induced nephrotoxicity. Free Radic. Biol. Med 2007, 43, 528–534. [Google Scholar]
- Weinreb, O; Amit, T; Bar-Am, O; Youdim, MB. Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol 2010, 92, 330–344. [Google Scholar]
- Lew, MF; Hauser, RA; Hurtig, HI; Ondo, WG; Wojcieszek, J; Goren, T; Fitzer-Attas, CJ. Long-term efficacy of rasagiline in early Parkinson’s disease. Int. J. Neurosci 2010, 120, 404–408. [Google Scholar]
- Weinreb, O; Amit, T; Bar-Am, O; Chillag-Talmor, O; Youdim, MB. Novel neuroprotective mechanism of action of rasagiline is associated with its propargyl moiety: interaction of Bcl-2 family members with PKC pathway. Ann. N. Y. Acad. Sci 2005, 1053, 348–355. [Google Scholar]
- Zheng, H; Gal, S; Weiner, LM; Bar-Am, O; Warshawsky, A; Fridkin, M; Youdim, MB. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: in vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem 2005, 95, 68–78. [Google Scholar]
- Youdim, MB; Fridkin, M; Zheng, H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech. Ageing Dev 2005, 126, 317–326. [Google Scholar]
- Mandel, SA; Avramovich-Tirosh, Y; Reznichenko, L; Zheng, H; Weinreb, O; Amit, T; Youdim, MB. Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 2005, 14, 46–60. [Google Scholar]
- Mandel, S; Maor, G; Youdim, MB. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J. Mol. Neurosci 2004, 24, 401–416. [Google Scholar]
- Cleren, C; Calingasan, NY; Chen, J; Beal, MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem 2005, 94, 995–1004. [Google Scholar]
- Klivenyi, P; Andreassen, OA; Ferrante, RJ; Lancelot, E; Reif, D; Beal, MF. Inhibition of neuronal nitric oxide synthase protects against MPTP toxicity. Neuroreport 2000, 11, 1265–1268. [Google Scholar]
- Watanabe, H; Muramatsu, Y; Kurosaki, R; Michimata, M; Matsubara, M; Imai, Y; Araki, T. Protective effects of neuronal nitric oxide synthase inhibitor in mouse brain against MPTP neurotoxicity: an immunohistological study. Eur. Neuropsychopharmacol 2004, 14, 93–104. [Google Scholar]
- Wang, W; Shi, L; Xie, Y; Ma, C; Li, W; Su, X; Huang, S; Chen, R; Zhu, Z; Mao, Z; Han, Y; Li, M. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res 2004, 48, 195–202. [Google Scholar]
- Teismann, P; Tieu, K; Choi, DK; Wu, DC; Naini, A; Hunot, S; Vila, M; Jackson-Lewis, V; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci USA 2003, 100, 5473–5478. [Google Scholar]
- Kuan, CY; Burke, RE. Targeting the JNK signaling pathway for stroke and Parkinson’s diseases therapy. Curr. Drug Targets CNS Neurol. Disord 2005, 4, 63–67. [Google Scholar]
- Silva, RM; Kuan, CY; Rakic, P; Burke, RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov. Disord 2005, 20, 653–664. [Google Scholar]
- Testa, CM; Sherer, TB; Greenamyre, JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res. Mol. Brain Res 2005, 134, 109–118. [Google Scholar]
- Virmani, A; Gaetani, F; Binienda, Z. Effects of metabolic modifiers such as carnitines, coenzyme Q10, and PUFAs against different forms of neurotoxic insults: metabolic inhibitors, MPTP, and methamphetamine. Ann. N. Y. Acad. Sci 2005, 1053, 183–191. [Google Scholar]
- Bhat, V; Weiner, WJ. Parkinson’s disease. Diagnosis and the initiation of therapy. Minerva Med 2005, 96, 145–154. [Google Scholar]
- Shults, CW. Therapeutic role of coenzyme Q(10) in Parkinson’s disease. Pharmacol. Ther 2005, 107, 120–130. [Google Scholar]
- Etminan, M; Gill, SS; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol 2005, 4, 362–365. [Google Scholar]
- Andres, RH; Huber, AW; Schlattner, U; Pérez-Bouza, A; Krebs, SH; Seiler, RW; Wallimann, T; Widmer, HR. Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience 2005, 133, 701–713. [Google Scholar]
- Klivenyi, P; Gardian, G; Calingasan, NY; Yang, L; Beal, MF. Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Mol. Neurosci 2003, 21, 191–198. [Google Scholar]
- Borah, A; Mohanakumar, KP. Melatonin inhibits 6-hydroxydopamine production in the brain to protect against experimental Parkinsonism in rodents. J. Pineal Res 2009, 47, 293–300. [Google Scholar]
- Bi, J; Wang, XB; Chen, L; Hao, S; An, LJ; Jiang, B; Guo, L. Catalpol protects mesencephalic neurons against MPTP induced neurotoxicity via attenuation of mitochondrial dysfunction and MAO-B activity. Toxicol In Vitro 2008, 22, 1883–1889. [Google Scholar]
- Bahat-Stroomza, M; Gilgun-Sherki, Y; Offen, D; Panet, H; Saada, A; Krool-Galron, N; Barzilai, A; Atlas, D; Melamed, E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur. J. Neurosci 2005, 21, 637–646. [Google Scholar]
- Levy, YS; Gilgun-Sherki, Y; Melamed, E; Offen, D. Therapeutic potential of neurotrophic factors in neurodegenerative diseases. BioDrugs 2005, 19, 97–127. [Google Scholar]
- Slevin, JT; Gerhardt, GA; Smith, CD; Gash, DM; Kryscio, R; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg 2005, 102, 216–222. [Google Scholar]
- D’Astous, M; Morissette, M; Tanguay, B; Callier, S; Di Paolo, T. Dehydroepiandrosterone (DHEA) such as 17beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse 2003, 47, 10–14. [Google Scholar]
- D’Astous, M; Morissette, M; Di Paolo, T. Effect of estrogen receptor agonists treatment in MPTP mice: evidence of neuroprotection by an ER alpha agonist. Neuropharmacology 2004, 47, 1180–1188. [Google Scholar]
- Morelli, M; Carta, AR; Kachroo, A; Schwarzschild, MA. Pathophysiological roles for purines: adenosine, caffeine and urate. Prog. Brain Res 2010, 183, 183–208. [Google Scholar]
- Ikeda, K; Kurokawa, M; Aoyama, S; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem 2002, 80, 262–270. [Google Scholar]
- Xu, K; Bastia, E; Schwarzschild, M. Therapeutic potential of adenosine A2A receptor antagonists in Parkinson’s disease. Pharmacol. Ther 2005, 105, 267–310. [Google Scholar]
- Azam, F; Ibn-Rajab, IA; Alruiad, AA. Adenosine A2A receptor antagonists as novel anti- Parkinsonian agents: a review of structure-activity relationships. Pharmazie 2009, 64, 771–795. [Google Scholar]
- García, E; Villeda-Hernández, J; Pedraza-Chaverrí, J; Maldonado, PD; Santamaría, A. S-allylcysteine reduces the MPTP-induced striatal cell damage via inhibition of pro-inflammatory cytokine tumor necrosis factor-α and inducible nitric oxide synthase expressions in mice. Phytomedicine 2010, 18, 65–73. [Google Scholar]
- Battaglia, G; Busceti, CL; Pontarelli, F; Biagioni, F; Fornai, F; Paparelli, A; Bruno, V; Ruggieri, S; Nicoletti, F. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology 2003, 45, 155–166. [Google Scholar]
- Kim, YK; Lim, HH; Song, YK; Lee, HH; Lim, S; Han, SM; Kim, CJ. Effect of acupuncture on 6-hydroxydopamine-induced nigrostratal dopaminergic neuronal cell death in rats. Neurosci. Lett 2005, 384, 133–138. [Google Scholar]
- Li, XM; Ma, HB; Ma, ZQ; Li, LF; Xu, CL; Qu, R; Ma, SP. Ameliorative and neuroprotective effect in MPTP model of Parkinson’s disease by Zhen-Wu-Tang (ZWT), a traditional Chinese medicine. J. Ethnopharmacol 2010, 130, 19–27. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Kato, H; Watanabe, Y; Imai, Y; Itoyama, Y; Araki, T. Effect of angiotensin-converting enzyme inhibitor perindopril on interneurons in MPTP-treated mice. Eur. Neuropsychopharmacol 2005, 15, 57–67. [Google Scholar]
- Singh, N; Pillay, V; Choonara, YE. Advances in the treatment of Parkinson’s disease. Prog. Neurobiol 2007, 81, 29–44. [Google Scholar]
- Kotake, Y; Ohta, S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr. Med. Chem 2003, 10, 2507–2516. [Google Scholar]
- Nagatsu, T. Isoquinoline neurotoxins in the brain and Parkinson’s disease. Neurosci. Res 1997, 29, 99–111. [Google Scholar]
- Mazzio, EA; Soliman, KF. Effects of enhancing mitochondrial oxidative phosphorylation with reducing equivalents and ubiquinone on 1-methyl-4-phenylpyridinium toxicity and complex I-IV damage in neuroblastoma cells. Biochem. Pharmacol 2004, 67, 1167–1184. [Google Scholar]
- Miller, GW; Gainetdinov, RR; Levey, AI; Caron, MG. Dopamine transporters and neuronal injury. Trends Pharmacol. Sci 1999, 20, 424–429. [Google Scholar]
- Del Zompo, M; Piccardi, MP; Ruiu, S; Quartu, M; Gessa, GL; Vaccari, A. Selective MPP+ uptake into synaptic dopamine vesicles: possible involvement in MPTP neurotoxicity. Br. J. Pharmacol 1993, 109, 411–414. [Google Scholar]
- Mazzio, E; Soliman, KF. d-(+)-glucose rescue against 1-methyl-4-phenylpyridinium toxicity through anaerobic glycolysis in neuroblastoma cells. Brain Res 2003, 962, 48–60. [Google Scholar]
- Palacios, JM; Wiederhold, KH. Acute administration of 1-N-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP), a compound producing parkinsonism in humans, stimulates [2–14C]deoxyglucose uptake in the regions of the catecholaminergic cell bodies in the rat and guinea pig brains. Brain Res 1984, 301, 187–191. [Google Scholar]
- Palombo, E; Porrino, LJ; Bankiewicz, KS; Crane, AM; Kopin, IJ; Sokoloff, L. Administration of MPTP acutely increases glucose utilization in the substantia nigra of primates. Brain Res 1988, 453, 227–234. [Google Scholar]
- Schwartzman, RJ; Alexander, GM. Changes in the local cerebral metabolic rate for glucose in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) primate model of Parkinson’s disease. Brain Res 1985, 358, 137–143. [Google Scholar]
- Schwartzman, RJ; Alexander, GM; Ferraro, TN; Grothusen, JR; Stahl, SM. Cerebral metabolism of parkinsonian primates 21 days after MPTP. Exp Neurol 1988, 102, 307–313. [Google Scholar]
- Lagrue, E; Abert, B; Nadal, L; Tabone, L; Bodard, S; Medja, F; Lombes, A; Chalon, S; Castelnau, P. MPTP intoxication in mice: a useful model of Leigh syndrome to study mitochondrial diseases in childhood. Metab. Brain Dis 2009, 24, 321–335. [Google Scholar]
- Drolet, RE; Behrouz, B; Lookingland, KJ; Goudreau, JL. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology 2004, 25, 761–769. [Google Scholar]
- Chan, P; DeLanney, LE; Irwin, I; Langston, JW; Di Monte, D. MPTP-induced ATP loss in mouse brain. Ann. N. Y. Acad. Sci 1992, 648, 306–308. [Google Scholar]
- Koga, K; Mori, A; Ohashi, S; Kurihara, N; Kitagawa, H; Ishikawa, M; Mitsumoto, Y; Nakai, M. 1H MRS identifies lactate rise in the striatum of MPTP-treated C57BL/6 mice. Eur. J. Neurosci 2006, 23, 1077–1081. [Google Scholar]
- Brownell, AL; Jenkins, BG; Elmaleh, DR; Deacon, TW; Spealman, RD; Isacson, O. Combined PET/MRS brain studies show dynamic and long-term physiological changes in a primate model of Parkinson disease. Nat. Med 1998, 4, 1308–1312. [Google Scholar]
- Pastoris, O; Dossena, M; Foppa, P; Catapano, M; Ferrari, R; Dagani, F. Biochemical evaluations in skeletal muscles of primates with MPTP Parkinson-like syndrome. Pharmacol. Res 1995, 31, 361–369. [Google Scholar]
- Singh, Y; Swanson, E; Sokoloski, E; Kutty, RK; Krishna, G. MPTP and MPTP analogs induced cell death in cultured rat hepatocytes involving the formation of pyridinium metabolites. Toxicol. Appl. Pharmacol 1988, 96, 347–359. [Google Scholar]
- Singer, TP; Ramsay, RR; McKeown, K; Trevor, A; Castagnoli, NE, Jr. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+) the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar]
- Scotcher, KP; Irwin, I; DeLanney, LE; Langston, JW; Di Monte, D. Effects of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on ATP levels of mouse brain synaptosomes. J. Neurochem 1990, 54, 1295–1301. [Google Scholar]
- Di Monte, DA; Wu, EY; Delanney, LE; Irwin, I; Langston, JW. Toxicity of 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine in primary cultures of mouse astrocytes. J. Pharmacol. Exp. Ther 1992, 261, 44–49. [Google Scholar]
- Mazzio, EA; Soliman, YI; Soliman, KF. Variable toxicological response to the loss of OXPHOS through 1-methyl-4-phenylpyridinium-induced mitochondrial damage and anoxia in diverse neural immortal cell lines. Cell Biol. Toxicol 2010, 26, 527–539. [Google Scholar]
- Maruoka, N; Murata, T; Omata, N; Takashima, Y; Fujibayashi, Y; Wada, Y. Topological and chronological features of the impairment of glucose metabolism induced by 1-methyl-4-phenylpyridinium ion (MPP+) in rat brain slices. J. Neural. Transm 2007, 114, 1155–1159. [Google Scholar]
- Clausen, T; Khaldi, A; Zauner, A; Reinert, M; Doppenberg, E; Menzel, M; Soukup, J; Alves, OL; Bullock, MR. Cerebral acid-base homeostasis after severe traumatic brain injury. J. Neurosurg 2005, 103, 597–607. [Google Scholar]
- Woo, CW; Lee, BS; Kim, ST; Kim, KS. Correlation between lactate and neuronal cell damage in the rat brain after focal ischemia: An in vivo 1H magnetic resonance spectroscopic (1H-MRS) study. Acta Radiol 2010, 51, 344–350. [Google Scholar]
- Chow, SL; Rooney, ZJ; Cleary, MA; Clayton, PT; Leonard, JV. The significance of elevated CSF lactate. Arch. Dis. Child 2005, 90, 1188–1189. [Google Scholar]
- Makoroff, KL; Cecil, KM; Care, M; Ball, WS, Jr. Elevated lactate as an early marker of brain injury in inflicted traumatic brain injury. Pediatr. Radiol 2005, 35, 668–676. [Google Scholar]
- Cavus, I; Kasoff, WS; Cassaday, MP; Jacob, R; Gueorguieva, R; Sherwin, RS; Krystal, JH; Spencer, DD; Abi-Saab, WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol 2005, 57, 226–235. [Google Scholar]
- Brooks, DJ. Imaging approaches to Parkinson disease. J. Nucl. Med 2010, 51, 596–609. [Google Scholar]
- Noda, A; Ohba, H; Kakiuchi, T; Futatsubashi, M; Tsukada, H; Nishimura, S. Age-related changes in cerebral blood flow and glucose metabolism in conscious rhesus monkeys. Brain Res 2002, 936, 76–81. [Google Scholar]
- Cunnane, S; Nugent, S; Roy, M; Courchesne-Loyer, A; Croteau, E; Tremblay, S; Castellano, A; Pifferi, F; Bocti, C; Paquet, N; Begdouri, H; Bentourkia, M; Turcotte, E; Allard, M; Barberger-Gateau, P; Fulop, T; Rapoport, SI. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar]
- Meredith, GE; Totterdell, S; Beales, M; Meshul, CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol 2009, 219, 334–340. [Google Scholar]
- Halestrap, AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans 2010, 38, 841–860. [Google Scholar]
- Bisaglia, M; Soriano, ME; Arduini, I; Mammi, S; Bubacco, L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: implications for mitochondrial dysfunction in Parkinson disease. Biochim. Biophys Acta 2010, 1802, 699–706. [Google Scholar]
- Liou, AK; Zhou, Z; Pei, W; Lim, TM; Yin, XM; Chen, J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J 2005, 19, 1350–1352. [Google Scholar]
- Halestrap, AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem. Soc. Trans 2006, 34, 232–237. [Google Scholar]
- Bo, J; Ming, BY; Gang, LZ; Lei, C; Jia, AL. Protection by puerarin against MPP+-induced neurotoxicity in PC12 cells mediated by inhibiting mitochondrial dysfunction and caspase-3-like activation. Neurosci. Res 2005, 53, 183–188. [Google Scholar]
- Cappelletti, G; Surrey, T; Maci, R. The Parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilising factor. FEBS Lett 2005, 579, 4781–4786. [Google Scholar]
- Thomas, B; Beal, MF. Parkinson’s disease. Hum. Mol. Genet 2007, 16, R183–R194. [Google Scholar]
- Rollema, H; de Vries, JB; Damsma, G; Westerink, BH; Kranenborg, GL; Kuhr, WG; Horn, AS. The use of in vivo brain dialysis of dopamine, acetylcholine, amino acids and lactic acid in studies on the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 503–511. [Google Scholar]
- Ofori, S; Heikkila, RE; Nicklas, WJ. Attenuation by dopamine uptake blockers of the inhibitory effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and some of its analogs on NADH-linked metabolism in mouse neostriatal slices. J. Pharmacol. Exp. Ther 1989, 251, 258–266. [Google Scholar]
- Guillot, TS; Miller, GW. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol 2009, 39, 149–170. [Google Scholar]
- Tanaka, R; Asaga, H; Takeda, M. Nucleoside triphosphate and cation requirement for dopamine uptake by plain synaptic vesicles isolated from rat cerebrums. Brain Res 1976, 115, 273–283. [Google Scholar]
- Hossain, MM; Filipov, NM. Alteration of dopamine uptake into rat striatal vesicles and synaptosomes caused by an in vitro exposure to atrazine and some of its metabolites. Toxicology 2008, 248, 52–58. [Google Scholar]
- Choi, HJ; Lee, SY; Cho, Y; Hwang, O. Inhibition of vesicular monoamine transporter enhances vulnerability of dopaminergic cells: relevance to Parkinson’s disease. Neurochem. Int 2005, 46, 329–335. [Google Scholar]
- Ren, Y; Liu, W; Jiang, H; Jiang, Q; Feng, J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem 2005, 280, 34105–34112. [Google Scholar]
- Chang, GD; Ramirez, VD. The mechanism of action of MPTP and MPP+ on endogenous dopamine release from the rat corpus striatum superfused in vitro. Brain Res 1986, 368, 134–140. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Watanabe, H; Michimata, M; Matsubara, M; Imai, Y; Araki, T. Role of dopamine transporter against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity in mice. Metab. Brain Dis 2003, 18, 139–146. [Google Scholar]
- Jourdain, S; Morissette, M; Morin, N; Di Paolo, T. Oestrogens prevent loss of dopamine transporter (DAT) and vesicular monoamine transporter (VMAT2) in substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. J. Neuroendocrinol 2005, 17, 509–517. [Google Scholar]
- Hogan, KA; Staal, RG; Sonsalla, PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J. Neurochem 2000, 74, 2217–2220. [Google Scholar]
- Harrington, KA; Augood, SJ; Kingsbury, AE; Foster, OJ; Emson, PC. Dopamine transporter (Dat) and synaptic vesicle amine transporter (VMAT2) gene expression in the substantia nigra of control and Parkinson’s disease. Brain Res. Mol. Brain Res 1996, 36, 157–162. [Google Scholar]
- Frey, KA; Koeppe, RA; Kilbourn, MR; van der Borght, TM; Albin, RL; Gilman, S; Kuhl, DE. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann. Neurol 1996, 40, 873–884. [Google Scholar]
- Reinhard, JF, Jr; Carmichael, SW; Daniels, AJ. Mechanisms of toxicity and cellular resistance to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium in adrenomedullary chromaffin cell cultures. J. Neurochem 1990, 55, 311–320. [Google Scholar]
- Wimalasena, DS; Perera, RP; Heyen, BJ; Balasooriya, IS; Wimalasena, K. Vesicular monoamine transporter substrate/inhibitor activity of MPTP/MPP+ derivatives: A structure-activity study. J. Med. Chem 2008, 51, 760–768. [Google Scholar]
- Przedborski, S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat. Disord 2005, 11, S3–S7. [Google Scholar]
- Sala, G; Brighina, L; Saracchi, E; Fermi, S; Riva, C; Carrozza, V; Pirovano, M; Ferrarese, C. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson’s disease. J. Neural. Transm 2010, 117, 1093–1098. [Google Scholar]
- Serra, PA; Sciola, L; Delogu, MR; Spano, A; Monaco, G; Miele, E; Rocchitta, G; Miele, M; Migheli, R; Desole, MS. The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induces apoptosis in mouse nigrostriatal glia. Relevance to nigral neuronal death and striatal neurochemical changes. J. Biol. Chem 2002, 277, 34451–34461. [Google Scholar]
- Asanuma, M; Miyazaki, I; Diaz-Corrales, FJ; Ogawa, N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson’s disease and neurotoxin-induced parkinsonism. Acta Med Okayama 2004, 58, 221–233. [Google Scholar]
- Antunes, F; Nunes, C; Laranjinha, J; Cadenas, E. Redox interactions of nitric oxide with dopamine and its derivatives. Toxicology 2005, 208, 207–212. [Google Scholar]
- Li, HT; Lin, DH; Luo, XY; Zhang, F; Ji, LN; Du, HN; Song, GQ; Hu, J; Zhou, JW; Hu, HY. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J 2005, 272, 3661–3672. [Google Scholar]
- Akagawa, M; Ishii, Y; Ishii, T; Shibata, T; Yotsu-Yamashita, M; Suyama, K; Uchida, K. Metal-catalyzed oxidation of protein-bound dopamine. Biochemistry 2006, 45, 15120–15128. [Google Scholar]
- Smythies, J; Galzigna, L. The oxidative metabolism of catecholamines in the brain: a review. Biochim. Biophys Acta 1998, 1380, 159–162. [Google Scholar]
- Zecca, L; Fariello, R; Riederer, P; Sulzer, D; Gatti, A; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett 2002, 510, 216–220. [Google Scholar]
- Khan, FH; Sen, T; Maiti, AK; Jana, S; Chatterjee, U; Chakrabarti, S. Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: implications for Parkinson’s disease. Biochim. Biophys Acta 2005, 1741, 65–74. [Google Scholar]
- Halliday, GM; Ophof, A; Broe, M; Jensen, PH; Kettle, E; Fedorow, H; Cartwright, MI; Griffiths, FM; Shepherd, CE; Double, KL. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson’s disease. Brain 2005, 28, 2654–2664. [Google Scholar]
- García-Molina, F; Fenoll, LG; Morote, JC; García-Ruiz, PA; Rodríguez-López, JN; García-Cánovas, F; Tudela, J. Opposite effects of peroxidase in the initial stages of tyrosinase-catalysed melanin biosynthesis. Int. J. Biochem. Cell Biol 2005, 37, 1179–1196. [Google Scholar]
- Alexi, T; Borlongan, CV; Faull, RL; Williams, CE; Clark, RG; Gluckman, PD; Hughes, PE. Neuroprotective strategies for basal ganglia degeneration: Parkinson’s and Huntington’s diseases. Prog. Neurobiol 2000, 60, 409–740. [Google Scholar]
- Double, KL; Ben-Shachar, D; Youdim, MB; Zecca, L; Riederer, P; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol 2002, 24, 621–628. [Google Scholar]
- Gentile, V; Cooper, AJ. Transglutaminases—possible drug targets in human diseases. Curr. Drug Targets CNS Neurol. Disord 2004, 3, 99–104. [Google Scholar]
- Caccamo, D; Currò, M; Condello, S; Ferlazzo, N; Ientile, R. Critical role of transglutaminase and other stress proteins during neurodegenerative processes. Amino Acids 2010, 38, 653–658. [Google Scholar]
- Klivenyi, P; Beal, MF; Ferrante, RJ; Andreassen, OA; Wermer, M; Chin, MR; Bonventre, JV. Mice deficient in group IV cytosolic phospholipase A2 are resistant to MPTP neurotoxicity. J. Neurochem 1998, 71, 2634–2637. [Google Scholar]
- Feng, ZH; Wang, TG; Li, DD; Fung, P; Wilson, BC; Liu, B; Ali, SF; Langenbach, R; Hong, JS. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1,2,3,6- tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci. Lett 2002, 329, 354–358. [Google Scholar]
- Zhang, J; Graham, DG; Montine, TJ; Ho, YS. Enhanced N-methyl-4-phenyl-1,2,3,6- tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione peroxidase. J. Neuropathol. Exp. Neurol 2000, 59, 53–61. [Google Scholar]
- St-Pierre, J; Drori, S; Uldry, M; Silvaggi, JM; Rhee, J; Jäger, S; Handschin, C; Zheng, K; Lin, J; Yang, W; Simon, DK; Bachoo, R; Spiegelman, BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Mohanakumar, KP; Muralikrishnan, D; Thomas, B. Neuroprotection by sodium salicylate against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. Brain Res 2000, 864, 281–290. [Google Scholar]
- Gupta, A; Dhir, A; Kumar, A; Kulkarni, SK. Effect of preferential cyclooxygenase-2 (COX-2) inhibitor against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced striatal lesions in rats: behavioral, biochemical and histological evidences. Indian J. Exp. Biol 2010, 48, 577–585. [Google Scholar]
- Teismann, P; Ferger, B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson’s disease. Synapse 2001, 39, 167–174. [Google Scholar]
- Tariq, M; Khan, HA; Al Moutaery, K; Al Deeb, S. Protective effect of quinacrine on striatal dopamine levels in 6-OHDA and MPTP models of Parkinsonism in rodents. Brain Res Bullutin 2001, 54, 77–82. [Google Scholar]
- Nishino, Y; Ando, M; Makino, R; Ueda, K; Okamoto, Y; Kojima, N. Different mechanisms between copper and iron in catecholamines-mediated oxidative DNA damage and disruption of gene expression in vitro. Neurotox Res 2010, in press. [Google Scholar]
- Sayre, LM; Perry, G; Smith, MA. Redox metals and neurodegenerative disease. Curr. Opin. Chem. Biol 1999, 3, 220–225. [Google Scholar]
- Blum, D; Torch, S; Lambeng, N; Nissou, M; Benabid, AL; Sadoul, R; Verna, JM. Molecular pathways involved in the neurotoxicity of 6-OHDA., dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol 2001, 65, 135–172. [Google Scholar]
- Ebadi, M; Srinivasan, SK; Baxi, MD. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol 1996, 48, 1–19. [Google Scholar]
- Annepu, J; Ravindranath, V. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced complex I inhibition is reversed by disulfide reductant, dithiothreitol in mouse brain. Neurosci. Lett 2000, 289, 209–212. [Google Scholar]
- Asanuma, M; Miyazaki, I; Diaz-Corrales, FJ; Ogawa, N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson’s disease and neurotoxin-induced parkinsonism. Acta Med Okayama 2004, 58, 221–233. [Google Scholar]
- Hare, DJ; George, JL; Grimm, R; Wilkins, S; Adlard, PA; Cherny, RA; Bush, AI; Finkelstein, DI; Doble, P. Three-dimensional elemental bio-imaging of Fe, Zn, Cu, Mn and P in a 6-hydroxydopamine lesioned mouse brain. Metallomics 2010, 2, 745–753. [Google Scholar]
- Barnham, KJ; Bush, AI. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol 2008, 12, 222–228. [Google Scholar]
- Hirsch, EC. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord 2009, 15, S209–S211. [Google Scholar]
- Jameson, GN; Jameson, RF; Linert, W. New insights into iron release from ferritin: direct observation of the neurotoxin 6-hydroxydopamine entering ferritin and reaching redox equilibrium with the iron core. Org. Biomol. Chem 2004, 2, 2346–2351. [Google Scholar]
- Kobayashi, H; Oikawa, S; Umemura, S; Hirosawa, I; Kawanishi, S. Mechanism of metalmediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free Radic. Res 2008, 42, 651–660. [Google Scholar]
- Gauthier, MA; Eibl, JK; Crispo, JA; Ross, GM. Covalent arylation of metallothionein by oxidized dopamine products: a possible mechanism for zinc-mediated enhancement of dopaminergic neuron survival. Neurotox. Res 2008, 14, 317–328. [Google Scholar]
- Jiang, H; Song, N; Xu, H; Zhang, S; Wang, J; Xie, J. Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res 2010, 20, 345–356. [Google Scholar]
- Nicolaus, BJ. A critical review of the function of neuromelanin and an attempt to provide a unified theory. Med Hypotheses 2005, 65, 791–796. [Google Scholar]
- Koeppen, AH. The history of iron in the brain. J. Neurol. Sci 1995, 134, 1–9. [Google Scholar]
- Kaur, D; Andersen, J. Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res. Rev 2004, 3, 327–343. [Google Scholar]
- Takanashi, M; Mochizuki, H; Yokomizo, K; Hattori, N; Mori, H; Yamamura, Y; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Parkinsonism Relat. Disord 2001, 7, 311–314. [Google Scholar]
- Faucheux, BA; Nillesse, N; Damier, P; Spik, G; Mouatt-Prigent, A; Pierce, A; Leveugle, B; Kubis, N; Hauw, JJ; Agid, Y; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci USA 1995, 92, 9603–9607. [Google Scholar]
- Jiang, H; Qian, ZM; Xie, JX. Increased DMT1 expression and iron content in MPTP-treated C57BL/6 mice. Sheng Li Xue Bao 2003, 55, 571–576. [Google Scholar]
- Zucca, FA; Giaveri, G; Gallorini, M; Albertini, A; Toscani, M; Pezzoli, G; Lucius, R; Wilms, H; Sulzer, D; Ito, S; Wakamatsu, K; Zecca, L. The neuromelanin of human substantia nigra: physiological and pathogenic aspects. Pigment Cell Res 2004, 17, 610–617. [Google Scholar]
- Zhang, J; Zhang, Y; Wang, J; Cai, P; Luo, C; Qian, Z; Dai, Y; Feng, H. Characterizing iron deposition in Parkinson’s disease using susceptibility-weighted imaging: an in vivo MR study. Brain Res 2010, 1330, 124–130. [Google Scholar]
- Andersen, JK. Iron dysregulation and Parkinson’s disease. J. Alzheimers Dis 2004, 6, S47–S52. [Google Scholar]
- Bou-Abdallah, F; McNally, J; Liu, XX; Melman, A. Oxygen catalyzed mobilization of iron from ferritin by iron(iii) chelate ligands. Chem. Commun 2011, 47, 731–733. [Google Scholar]
- Qian, ZM; Wang, Q. Expression of iron transport proteins and excessive iron accumulation in the brain in neurodegenerative disorders. Brain Res. Rev 1998, 27, 257–267. [Google Scholar]
- Ke, Y; Ming Qian, Z. Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol 2003, 2, 246–253. [Google Scholar]
- Schipper, HM; Liberman, A; Stopa, EG. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol 1998, 150, 60–68. [Google Scholar]
- Shamoto-Nagai, M; Maruyama, W; Yi, H; Akao, Y; Tribl, F; Gerlach, M; Osawa, T; Riederer, P; Naoi, M. Neuromelanin induces oxidative stress in mitochondria through release of iron: mechanism behind the inhibition of 26S proteasome. J. Neural. Transm 2006, 113, 633–644. [Google Scholar]
- Hirsch, EC. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord 2009, 15, S209–S211. [Google Scholar]
- Levenson, CW; Cutler, RG; Ladenheim, B; Cadet, JL; Hare, J; Mattson, MP. Role of dietary iron restriction in a mouse model of Parkinson’s disease. Exp. Neurol 2004, 190, 506–514. [Google Scholar]
- Gal, S; Fridkin, M; Amit, T; Zheng, H; Youdim, MB. M30, a novel multifunctional neuroprotective drug with potent iron chelating and brain selective monoamine oxidase-ab inhibitory activity for Parkinson’s disease. J. Neural. Transm. Suppl 2006, 70, 447–456. [Google Scholar]
- Kaur, D; Yantiri, F; Rajagopalan, S; Kumar, J; Mo, JQ; Boonplueang, R; Viswanath, V; Jacobs, R; Yang, L; Beal, MF; DiMonte, D; Volitaskis, I; Ellerby, L; Cherny, RA; Bush, AI; Andersen, JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar]
- Zecca, L; Berg, D; Arzberger, T; Ruprecht, P; Rausch, WD; Musicco, M; Tampellini, D; Riederer, P; Gerlach, M; Becker, G. In vivo detection of iron and neuromelanin by transcranial sonography: a new approach for early detection of substantia nigra damage. Mov. Disord 2005, 20, 1278–1285. [Google Scholar]
- Naoi, M; Maruyama, W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des 2010, 16, 2799–2817. [Google Scholar]
- Bortolato, M; Chen, K; Shih, JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv. Drug Deliv. Rev 2008, 60, 1527–1533. [Google Scholar]
- Li, SW; Lin, TS; Minteer, S; Burke, WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson’s disease pathogenesis. Brain Res. Mol. Brain Res 2001, 93, 1–7. [Google Scholar]
- Tabner, BJ; Turnbull, S; El-Agnaf, OM; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med 2002, 32, 1076–1083. [Google Scholar]
- Matsubara, K; Aoyama, K; Suno, M; Awaya, T. N-methylation underlying Parkinson’s disease. Neurotoxicol. Teratol 2002, 24, 593–598. [Google Scholar]
- Parsons, RB; Smith, SW; Waring, RH; Williams, AC; Ramsden, DB. High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson’s disease. Neurosci. Lett 2003, 342, 13–16. [Google Scholar]
- Naoi, M; Maruyama, W; Nagy, GM. Dopamine-derived salsolinol derivatives as endogenous monoamine oxidase inhibitors: occurrence, metabolism and function in human brains. Neurotoxicology 2004, 25, 193–204. [Google Scholar]
- DeCuypere, M; Lu, Y; Miller, DD; LeDoux, MS. Regional distribution of tetrahydroisoquinoline derivatives in rodent, human, and Parkinson’s disease brain. J. Neurochem 2008, 107, 1398–1413. [Google Scholar]
- Antkiewicz-Michaluk, L. Endogenous risk factors in Parkinson’s disease: dopamine and tetrahydroisoquinolines. Pol. J. Pharmacol 2002, 54, 567–572. [Google Scholar]
- Soto-Otero, R; Méndez-Alvarez, E; Sánchez-Sellero, I; Cruz-Landeira, A; López-Rivadulla Lamas, M. Reduction of rat brain levels of the endogenous dopaminergic proneurotoxins 1,2,3,4- tetrahydroisoquinoline and 1,2,3,4-tetrahydro-beta-carboline by cigarette smoke. Neurosci. Lett 2001, 298, 187–190. [Google Scholar]
- Gearhart, DA; Neafsey, EJ; Collins, MA. Phenylethanolamine N-methyltransferase has beta-carboline 2N-methyltransferase activity: hypothetical relevance to Parkinson’s disease. Neurochem. Int 2002, 40, 611–620. [Google Scholar]
- Wu, YN; Johnson, SW. Rotenone potentiates NMDA currents in substantia nigra dopamine neurons. Neurosci. Lett 2007, 421, 96–100. [Google Scholar]
- Gao, WJ; Goldman-Rakic, PS. NMDA receptor-mediated epileptiform persistent activity requires calcium release from intracellular stores in prefrontal neurons. Exp. Neurol 2006, 197, 495–504. [Google Scholar]
- Nizami, S; Lee, VW; Davies, J; Long, P; Jovanovic, JN; Sihra, TS. Presynaptic roles of intracellular Ca(2+) stores in signalling and exocytosis. Biochem. Soc. Trans 2010, 38, 529–535. [Google Scholar]
- Maurois, P; Pages, N; Bac, P; German-Fattal, M; Agnani, G; Delplanque, B; Durlach, J. Threshold to N-methyl-d-aspartate-induced seizures in mice undergoing chronic nutritional magnesium deprivation is lowered in a way partly responsive to acute magnesium and antioxidant administrations. Br. J. Nutr 2009, 101, 317–321. [Google Scholar]
- Gu, Z; Nakamura, T; Lipton, SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol 2010, 41, 55–72. [Google Scholar]
- Martínez, A; Portero-Otin, M; Pamplona, R; Ferrer, I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 2010, 20, 281–297. [Google Scholar]
- Adamczyk, A; KaŸmierczak, A; Czapski, GA; Strosznajder, JB. Alpha-synuclein induced cell death in mouse hippocampal (HT22) cells is mediated by nitric oxide-dependent activation of caspase-3. FEBS Lett 2010, 584, 3504–3508. [Google Scholar]
- Adamczyk, A; Czapski, GA; KaŸmierczak, A; Strosznajder, JB. Effect of N-methyl-d-aspartate (NMDA) receptor antagonists on alpha-synuclein-evoked neuronal nitric oxide synthase activation in the rat brain. Pharmacol. Rep 2009, 61, 1078–1085. [Google Scholar]
- Meredith, GE; Totterdell, S; Beales, M; Meshul, CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol 2009, 219, 334–340. [Google Scholar]
- Nimmrich, V; Reymann, KG; Strassburger, M; Schöder, UH; Gross, G; Hahn, A; Schoemaker, H; Wicke, K; Möller, A. Inhibition of calpain prevents NMDA-induced cell death and beta-amyloid-induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol 2010, 159 , 1523–1531. [Google Scholar]
- Ma, T; Zhao, Y; Kwak, YD; Yang, Z; Thompson, R; Luo, Z; Xu, H; Liao, FF. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J. Neurosci 2009, 29, 11226–11236. [Google Scholar]
- Pan, J; Xiao, Q; Sheng, CY; Hong, Z; Yang, HQ; Wang, G; Ding, JQ; Chen, SD. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem. Int 2009, 54, 418–425. [Google Scholar]
- Wang, AL; Liou, YM; Pawlak, CR; Ho, YJ. Involvement of NMDA receptors in both MPTP-induced neuroinflammation and deficits in episodic-like memory in Wistar rats. Behav. Brain Res 2010, 208, 38–46. [Google Scholar]
- Hald, A; van Beek, J; Lotharius, J. Inflammation in Parkinson’s disease: causative or epiphenomenal? Subcell. Biochem 2007, 42, 249–279. [Google Scholar]
- Kurkowska-Jastrzebska, I; Wrońska, A; Kohutnicka, M; Członkowski, A; Członkowska, A. MHC class II positive microglia and lymphocytic infiltration are present in the substantia nigra and striatum in mouse model of Parkinson’s disease. Acta Neurobiol. Exp 1999, 59, 1–8. [Google Scholar]
- Ouchi, Y; Yoshikawa, E; Sekine, Y; Futatsubashi, M; Kanno, T; Ogusu, T; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol 2005, 57, 168–175. [Google Scholar]
- Nagatsu, T; Sawada, M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr. Pharm. Des 2005, 11, 999–1016. [Google Scholar]
- McGeer, PL; McGeer, EG. Inflammation and the degenerative diseases of aging. Ann. N. Y. Acad. Sci 2004, 1035, 104–116. [Google Scholar]
- Nagatsu, T; Mogi, M; Ichinose, H; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural. Transm. Suppl 2000, 60, 277–290. [Google Scholar]
- Sriram, K; Matheson, JM; Benkovic, SA; Miller, DB; Luster, MI; O’Callaghan, JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J 2002, 16, 1474–1476. [Google Scholar]
- Onyango, IG; Tuttle, JB; Bennett, JP, Jr. Activation of p38 and N-acetylcysteine-sensitive c-Jun NH2-terminal kinase signaling cascades is required for induction of apoptosis in Parkinson’s disease cybrids. Mol. Cell Neurosci 2005, 28, 452–461. [Google Scholar]
- Hirsch, EC; Hunot, S; Hartmann, A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism Relat. Disord 2005, 11, S9–S15. [Google Scholar]
- Tansey, MG; Goldberg, MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis 2010, 37, 510–518. [Google Scholar]
- Zhang, W; Wang, T; Pei, Z; Miller, DS; Wu, X; Block, ML; Wilson, B; Zhang, W; Zhou, Y; Hong, JS; Zhang, J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 2005, 19, 533–542. [Google Scholar]
- Hirsch, EC; Hunot, S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 2009, 8, 382–397. [Google Scholar]
- Ji, H; Wang, H; Zhang, F; Li, X; Xiang, L; Aiguo, S. PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm. Res 2010, 59, 921–929. [Google Scholar]
- Kim, SH; Kim, J; Sharma, RP. Inhibition of p38 and ERK MAP kinases blocks endotoxininduced nitric oxide production and differentially modulates cytokine expression. Pharmacol. Res 2004, 49, 433–439. [Google Scholar]
- Thomas, T; Timmer, M; Cesnulevicius, K; Hitti, E; Kotlyarov, A; Gaestel, M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation—relevance in a mouse model of Parkinson’s disease. J. Neurochem 2008, 105, 2039–2052. [Google Scholar]
- Willesen, MG; Gammeltoft, S; Vaudano, E. Activation of the c-Jun N terminal kinase pathway in an animal model of Parkinson’s disease. Ann. N. Y. Acad. Sci 2002, 973, 237–240. [Google Scholar]
- Lee, DY; Oh, YJ; Jin, BK. Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia 2005, 51, 98–110. [Google Scholar]
- Kao, SJ; Lei, HC; Kuo, CT; Chang, MS; Chen, BC; Chang, YC; Chiu, WT; Lin, CH. Lipoteichoic acid induces nuclear factor-kappaB activation and nitric oxide synthase expression via phosphatidylinositol 3-kinase, Akt, and p38 MAPK in RAW 264.7 macrophages. Imunology 2005, 115, 366–374. [Google Scholar]
- Chio, CC; Chang, YH; Hsu, YW; Chi, KH; Lin, WW. PKA-dependent activation of PKC.; p38 MAPK and IKK in macrophage: implication in the induction of inducible nitric oxide synthase and interleukin-6 by dibutyryl cAMP. Cell Signal 2004, 16, 565–575. [Google Scholar]
- Lee, JK; Choi, SS; Won, JS; Suh, HW. The regulation of inducible nitric oxide synthase gene expression induced by lipopolysaccharide and tumor necrosis factor-alpha in C6 cells: involvement of AP-1 and NFkappaB. Life Sci 2003, 73, 595–609. [Google Scholar]
- Wang, MJ; Lin, WW; Chen, HL; Chang, YH; Ou, HC; Kuo, JS; Hong, JS; Jeng, KC. Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. Eur. J. Neurosci 2002, 16, 2103–2112. [Google Scholar]
- Hua, LL; Zhao, ML; Cosenza, M; Kim, MO; Huang, H; Tanowitz, HB; Brosnan, CF; Lee, SC. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J. Neuroimmunol 2002, 126, 180–189. [Google Scholar]
- Gerhard, A; Pavese, N; Hotton, G; Turkheimer, F; Es, M; Hammers, A; Eggert, K; Oertel, W; Banati, RB; Brooks, DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis 2006, 21, 404–412. [Google Scholar]
- Ban, HS; Suzuki, K; Lim, SS; Jung, SH; Lee, S; Ji, J; Lee, HS; Lee, YS; Shin, KH; Ohuchi, K. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase and tumor necrosis factor-alpha by 2′-hydroxychalcone derivatives in RAW 264.7 cells. Biochem. Pharmacol 2004, 67, 1549–1557. [Google Scholar]
- Wang, W; Ma, C; Mao, Z; Li, M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect 2004, 17, 646–654. [Google Scholar]
- Saporito, MS; Thomas, BA; Scott, RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem 2000, 75, 1200–1208. [Google Scholar]
- Saporito, MS; Brown, EM; Miller, MS; Carswell, S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J. Pharmacol. Exp. Ther 1999, 288, 421–427. [Google Scholar]
- Kurkowska-Jastrzebska, I; Babiuch, M; Joniec, I; Przybyłkowski, A; Członkowski, A; Członkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol 2002, 2, 1213–1218. [Google Scholar]
- Fahrig, T; Gerlach, I; Horváth, E. A synthetic derivative of the natural product rocaglaol is a potent inhibitor of cytokine-mediated signaling and shows neuroprotective activity in vitro and in animal models of Parkinson’s disease and traumatic brain injury. Mol. Pharmacol 2005, 67, 1544–1555. [Google Scholar]
- Wu, DC; Jackson-Lewis, V; Vila, M; Tieu, K; Teismann, P; Vadseth, C; Choi, DK; Ischiropoulos, H; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci 2002, 22, 1763–1771. [Google Scholar]
- Du, Y; Ma, Z; Lin, S; Dodel, RC; Gao, F; Bales, KR; Triarhou, LC; Chernet, E; Perry, KW; Nelson, DL; Luecke, S; Phebus, LA; Bymaster, FP; Paul, SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci USA 2001, 98, 14669–14674. [Google Scholar]
- Lee, HG; Kim, H; Oh, WK; Yu, KA; Choe, YK; Ahn, JS; Kim, DS; Kim, SH; Dinarello, CA; Kim, K; Yoon, DY. Tetramethoxy hydroxyflavone p7F downregulates inflammatory mediators via the inhibition of nuclear factor kappaB. Ann. N. Y. Acad. Sci 2004, 1030, 555–568. [Google Scholar]
- Anwar, AA; Li, FY; Leake, DS; Ishii, T; Mann, GE; Siow, RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med 2005, 39, 227–236. [Google Scholar]
- Tieu, K; Ischiropoulos, H; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar]
- Wu, DC; Teismann, P; Tieu, K; Vila, M; Jackson-Lewis, V; Ischiropoulos, H; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci USA 2003, 100, 6145–6150. [Google Scholar]
- Dehmer, T; Lindenau, J; Haid, S; Dichgans, J; Schulz, JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem 2000, 74, 2213–2216. [Google Scholar]
- Klivenyi, P; St Clair, D; Wermer, M; Yen, HC; Oberley, T; Yang, L; Beal, MF. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol. Dis 1998, 5, 253–258. [Google Scholar]
- Andreassen, OA; Ferrante, RJ; Dedeoglu, A; Albers, DW; Klivenyi, P; Carlson, EJ; Epstein, CJ; Beal, MF. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp. Neurol 2001, 167, 189–195. [Google Scholar]
- Callio, J; Oury, TD; Chu, CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J. Biol. Chem 2005, 280, 18536–18542. [Google Scholar]
- Levites, Y; Weinreb, O; Maor, G; Youdim, MB; Mandel, S. Green tea polyphenol (−)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem 2001, 78, 1073–1082. [Google Scholar]
- Kurosaki, R; Muramatsu, Y; Michimata, M; Matsubara, M; Kato, H; Imai, Y; Itoyama, Y; Araki, T. Role of nitric oxide synthase against MPTP neurotoxicity in mice. Neurol. Res 2002, 24, 655–662. [Google Scholar]
- Choi, JY; Park, CS; Kim, DJ; Cho, MH; Jin, BK; Pie, JE; Chung, WG. Prevention of nitric oxide-mediated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease in mice by tea phenolic epigallocatechin 3-gallate. Neurotoxicology 2002, 23, 367–374. [Google Scholar]
- Mazzio, EA; Soliman, KF. Cytoprotection of pyruvic acid and reduced beta-nicotinamide adenine dinucleotide against hydrogen peroxide toxicity in neuroblastoma cells. Neurochem. Res 2003, 28, 733–741. [Google Scholar]
- Mazzio, EA; Reams, RR; Soliman, KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 2004, 1004, 29–44. [Google Scholar]
- Auer, RN. Hypoglycemic brain damage. Metab Brain Dis 2004, 19, 169–175. [Google Scholar]
- Mazzio, E; Soliman, KF. Pyruvic acid cytoprotection against 1-methyl-4-phenylpyridinium, 6-hydroxydopamine and hydrogen peroxide toxicities in vitro. Neurosci. Lett 2003, 337, 77–80. [Google Scholar]
- Gonzalez, SV; Nguyen, NH; Rise, F; Hassel, B. Brain metabolism of exogenous pyruvate. J. Neurochem 2005, 95, 284–293. [Google Scholar]
- Lee, JY; Kim, YH; Koh, JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci 2001, 21, RC171. [Google Scholar]
- Izumi, Y; Zorumski, CF. Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci. Lett 2010, 478, 131–135. [Google Scholar]
- Cosi, C; Marien, M. Decreases in mouse brain NAD+ and ATP induced by 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP): prevention by the poly(ADP-ribose) polymerase inhibitor, benzamide. Brain Res 1998, 809, 58–67. [Google Scholar]
- Cosi, C; Marien, M. Implication of poly (ADP-ribose) polymerase (PARP) in neurodegeneration and brain energy metabolism. Decreases in mouse brain NAD+ and ATP caused by MPTP are prevented by the PARP inhibitor benzamide. Ann. N. Y. Acad. Sci 1999, 890, 227–239. [Google Scholar]
- Iwashita, A; Yamazaki, S; Mihara, K; Hattori, K; Yamamoto, H; Ishida, J; Matsuoka, N; Mutoh, S. Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, 2-[3-[4-(4-chlorophenyl)-1-piperazinyl] propyl]-4(3H)-quinazolinone (FR255595), in an in vitro model of cell death and in mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J. Pharmacol. Exp. Ther 2004, 309, 1067–1078. [Google Scholar]
- Yokoyama, H; Kuroiwa, H; Tsukada, T; Uchida, H; Kato, H; Araki, T. Poly(ADPribose) polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-induced neurotoxicity in mice. J. Neurosci. Res 2010, 88, 1522–1536. [Google Scholar]
- Anderson, DW; Bradbury, KA; Schneider, JS. Broad neuroprotective profile of nicotinamide in different mouse models of MPTP-induced Parkinsonism. Eur. J. Neurosci 2008, 28, 610–617. [Google Scholar]
- Mukherjee, SK; Klaidman, LK; Yasharel, R; Adams, JD, Jr. Increased brain NAD prevents neuronal apoptosis in vivo. Eur. J. Pharmacol 1997, 330, 27–34. [Google Scholar]
- Yokoyama, H; Kuroiwa, H; Tsukada, T; Uchida, H; Kato, H; Araki, T. Poly(ADPribose) polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-induced neurotoxicity in mice. J. Neurosci. Res 2010, 88, 1522–1536. [Google Scholar]
- Mandir, AS; Simbulan-Rosenthal, CM; Poitras, MF; Lumpkin, JR; Dawson, VL; Smulson, ME; Dawson, TM. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J. Neurochem 2002, 83, 186–192. [Google Scholar]
- Duan, W; Zhu, X; Ladenheim, B; Yu, QS; Guo, Z; Oyler, J; Cutler, RG; Cadet, JL; Greig, NH; Mattson, MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol 2002, 52, 597–606. [Google Scholar]
- Fukushima, T; Ohta, M; Tanaka, K; Kaneko, S-Y; Maeda, T; Sasaki, A. Niacin metabolism and Parkinson’s disease. Asia Pac. J. Clin. Nutr 2004, 13, S176. [Google Scholar]
- Alston, TA; Abeles, RH. Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch. Biochem. Biophys 1988, 260, 601–618. [Google Scholar]
- Ashihara, H; Crozier, A. Caffeine: a well known but little mentioned compound in plant science. Trends Plant Sci 2001, 6, 407–413. [Google Scholar]
- Koshiishi, C; Kato, A; Yama, S; Crozier, A; Ashihara, H. A new caffeine biosynthetic pathway in tea leaves: utilisation of adenosine released from the S-adenosyl-l-methionine cycle. FEBS Lett 2001, 499, 50–54. [Google Scholar]
- Góngora-Alfaro, JL. Caffeine as a preventive drug for Parkinson’s disease: epidemiologic evidence and experimental support. Rev. Neurol 2010, 50, 221–229. [Google Scholar]
- Singh, K; Singh, S; Singhal, NK; Sharma, A; Parmar, D; Singh, MP. Nicotine- and caffeinemediated changes in gene expression patterns of MPTP-lesioned mouse striatum: Implications in neuroprotection mechanism. Chem. Biol. Interact 2010, 185, 81–93. [Google Scholar]
- Upmeier, B; Gross, W; Köster, S; Barz, W. Purification and properties of S-adenosyl-l-methionine: nicotinic acid-N-methyltransferase from cell suspension cultures of Glycine max L. Arch. Biochem. Biophys 1988, 262, 445–454. [Google Scholar]
- Oyanagi, K. The nature of the parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam and magnesium deficiency. Parkinsonism Relat. Disord 2005, 11, S17–S23. [Google Scholar]
- Barbiroli, B; Martinelli, P; Patuelli, A; Lodi, R; Iotti, S; Cortelli, P; Montagna, P. Phosphorus magnetic resonance spectroscopy in multiple system atrophy and Parkinson’s disease. Mov. Disord 1999, 14, 430–435. [Google Scholar]
- Philippu, A; Matthaei, H; Lentzen, H. Uptake of dopamine into fractions of pig caudate nucleus homogenates. Naunyn Schmiedebergs Arch. Pharmacol 1975, 287, 181–190. [Google Scholar]
- Schümann, HJ; Althoff, B. Effects of calcium and phosphate on catecholamines, ATP and dopamine beta-hydroxylase of chromaffin medullary granules. Naunyn Schmiedebergs Arch. Pharmacol 1976, 293, 67–74. [Google Scholar]
- Baker, PF; Knight, DE. Gaining access to the site of exocytosis in bovine adrenal medullary cells. J. Physiol 1980, 76, 497–504. [Google Scholar]
- Yang, YC; Lee, CH; Kuo, CC. Ionic flow enhances low-affinity binding: a revised mechanistic view into Mg2+ block of NMDA receptors. J. Physiol 2010, 588, 633–650. [Google Scholar]
- Safar, MM; Abdallah, DM; Arafa, NM; Abdel-Aziz, MT. Magnesium supplementation enhances the anticonvulsant potential of valproate in pentylenetetrazol-treated rats. Brain Res 2010, 1334, 58–64. [Google Scholar]
- Lin, JY; Chung, SY; Lin, M; Cheng, FC. Effects of magnesium sulfate on energy metabolites and glutamate in the cortex during focal cerebral ischemia and reperfusion in the gerbil monitored by a dual-probe microdialysis technique. Life Sci 2002, 71, 803–811. [Google Scholar]
- Johnson, S. Micronutrient accumulation and depletion in schizophrenia, epilepsy, autism and Parkinson’s disease? Med Hypotheses 2001, 56, 641–645. [Google Scholar]
- Brosnan, JT; Jacobs, RL; Stead, LM; Brosnan, ME. Methylation demand: a key determinant of homocysteine metabolism. Acta Biochim. Pol 2004, 51, 405–413. [Google Scholar]
- Zoccolella, S; Lamberti, P; Armenise, E; de Mari, M; Lamberti, SV; Mastronardi, R; Fraddosio, A; Iliceto, G; Livrea, P. Plasma homocysteine levels in Parkinson’s disease: role of antiparkinsonian medications. Parkinsonism Relat. Disord 2005, 11, 131–133. [Google Scholar]
- Lamberti, P; Zoccolella, S; Armenise, E; Lamberti, SV; Fraddosio, A; de Mari, M; Iliceto, G; Livrea, P. Hyperhomocysteinemia in L-dopa treated Parkinson’s disease patients: effect of cobalamin and folate administration. Eur. J. Neurol 2005, 12, 365–368. [Google Scholar]
- McCully, KS. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann. Clin. Lab. Sci 2009, 39, 219–232. [Google Scholar]
- dos Santos, EF; Busanello, EN; Miglioranza, A; Zanatta, A; Barchak, AG; Vargas, CR; Saute, J; Rosa, C; Carrion, MJ; Camargo, D; Dalbem, A; da Costa, JC; de Sousa Miguel, SR; de Mello Rieder, CR; Wajner, M. Evidence that folic acid deficiency is a major determinant of hyperhomocysteinemia in Parkinson’s disease. Metab. Brain Dis 2009, 24, 257–269. [Google Scholar]
- Miller, JW. Homocysteine, folate deficiency, and Parkinson’s disease. Nutr. Rev 2002, 60, 410–413. [Google Scholar]
- Duan, W; Ladenheim, B; Cutler, RG; Kruman, II; Cadet, JL; Mattson, MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem 2002, 80, 101–110. [Google Scholar]
- Yagisawa, M; Okawa, N; Shigematsu, N; Nakata, R. Effects of intravenous betaine on methionine-loading-induced plasma homocysteine elevation in rats. J. Nutr. Biochem 2004, 15, 666–671. [Google Scholar]
- Kharbanda, KK; Rogers, DD, II; Mailliard, ME; Siford, GL; Barak, AJ; Beckenhauer, HC; Sorrell, MF; Tuma, DJ. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: protection by betaine. Biochem. Pharmacol 2005, 70, 1883–1890. [Google Scholar]
- Yeh, Y-Y; Lim, H-S; Yeh, S-M; Picciano, MF. Garlic extract attenuates hyperhomocysteinemia caused by folic acid deficiency in the rat. Nutr. Res 2005, 25, 93–102. [Google Scholar]
- Sled, VD; Rudnitzky, NI; Hatefi, Y; Ohnishi, T. Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 1994, 33, 10069–10075. [Google Scholar]
- Gerards, M; van den Bosch, BJ; Danhauser, K; Serre, V; van Weeghel, M; Wanders, RJ; Nicolaes, GA; Sluiter, W; Schoonderwoerd, K; Scholte, HR; Prokisch, H; Rötig, A; de Coo, IF; Smeets, HJ. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain 2011, 134, 210–219. [Google Scholar]
- Bar-Meir, M; Elpeleg, ON; Saada, A. Effect of various agents on adenosine triphosphate synthesis in mitochondrial complex I deficiency. J. Pediatr 2001, 139, 868–870. [Google Scholar]
- Griebel, V; Krägeloh-Mann, I; Ruitenbeek, W; Trijbels, JM; Paulus, W. A mitochondrial myopathy in an infant with lactic acidosis. Dev. Med. Child Neurol 1990, 32, 528–531. [Google Scholar]
- Antozzi, C; Garavaglia, B; Mora, M; Rimoldi, M; Morandi, L; Ursino, E; DiDonato, S. Late-onset riboflavin-responsive myopathy with combined multiple acyl coenzyme A dehydrogenase and respiratory chain deficiency. Neurology 1994, 44, 2153–2158. [Google Scholar]
- Ogle, RF; Christodoulou, J; Fagan, E; Blok, RB; Kirby, DM; Seller, KL; Dahl, HH; Thorburn, DR. Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I deficiency responsive to riboflavin. J. Pediatr 1997, 130, 138–145. [Google Scholar]
- Kerr, DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol. Genet. Metab 2010, 99, 246–255. [Google Scholar]
- Jia, H; Liu, Z; Li, X; Feng, Z; Hao, J; Li, X; Shen, W; Zhang, H; Liu, J. Synergistic anti-Parkinsonism activity of high doses of B vitamins in a chronic cellular model. Neurobiol Aging 2010, 31, 636–646. [Google Scholar]
- Brownell, AL; Jenkins, BG; Isacson, O. Dopamine imaging markers and predictive mathematical models for progressive degeneration in Parkinson’s disease. Biomed. Pharmacother 1999, 53, 131–140. [Google Scholar]
- Matthews, RT; Ferrante, RJ; Klivenyi, P; Yang, L; Klein, AM; Mueller, G; Kaddurah- Daouk, R; Beal, MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol 1999, 157, 142–149. [Google Scholar]
- McCarty, MF. The therapeutic potential of glucose tolerance factor. Med Hypotheses 1980, 6, 1177–1189. [Google Scholar]
- McCarty, MF. High-dose biotin, an inducer of glucokinase expression, may synergize with chromium picolinate to enable a definitive nutritional therapy for type II diabetes. Med Hypotheses 1999, 52, 401–406. [Google Scholar]
- Aguilar, MV; Jiménez-Jiménez, FJ; Molina, JA; Meseguer, I; Mateos-Vega, CJ; González- Muñoz, MJ; de Bustos, F; Gómez-Escalonilla, C; Ort-Pareja, M; Zurdo, M; Martínez-Para, MC. Cerebrospinal fluid selenium and chromium levels in patients with Parkinson’s disease. J. Neural Transm 1998, 105, 1245–1251. [Google Scholar]
- Greggio, E; Bergantino, E; Carter, D; Ahmad, R; Costin, GE; Hearing, VJ; Clarimon, J; Singleton, A; Eerola, J; Hellström, O; Tienari, PJ; Miller, DW; Beilina, A; Bubacco, L; Cookson, MR. Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease. J. Neurochem 2005, 93, 246–256. [Google Scholar]
- Boissy, RE; Visscher, M; DeLong, MA. DeoxyArbutin: a novel reversible tyrosinase inhibitor with effective in vivo skin lightening potency. Exp. Dermatol 2005, 14, 601–608. [Google Scholar]
- Galeazzi, MA. Behavior of polyphenoloxidases in food. Arch. Lat. Nutr 1984, 34, 269–289. [Google Scholar]
- Matheis, G; Belitz, HD. Studies on enzymic browning of potatoes (Solanum tuberosum). III. Kinetics of potato phenoloxidase (EC 1.14.18.1 monophenol, dihydroxyphenylalanine: oxygenoxidoreductase). Z. Lebensm. Unters. Forsch 1977, 163, 191–195. [Google Scholar]
- Henderson, HM; Eskin, NA; Pinsky, C; Bose, R; Ashique, AM. Pyridine and other coal tar constituents as inhibitors of potato polyphenol oxidase: a non-animal model for neurochemical studies. Life Sci 1992, 51, PL207–210. [Google Scholar]
- Khatib, S; Nerya, O; Musa, R; Shmuel, M; Tamir, S; Vaya, J. Chalcones as potent tyrosinase inhibitors: the importance of a 2,4-substituted resorcinol moiety. Bioorg. Med. Chem 2005, 13, 433–441. [Google Scholar]
- Nerya, O; Musa, R; Khatib, S; Tamir, S; Vaya, J. Chalcones as potent tyrosinase inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry 2004, 65, 1389–1395. [Google Scholar]
- Lee, NK; Son, KH; Chang, HW; Kang, SS; Park, H; Heo, MY; Kim, HP. Prenylated flavonoids as tyrosinase inhibitors. Arch. Pharm. Res 2004, 27, 1132–1135. [Google Scholar]
- Kim, SJ; Son, KH; Chang, HW; Kang, SS; Kim, HP. Tyrosinase inhibitory prenylated flavonoids from Sophora flavescens. Biol. Pharm Bullutin 2003, 26, 1348–1350. [Google Scholar]
- Son, JK; Park, JS; Kim, JA; Kim, Y; Chung, SR; Lee, SH. Prenylated flavonoids from the roots of Sophora flavescens with tyrosinase inhibitory activity. Planta Med 2003, 69, 559–561. [Google Scholar]
- Tan, C; Zhu, W; Lu, Y. Aloin, cinnamic acid and sophorcarpidine are potent inhibitors of tyrosinase. Chin. Med. J. (Engl ) 2002, 115, 1859–1862. [Google Scholar]
- Shi, Y; Chen, Q-X; Wang, Q; Song, KK; Qiu, L. Inhibitory effects of cinnamic acid and its derivatives on the diphenolase activity of mushroom (Agaricus bisporus) tyrosinase. Food Chem 2005, 92, 707–712. [Google Scholar]
- Nerya, O; Vaya, J; Musa, R; Izrael, S; Ben-Arie, R; Tamir, S. Glabrene and isoliquiritigenin as tyrosinase inhibitors from licorice roots. J. Agric. Food Chem 2003, 51, 1201–1207. [Google Scholar]
- Fu, B; Li, H; Wang, X; Lee, FS; Cui, S. Isolation and identification of flavonoids in licorice and a study of their inhibitory effects on tyrosinase. J. Agric. Food Chem 2005, 53, 7408–7414. [Google Scholar]
- Xie, LP; Chen, QX; Huang, H; Wang, HZ; Zhang, RQ. Inhibitory effects of some flavonoids on the activity of mushroom tyrosinase. Biochemistry 2003, 68, 487–491. [Google Scholar]
- Masamoto, Y; Murata, Y; Baba, K; Shimoishi, Y; Tada, M; Takahata, K. Inhibitory effects of esculetin on melanin biosynthesis. Biol. Pharm Bullutin 2004, 27, 422–425. [Google Scholar]
- Chen, QX; Ke, LN; Song, KK; Huang, H; Liu, XD. Inhibitory effects of hexylresorcinol and dodecylresorcinol on mushroom (Agaricus bisporus) tyrosinase. Protein J 2004, 23, 135–141. [Google Scholar]
- Shin, NH; Ryu, SY; Choi, EJ; Kang, SH; Chang, IM; Min, KR; Kim, Y. Oxyresveratrol as the potent inhibitor on dopa oxidase activity of mushroom tyrosinase. Biochem. Biophys. Res. Commun 1998, 243, 801–803. [Google Scholar]
- Ohguchi, K; Tanaka, T; Iliya, I; Ito, T; Iinuma, M; Matsumoto, K; Akao, Y; Nozawa, Y. Gnetol as a potent tyrosinase inhibitor from genus Gnetum. Biosci. Biotechnol. Biochem 2003, 67, 663–665. [Google Scholar]
- Kim, DS; Park, SH; Kwon, SB; Li, K; Youn, SW; Park, KC. (−)-Epigallocatechin-3- gallate and hinokitiol reduce melanin synthesis via decreased MITF production. Arch. Pharm. Res 2004, 27, 334–339. [Google Scholar]
- No, JK; Soung, DY; Kim, YJ; Shim, KH; Jun, YS; Rhee, SH; Yokozawa, T; Chung, HY. Inhibition of tyrosinase by green tea components. Life Sci 1999, 65, PL241–246. [Google Scholar]
- Zocca, F; Lomolino, G; Lante, A. Antibrowning potential of Brassicacaea processing water. Bioresour. Technol 2010, 101, 3791–3795. [Google Scholar]
- Negishi, O; Ozawa, T. Inhibition of enzymatic browning and protection of sulfhydryl enzymes by thiol compounds. Phytochemistry 2000, 54, 481–487. [Google Scholar]
- Nagai, T; Suzuki, N. Partial purification of polyphenol oxidase from Chinese cabbage Brassica rapa L. J. Agric. Food Chem 2001, 49, 3922–3926. [Google Scholar]
- Yang, CP; Fujita, S; Kohno, K; Kusubayashi, A; Ashrafuzzaman, M; Hayashi, N. Partial purification and characterization of polyphenol oxidase from banana (Musa sapientum L.) peel. J. Agric. Food Chem 2001, 49, 1446–1449. [Google Scholar]
- Pérez-Gilabert, M; García-Carmona, F. Dimethyl sulfide, a volatile flavor constituent, is a slow-binding inhibitor of tyrosinase. Biochem. Biophys. Res. Commun 2001, 285, 257–261. [Google Scholar]
- Graf, E; Empson, KL; Eaton, JW. Phytic acid. A natural antioxidant. J. Biol. Chem 1987, 262, 11647–11650. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Nihei, K; Soria, F; Takasaki, M; Calderón, JS; Céspedes, CL. Tyrosinase inhibitors from galls of Rhus javanica leaves and their effects on insects. Z. Naturforsch C 2003, 58, 719–725. [Google Scholar]
- Sasaki, K; Yoshizaki, F. Nobiletin as a tyrosinase inhibitor from the peel of Citrus fruit. Biol. Pharm Bullutin 2002, 25, 806–808. [Google Scholar]
- Kubo, I; Kinst-Hori, I. Flavonols from saffron flower: tyrosinase inhibitory activity and inhibition mechanism. J. Agric. Food Chem 1999, 47, 4121–4125. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Chaudhuri, SK; Kubo, Y; Sánchez, Y; Ogura, T. Flavonols from Heterotheca inuloides: tyrosinase inhibitory activity and structural criteria. Bioorg. Med. Chem 2000, 8, 1749–1755. [Google Scholar]
- Masuda, T; Yamashita, D; Takeda, Y; Yonemori, S. Screening for tyrosinase inhibitors among extracts of seashore plants and identification of potent inhibitors from Garcinia subelliptica. Biosci. Biotechnol. Biochem 2005, 69, 197–201. [Google Scholar]
- Gómez-Cordovés, C; Bartolomé, B; Vieira, W; Virador, VM. Effects of wine phenolics and sorghum tannins on tyrosinase activity and growth of melanoma cells. J. Agric. Food Chem 2001, 49, 1620–1624. [Google Scholar]
- An, BJ; Kwak, JH; Son, JH; Park, JM; Lee, JY; Park, TS; Kim, SY; Kim, YS; Jo, C; Byun, MW. Physiological activity of irradiated green tea polyphenol on the human skin. Am. J. Chin. Med 2005, 33, 535–546. [Google Scholar]
- Shoji, T; Masumoto, S; Moriichi, N; Kobori, M; Kanda, T; Shinmoto, H; Tsushida, T. Procyanidin trimers to pentamers fractionated from apple inhibit melanogenesis in B16 mouse melanoma cells. J. Agric. Food Chem 2005, 53, 6105–6111. [Google Scholar]
- Yamakoshi, J; Otsuka, F; Sano, A; Tokutake, S; Saito, M; Kikuchi, M; Kubota, Y. Lightening effect on ultraviolet-induced pigmentation of guinea pig skin by oral administration of a proanthocyanidin-rich extract from grape seeds. Pigment Cell Res 2003, 16, 629–638. [Google Scholar]
- Kubo, I; Kinst-Hori, I; Kubo, Y; Yamagiwa, Y; Kamikawa, T; Haraguchi, H. Molecular design of antibrowning agents. J. Agric. Food Chem 2000, 48, 1393–1399. [Google Scholar]
- Roh, JS; Han, JY; Kim, JH; Hwang, JK. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol. Pharm Bullutin 2004, 27, 1976–1978. [Google Scholar]
- Kubo, I; Chen, QX; Nihei, K; Calderón, JS; Céspedes, CL. Tyrosinase inhibition kinetics of anisic acid. Z. Naturforsch C 2003, 58, 713–718. [Google Scholar]
- Kubo, I; Kinst-Hori, I. Tyrosinase inhibitory activity of the olive oil flavor compounds. J. Agric. Food Chem 1999, 47, 4574–4578. [Google Scholar]
- Kim, HP; Son, KH; Chang, HW; Kang, SS. Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharmacol. Sci 2004, 96, 229–245. [Google Scholar]
- Francis, JA; Rumbeiha, W; Nair, MG. Constituents in Easter lily flowers with medicinal activity. Life Sci 2004, 76, 671–683. [Google Scholar]
- Hou, DX; Fujii, M; Terahara, N; Yoshimoto, M. Molecular Mechanisms Behind the Chemopreventive Effects of Anthocyanidins. J. Biomed. Biotechnol 2004, 5, 321–325. [Google Scholar]
- Seeram, NP; Zhang, Y; Nair, MG. Inhibition of proliferation of human cancer cells and cyclooxygenase enzymes by anthocyanidins and catechins. Nutr Cancer 2003, 46, 101–106. [Google Scholar]
- Woo, KJ; Jeong, YJ; Inoue, H; Park, JW; Kwon, TK. Chrysin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression through the inhibition of nuclear factor for IL-6 (NF-IL6) DNA-binding activity. FEBS Lett 2005, 579, 705–711. [Google Scholar]
- Liang, YC; Huang, YT; Tsai, SH; Lin-Shiau, SY; Chen, CF; Lin, JK. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis 1999, 20, 1945–1952. [Google Scholar]
- Raso, GM; Meli, R; Di Carlo, G; Pacilio, M; Di Carlo, R. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci 2001, 68, 921–931. [Google Scholar]
- Chi, YS; Cheon, BS; Kim, HP. Effect of wogonin, a plant flavone from Scutellaria radix, on the suppression of cyclooxygenase-2 and the induction of inducible nitric oxide synthase in lipopolysaccharide-treated RAW 264.7 cells. Biochem. Pharmacol 2001, 61, 1195–1203. [Google Scholar]
- O’Leary, KA; de Pascual-Tereasa, S; Needs, PW; Bao, YP; O’Brien, NM; Williamson, G. Effect of flavonoids and vitamin E on cyclooxygenase-2 (COX-2) transcription. Mutat. Res 2004, 551, 245–254. [Google Scholar]
- Mutoh, M; Takahashi, M; Fukuda, K; Komatsu, H; Enya, T; Matsushima-Hibiya, Y; Mutoh, H; Sugimura, T; Wakabayashi, K. Suppression by flavonoids of cyclooxygenase-2 promoterdependent transcriptional activity in colon cancer cells: structure-activity relationship. Jpn. J. Cancer Res 2000, 91, 686–691. [Google Scholar]
- Huss, U; Ringbom, T; Perera, P; Bohlin, L; Vasänge, M. Screening of ubiquitous plant constituents for COX-2 inhibition with a scintillation proximity based assay. J. Nat. Prod 2002, 65, 1517–1521. [Google Scholar]
- Seaver, B; Smith, JR. Inhibition of COX isoforms by nutraceuticals. J. Herb. Pharmacother 2004, 4, 11–18. [Google Scholar]
- Murias, M; Handler, N; Erker, T; Pleban, K; Ecker, G; Saiko, P; Szekeres, T; Jäger, W. Resveratrol analogues as selective cyclooxygenase-2 inhibitors: synthesis and structure-activity relationship. Bioorg. Med. Chem 2004, 12, 5571–5578. [Google Scholar]
- Chi, YS; Jong, HG; Son, KH; Chang, HW; Kang, SS; Kim, HP. Effects of naturally occurring prenylated flavonoids on enzymes metabolizing arachidonic acid: cyclooxygenases and lipoxygenases. Biochem. Pharmacol 2001, 62, 1185–1191. [Google Scholar]
- Selvam, C; Jachak, SM; Bhutani, KK. Cyclooxygenase inhibitory flavonoids from the stem bark of Semecarpus anacardium Linn. Phytother. Res 2004, 18, 582–584. [Google Scholar]
- Yoo, SW; Kim, JS; Kang, SS; Son, KH; Chang, HW; Kim, HP; Bae, K; Lee, CO. Constituents of the fruits and leaves of Euodia daniellii. Arch. Pharm. Res 2002, 25, 824–830. [Google Scholar]
- Kim, HP; Mani, I; Iversen, L; Ziboh, VA. Effects of naturally-occurring flavonoids and biflavonoids on epidermal cyclooxygenase and lipoxygenase from guinea-pigs. Prostaglandins Leukot. Essent Fatty Acids 1998, 58, 17–24. [Google Scholar]
- Chen, Y; Yang, L; Lee, TJ. Oroxylin A inhibition of lipopolysaccharide-induced iNOS and COX-2 gene expression via suppression of nuclear factor-kappaB activation. Biochem. Pharmacol 2000, 59, 1445–1457. [Google Scholar]
- Rossi, A; Ligresti, A; Longo, R; Russo, A; Borrelli, F; Sautebin, L. The inhibitory effect of propolis and caffeic acid phenethyl ester on cyclooxygenase activity in J774 macrophages. Phytomedicine 2002, 9, 530–535. [Google Scholar] [Green Version]
- You, KM; Jong, HG; Kim, HP. Inhibition of cyclooxygenase/lipoxygenase from human platelets by polyhydroxylated/methoxylated flavonoids isolated from medicinal plants. Arch. Pharm. Res 1999, 22, 18–24. [Google Scholar]
- Prasad, NS; Raghavendra, R; Lokesh, BR; Naidu, KA. Spice phenolics inhibit human PMNL 5-lipoxygenase. Prostaglandins Leukot. Essent Fatty Acids 2004, 70, 521–528. [Google Scholar]
- O’Prey, J; Brown, J; Fleming, J; Harrison, PR. Effects of dietary flavonoids on major signal transduction pathways in human epithelial cells. Biochem. Pharmacol 2003, 66, 2075–2088. [Google Scholar]
- Hsieh, RJ; German, JB; Kinsella, JE. Relative inhibitory potencies of flavonoids on 12-lipoxygenase of fish gill. Lipids 1988, 23, 322–326. [Google Scholar]
- Sekiya, K; Okuda, H; Arichi, S. Selective inhibition of platelet lipoxygenase by esculetin. Biochim. Biophys Acta 1982, 713, 68–72. [Google Scholar]
- Sadik, CD; Sies, H; Schewe, T. Inhibition of 15-lipoxygenases by flavonoids: structure-activity relations and mode of action. Biochem. Pharmacol 2003, 65, 773–781. [Google Scholar]
- Nakadate, T; Yamamoto, S; Aizu, E; Kato, R. Effects of flavonoids and antioxidants on 12-Otetradecanoyl- phorbol-13-acetate-caused epidermal ornithine decarboxylase induction and tumor promotion in relation to lipoxygenase inhibition by these compounds. Gann 1984, 75, 214–222. [Google Scholar]
- Fylaktakidou, KC; Hadjipavlou-Litina, DJ; Litinas, KE; Nicolaides, DN. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des 2004, 10, 3813–3833. [Google Scholar]
- Malterud, KE; Rydland, KM. Inhibitors of 15-lipoxygenase from orange peel. J. Agric. Food Chem 2000, 48, 5576–5580. [Google Scholar]
- Rui, YC. Advances in pharmacological studies of silymarin. Mem. Inst Oswaldo Cruz 1991, 86, 79–85. [Google Scholar]
- Robak, J; Duniec, Z; Rzadkowska-Bodalska, H; Olechnowicz-Stepień, W; Cisowski, W. The effect of some flavonoids on non-enzymatic lipid oxidation and enzymatic oxidation of arachidonic acid. Pol. J. Pharmacol. Pharm 1986, 38, 483–491. [Google Scholar]
- Ferrándiz, ML; Nair, AG; Alcaraz, MJ. Inhibition of sheep platelet arachidonate metabolism by flavonoids from Spanish and Indian medicinal herbs. Pharmazie 1990, 45, 206–208. [Google Scholar]
- Yoshimoto, T; Furukawa, M; Yamamoto, S; Horie, T; Watanabe-Kohno, S. Flavonoids: potent inhibitors of arachidonate 5-lipoxygenase. Biochem. Biophys. Res. Commun 1983, 116, 612–618. [Google Scholar]
- Schewe, T; Kühn, H; Sies, H. Flavonoids of cocoa inhibit recombinant human 5-lipoxygenase. J. Nutr 2002, 132, 1825–1829. [Google Scholar]
- Oomah, BD; Corbé, A; Balasubramanian, P. Antioxidant and anti-inflammatory activities of bean (Phaseolus vulgaris L.) hulls. J. Agric. Food. Chem 2010, 58, 8225–8230. [Google Scholar]
- Abad, MJ; Bermejo, P; Villar, A. The activity of flavonoids extracted from Tanacetum microphyllum DC. (Compositae) on soybean lipoxygenase and prostaglandin synthetase. Gen. Pharmacol 1995, 26, 815–819. [Google Scholar]
- Mattammal, MB; Strong, R; Lakshmi, VM; Chung, HD; Stephenson, AH. Prostaglandin H synthetase-mediated metabolism of dopamine: implication for Parkinson’s disease. J. Neurochem 1995, 64, 1645–1654. [Google Scholar]
- Zhou, LE; Wang, WJ; Bai, JY; Cheng, GF. Effects of ginkgolide B on arachidonic acid metabolizing enzymes and level of intracellular calcium in rat polymorphonuclear leukocytes. Yao Xue Xue Bao 2001, 36, 92–95. [Google Scholar]
- Kim, HR; Pham, HT; Ziboh, VA. Flavonoids differentially inhibit guinea pig epidermal cytosolic phospholipase A2. Prostaglandins Leukot. Essent Fatty Acids 2001, 65, 281–286. [Google Scholar]
- Grataroli, R; Léonardi, J; Charbonnier, M; Lafont, R; Lafont, H; Nalbone, G. Effects of dietary corn oil and salmon oil on lipids and prostaglandin E2 in rat gastric mucosa. Lipids 1988, 23, 666–670. [Google Scholar]
- Grataroli, R; Vamecq, J; Poupaert, JH; Léonardi, J; Termine, E; Lafont, H; Nalbone, G. Effects of dietary n−6/n−3 ratios on lipid and prostaglandin E2 metabolism in rat gastric mucosa. J. Lipid Mediat 1992, 5, 227–236. [Google Scholar]
- Han, CK; Son, MJ; Chang, HW; Chi, YS; Park, H; Kim, HP. Inhibition of prostaglandin production by a structurally-optimized flavonoid derivative, 2′,4′,7-trimethoxyflavone and cellular action mechanism. Biol. Pharm Bullutin 2005, 28, 1366–1370. [Google Scholar]
- Tanaka, S; Sato, T; Akimoto, N; Yano, M; Ito, A. Prevention of UVB-induced photoinflammation and photoaging by a polymethoxy flavonoid, nobiletin, in human keratinocytes in vivo and in vitro. Biochem. Pharmacol 2004, 68, 433–439. [Google Scholar]
- Ticli, FK; Hage, LI; Cambraia, RS; Pereira, PS; Magro, AJ; Fontes, MR; Stábeli, RG; Giglio, JR; França, SC; Soares, AM; Sampaio, SV. Rosmarinic acid, a new snake venom phospholipase A2 inhibitor from Cordia verbenacea (Boraginaceae): antiserum action potentiation and molecular interaction. Toxicon 2005, 46, 318–327. [Google Scholar]
- Adam, O. Dietary fatty acids and immune reactions in synovial tissue. Eur. J. Med. Res 2003, 8, 381–387. [Google Scholar]
- Shao, ZH; Vanden Hoek, TL; Li, CQ; Schumacker, PT; Becker, LB; Chan, KC; Qin, Y; Yin, JJ; Yuan, CS. Synergistic effect of Scutellaria baicalensis and grape seed proanthocyanidins on scavenging reactive oxygen species in vitro. Am. J. Chin. Med 2004, 32, 89–95. [Google Scholar]
- Dew, TP; Day, AJ; Morgan, MR. Xanthine oxidase activity in vitro: effects of food extracts and components. J. Agric. Food Chem 2005, 53, 6510–6515. [Google Scholar]
- Lin, JK; Chen, PC; Ho, CT; Lin-Shiau, SY. Inhibition of xanthine oxidase and suppression of intracellular reactive oxygen species in HL-60 cells by theaflavin-3,3′-digallate, (−)-epigallocatechin-3-gallate, and propyl gallate. J. Agric. Food Chem 2000, 48, 2736–2743. [Google Scholar]
- Kurisawa, M; Chung, JE; Uyama, H; Kobayashi, S. Oxidative coupling of epigallocatechin gallate amplifies antioxidant activity and inhibits xanthine oxidase activity. Chem. Commun 2004, 7, 294–295. [Google Scholar]
- van Hoorn, DE; Nijveldt, RJ; van Leeuwen, PA; Hofman, Z; M’Rabet, L; de Bont, DB; van Norren, K. Accurate prediction of xanthine oxidase inhibition based on the structure of flavonoids. Eur. J. Pharmacol 2002, 451, 111–118. [Google Scholar]
- Selloum, L; Reichl, S; Müller, M; Sebihi, L; Arnhold, J. Effects of flavonols on the generation of superoxide anion radicals by xanthine oxidase and stimulated neutrophils. Arch. Biochem. Biophys 2001, 395, 49–56. [Google Scholar]
- Nagao, A; Seki, M; Kobayashi, H. Inhibition of xanthine oxidase by flavonoids. Biosci. Biotechnol. Biochem 1999, 63, 1787–1790. [Google Scholar]
- Iio, M; Ono, Y; Kai, S; Fukumoto, M. Effects of flavonoids on xanthine oxidation as well as on cytochrome c reduction by milk xanthine oxidase. J. Nutr. Sci. Vitaminol 1986, 32, 635–642. [Google Scholar]
- Zhu, JX; Wang, Y; Kong, LD; Yang, C; Zhang, X. Effects of Biota orientalis extract and its flavonoid constituents, quercetin and rutin on serum uric acid levels in oxonate-induced mice and xanthine dehydrogenase and xanthine oxidase activities in mouse liver. J. Ethnopharmacol 2004, 93, 133–140. [Google Scholar]
- Moridani, MY; Pourahmad, J; Bui, H; Siraki, A; O’Brien, PJ. Dietary flavonoid iron complexes as cytoprotective superoxide radical scavengers. Free Radic. Biol. Med 2003, 34, 243–253. [Google Scholar]
- Foppoli, C; Coccia, R; Cini, C; Rosei, MA. Catecholamines oxidation by xanthine oxidase. Biochim. Biophys Acta 1997, 1334, 200–206. [Google Scholar]
- Day, AJ; Bao, Y; Morgan, MR; Williamson, G. Conjugation position of quercetin glucuronides and effect on biological activity. Free Radic. Biol. Med 2000, 29, 1234–1243. [Google Scholar]
- Lin, CM; Chen, CS; Chen, CT; Liang, YC; Lin, JK. Molecular modeling of flavonoids that inhibits xanthine oxidase. Biochem. Biophys. Res. Commun 2002, 294, 167–172. [Google Scholar]
- Beiler, JM; Martin, GJ. The inhibition of xanthine oxidase by flavonoids and related compounds. J. Biol. Chem 1951, 192, 831–834. [Google Scholar]
- Yoshizumi, K; Nishioka, N; Tsuji, T. Xanthine oxidase inhibitory activity and hypouricemia effect of propolis in rats. Yakugaku Zasshi 2005, 125, 315–321. [Google Scholar]
- Russo, A; Longo, R; Vanella, A. Antioxidant activity of propolis: role of caffeic acid phenethyl ester and galangin. Fitoterapia 2002, 73, S21–S29. [Google Scholar]
- Tapia, A; Rodriguez, J; Theoduloz, C; Lopez, S; Feresin, GE; Schmeda-Hirschmann, G. Free radical scavengers and antioxidants from Baccharis grisebachii. J. Ethnopharmacol 2004, 95, 155–161. [Google Scholar]
- Huang, Y; Tsang, SY; Yao, X; Chen, ZY. Biological properties of baicalein in cardiovascular system. Curr. Drug Targets Cardiovasc. Haematol. Disord 2005, 5, 177–184. [Google Scholar]
- Shieh, DE; Liu, LT; Lin, CC. Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res 2000, 20, 2861–2865. [Google Scholar]
- Chang, WS; Lee, YJ; Lu, FJ; Chiang, HC. Inhibitory effects of flavonoids on xanthine oxidase. Anticancer Res 1993, 13, 2165–2170. [Google Scholar]
- Varga, Z; Ujhelyi, L; Kiss, A; Balla, J; Czompa, A; Antus, S. Effect of silybin on phorbol myristate actetate-induced protein kinase C translocation NADPH oxidase activity and apoptosis in human neutrophils. Phytomedicine 2004, 11, 206–212. [Google Scholar]
- Sheu, SY; Lai, CH; Chiang, HC. Inhibition of xanthine oxidase by purpurogallin and silymarin group. Anticancer Res 1998, 18, 263–267. [Google Scholar]
- Łuczaj, W; Skrzydlewska, E. Antioxidative properties of black tea. Prev. Med 2005, 40, 910–918. [Google Scholar]
- Wang, Y; Zhu, JX; Kong, LD; Yang, C; Cheng, CH; Zhang, X. Administration of procyanidins from grape seeds reduces serum uric acid levels and decreases hepatic xanthine dehydrogenase/oxidase activities in oxonate-treated mice. Basic Clin. Pharmacol. Toxicol 2004, 94, 232–237. [Google Scholar]
- Packer, L; Rimbach, G; Virgili, F. Antioxidant activity and biologic properties of a procyanidin-rich extract from pine (Pinus maritima) bark, pycnogenol. Free Radic. Biol. Med 1999, 27, 704–724. [Google Scholar]
- Moini, H; Guo, Q; Packer, L. Enzyme inhibition and protein-binding action of the procyanidinrich french maritime pine bark extract, pycnogenol: effect on xanthine oxidase. J. Agric. Food Chem 2000, 48, 5630–5639. [Google Scholar]
- Moini, H; Guo, Q; Packe, L. Xanthine oxidase and xanthine dehydrogenase inhibition by the procyanidin-rich French maritime pine bark extract, pycnogenol: a protein binding effect. Adv. Exp. Med. Biol 2002, 505, 141–149. [Google Scholar]
- Acquaviva, R; Russo, A; Galvano, F; Galvano, G; Barcellona, ML; Li Volti, G; Vanella, A. Cyanidin and cyanidin 3-O-beta-d-glucoside as DNA cleavage protectors and antioxidants. Cell Biol. Toxicol 2003, 19, 243–252. [Google Scholar]
- Cioffi, G; D’Auria, M; Braca, A; Mendez, J; Castillo, A; Morelli, I; de Simone, F; de Tommasi, N. Antioxidant and free-radical scavenging activity of constituents of the leaves of Tachigalia paniculata. J. Nat. Prod 2002, 65, 1526–1529. [Google Scholar]
- Robak, J; Gryglewski, RJ. Flavonoids are scavengers of superoxide anions. Biochem. Pharmacol 1988, 37, 837–841. [Google Scholar]
- Ignatov, S; Shishniashvili, D; Ge, B; Scheller, FW; Lisdat, F. Amperometric biosensor based on a functionalized gold electrode for the detection of antioxidants. Biosens. Bioelectron 2002, 17, 191–199. [Google Scholar]
- Marfak, A; Trouillas, P; Allais, DP; Champavier, Y; Calliste, CA; Duroux, JL. Radiolysis of kaempferol in water/methanol mixtures. Evaluation of antioxidant activity of kaempferol and products formed. J. Agric. Food Chem 2003, 51, 1270–1277. [Google Scholar]
- Lu, Y; Foo, LY. Antioxidant activities of polyphenols from sage (Salvia officinalis). Food Chem 2001, 75, 197–202. [Google Scholar]
- Beyer, G; Melzig, MF. Effects of selected flavonoids and caffeic acid derivatives on hypoxanthine-xanthine oxidase-induced toxicity in cultivated human cells. Planta Med 2003, 69, 1125–1129. [Google Scholar]
- Park, YH; Han, DW; Suh, H; Ryu, GH; Hyon, SH; Cho, BK; Park, JC. Protective effects of green tea polyphenol against reactive oxygen species-induced oxidative stress in cultured rat calvarial osteoblast. Cell Biol. Toxicol 2003, 19, 325–337. [Google Scholar]
- Rah, DK; Han, DW; Baek, HS; Hyon, SH; Park, JC. Prevention of reactive oxygen species-induced oxidative stress in human microvascular endothelial cells by green tea polyphenol. Toxicol. Lett 2005, 155, 269–275. [Google Scholar]
- Liu, H; Yang, XL; Wang, Y; Tang, XQ; Jiang, DY; Xu, HB. Protective effects of scutellarin on superoxide-induced oxidative stress in rat cortical synaptosomes. Acta Pharmacol. Sin 2003, 24, 1113–1117. [Google Scholar]
- Taubert, D; Breitenbach, T; Lazar, A; Censarek, P; Harlfinger, S; Berkels, R; Klaus, W; Roesen, R. Reaction rate constants of superoxide scavenging by plant antioxidants. Free Radic. Biol. Med 2003, 35, 1599–1607. [Google Scholar]
- Cheel, J; Theoduloz, C; Rodríguez, J; Schmeda-Hirschmann, G. Free radical scavengers and antioxidants from Lemongrass (Cymbopogon citratus (DC.) Stapf.). J. Agric Food Chem 2005, 53, 2511–2517. [Google Scholar]
- Moridani, MY; O’Brien, PJ. Iron complexes of deferiprone and dietary plant catechols as cytoprotective superoxide radical scavengers. Biochem. Pharmacol 2001, 62, 1579–1585. [Google Scholar]
- Shi, H; Zhao, B; Xin, W. Scavenging effects of baicalin on free radicals and its protection on erythrocyte membrane from free radical injury. Biochem. Mol. Biol. Int 1995, 35, 981–994. [Google Scholar]
- Toyo’oka, T; Kashiwazaki, T; Kato, M. On-line screening methods for antioxidants scavenging superoxide anion radical and hydrogen peroxide by liquid chromatography with indirect chemiluminescence detection. Talanta 2003, 60, 467–475. [Google Scholar]
- Stinefelt, B; Leonard, SS; Blemings, KP; Shi, X; Klandorf, H. Free radical scavenging, DNA protection, and inhibition of lipid peroxidation mediated by uric acid. Ann. Clin. Lab. Sci 2005, 35, 37–45. [Google Scholar]
- Mishra, B; Priyadarsini, KI; Kumar, MS; Unnikrishnan, MK; Mohan, H. Effect of O-glycosilation on the antioxidant activity and free radical reactions of a plant flavonoid, chrysoeriol. Bioorg. Med. Chem 2003, 11, 2677–2685. [Google Scholar]
- Candan, F. Effect of Rhus coriaria L. (Anacardiaceae) on superoxide radical scavenging and xanthine oxidase activity. J. Enzyme Inhib. Med. Chem 2003, 18, 59–62. [Google Scholar]
- Ozgová, S; Hermánek, J; Gut, I. Different antioxidant effects of polyphenols on lipid peroxidation and hydroxyl radicals in the NADPH-, Fe-ascorbate- and Fe-microsomal systems. Biochem. Pharmacol 2003, 66, 1127–1137. [Google Scholar]
- Wei, IH; Wu, YC; Wen, CY; Shieh, JY. Green tea polyphenol (−)-epigallocatechin gallate attenuates the neuronal NADPH-d/nNOS expression in the nodose ganglion of acute hypoxic rats. Brain Res 2004, 999, 73–80. [Google Scholar]
- Chen, S; Deng, PS; Swiderek, K; Li, M; Chan, SI. Interaction of flavones and their bromoacetyl derivatives with NAD(P)H:quinone acceptor oxidoreductase. Mol. Pharmacol 1995, 47, 419–424. [Google Scholar]
- Liu, XF; Liu, ML; Iyanagi, T; Legesse, K; Lee, TD; Chen, SA. Inhibition of rat liver NAD(P)H:quinone acceptor oxidoreductase (DT-diaphorase) by flavonoids isolated from the Chinese herb scutellariae radix (Huang Qin). Mol. Pharmacol 1990, 37, 911–915. [Google Scholar]
- Tamura, M; Kagawa, S; Tsuruo, Y; Ishimura, K; Morita, K. Effects of flavonoid compounds on the activity of NADPH diaphorase prepared from the mouse brain. Jpn. J. Pharmacol 1994, 65, 371–373. [Google Scholar]
- Terland, O; Flatmark, T; Tangerås, A; Grønberg, M. Dopamine oxidation generates an oxidative stress mediated by dopamine semiquinone and unrelated to reactive oxygen species. J. Mol. Cell Cardiol 1997, 29, 1731–1738. [Google Scholar]
- Mocchegiani, E; Bertoni-Freddari, C; Marcellini, F; Malavolta, M. Brain, aging and neurodegeneration: role of zinc ion availability. Prog. Neurobiol 2005, 75, 367–390. [Google Scholar]
- Kukreja, RC; Loesser, KE; Kearns, AA; Naseem, SA; Hess, ML. Protective effects of histidine during ischemia-reperfusion in isolated perfused rat hearts. Am. J. Physiol 1993, 264, H1370–H1381. [Google Scholar]
- Obata, T; Aomine, M; Yamanaka, Y. Protective effect of histidine on iron (II)-induced hydroxyl radical generation in rat hearts. J. Physiol Paris 1999, 93, 213–218. [Google Scholar]
- Obata, T; Inada, T. Protective effect of histidine on MPP+-induced hydroxyl radical generation in rat striatum. Brain Res 1999, 817, 206–208. [Google Scholar]
- Lundvig, D; Lindersson, E; Jensen, PH. Pathogenic effects of alpha-synuclein aggregation. Brain Res. Mol. Brain Res 2005, 134, 3–17. [Google Scholar]
- O’Dowd, Y; Driss, F; Dang, PM; Elbim, C; Gougerot-Pocidalo, MA; Pasquier, C; El-Benna, J. Antioxidant effect of hydroxytyrosol, a polyphenol from olive oil: scavenging of hydrogen peroxide but not superoxide anion produced by human neutrophils. Biochem. Pharmacol 2004, 68, 2003–2008. [Google Scholar]
- Everse, J; Coates, PW. Role of peroxidases in Parkinson disease: a hypothesis. Free Radic. Biol. Med 2005, 38, 1296–1310. [Google Scholar]
- Thiruchelvam, M; Prokopenko, O; Cory-Slechta, DA; Buckley, B; Mirochnitchenko, O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J. Biol. Chem 2005, 280, 22530–22539. [Google Scholar]
- Fornai, F; Carrì, MT; Ferri, A; Paolucci, E; Prisco, S; Bernardi, G; Rotilio, G; Mercuri, NB. Resistance to striatal dopamine depletion induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice expressing human mutant Cu,Zn superoxide dismutase. Neurosci. Lett 2002, 325, 124–128. [Google Scholar]
- Zhao, Y; Gao, Z; Li, H; Xu, H. Hemin/nitrite/H2O2 induces brain homogenate oxidation and nitration: effects of some flavonoids. Biochim. Biophys Acta 2004, 1675, 105–112. [Google Scholar]
- Klivenyi, P; Andreassen, OA; Ferrante, RJ; Dedeoglu, A; Mueller, G; Lancelot, E; Bogdanov, M; Andersen, JK; Jiang, D; Beal, MF. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4- phenyl-1,2,5,6-tetrahydropyridine. J. Neurosci 2000, 20, 1–7. [Google Scholar]
- Biswas, SK; McClure, D; Jimenez, LA; Megson, IL; Rahman, I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid. Redox Signal 2005, 7, 32–41. [Google Scholar]
- Ferrari, CK. Functional foods, herbs and nutraceuticals: towards biochemical mechanisms of healthy aging. Biogerontology 2004, 5, 275–289. [Google Scholar]
- Mansouri, A; Makris, DP; Kefalas, P. Determination of hydrogen peroxide scavenging activity of cinnamic and benzoic acids employing a highly sensitive peroxyoxalate chemiluminescencebased assay: structure-activity relationships. J. Pharm. Biomed. Anal 2005, 39, 22–26. [Google Scholar]
- Yilmaz, Y; Toledo, RT. Major flavonoids in grape seeds and skins: antioxidant capacity of catechin, epicatechin, and gallic acid. J. Agric. Food Chem 2004, 52, 255–260. [Google Scholar]
- Sroka, Z; Cisowski, W. Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food Chem. Toxicol 2003, 41, 753–758. [Google Scholar]
- Vanzani, P; Rossetto, M; Rigo, A; Vrhovsek, U; Mattivi, F; D’Amato, E; Scarpa, M. Major phytochemicals in apple cultivars: contribution to peroxyl radical trapping efficiency. J. Agric. Food Chem 2005, 53, 3377–3382. [Google Scholar]
- López-Alarcón, C; Lissi, E. Interaction of pyrogallol red with peroxyl radicals. A basis for a simple methodology for the evaluation of antioxidant capabilities. Free Radic. Res 2005, 39, 729–736. [Google Scholar]
- Mazzio, EA; Reams, RR; Soliman, KF. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 2004, 1004, 29–44. [Google Scholar]
- Kabuto, H; Nishizawa, M; Tada, M; Higashio, C; Shishibori, T; Kohno, M. Zingerone [4-(4-hydroxy-3-methoxyphenyl)-2-butanone] prevents 6-hydroxydopamine-induced dopamine depression in mouse striatum and increases superoxide scavenging activity in serum. Neurochem. Res 2005, 30, 325–332. [Google Scholar]
- Caillet, S; Yu, H; Lessard, S; Lamoureux, G; Ajdukovic, D; Lacroix, M. Fenton reaction applied for screening natural antioxidants. Food Chem 2007, 100, 542–552. [Google Scholar]
- van Acker, SA; van den Berg, DJ; Tromp, MN; Griffioen, DH; van Bennekom, WP; van der Vijgh, WJ; Bast, A. Structural aspects of antioxidant activity of flavonoids. Free Radic. Biol. Med 1996, 20, 331–342. [Google Scholar]
- Brown, JE; Khodr, H; Hider, RC; Rice-Evans, CA. Structural dependence of flavonoid interactions with Cu2+ ions: implications for their antioxidant properties. Biochem. J 1998, 330, 1173–1178. [Google Scholar]
- Arora, A; Nair, MG; Strasburg, GM. Structure-activity relationships for antioxidant activities of a series of flavonoids in a liposomal system. Free Radic. Biol. Med 1998, 24, 1355–1363. [Google Scholar]
- Fernandez, MT; Mira, ML; Florêncio, MH; Jennings, KR. Iron and copper chelation by flavonoids: an electrospray mass spectrometry study. J. Inorg. Biochem 2002, 92, 105–111. [Google Scholar]
- Cheng, IF; Breen, K. On the ability of four flavonoids, baicilein, luteolin, naringenin, and quercetin, to suppress the Fenton reaction of the iron-ATP complex. Biometals 2000, 13, 77–83. [Google Scholar]
- Aherne, SA; O’Brien, NM. Mechanism of protection by the flavonoids, quercetin and rutin, against tert-butylhydroperoxide- and menadione-induced DNA single strand breaks in Caco-2 cells. Free Radic. Biol. Med 2000, 29, 507–514. [Google Scholar]
- Mahakunakorn, P; Tohda, M; Murakami, Y; Matsumoto, K; Watanabe, H. Antioxidant and free radical-scavenging activity of Choto-san and its related constituents. Biol. Pharm Bullutin 2004, 27, 38–46. [Google Scholar]
- Yoshida, H; Ishikawa, T; Hosoai, H; Suzukawa, M; Ayaori, M; Hisada, T; Sawada, S; Yonemura, A; Higashi, K; Ito, T; Nakajima, K; Yamashita, T; Tomiyasu, K; Nishiwaki, M; Ohsuzu, F; Nakamura, H. Inhibitory effect of tea flavonoids on the ability of cells to oxidize low density lipoprotein. Biochem. Pharmacol 1999, 58, 1695–1703. [Google Scholar]
- O’Coinceanainn, M; Bonnely, S; Baderschneider, B; Hynes, MJ. Reaction of iron(III) with theaflavin: complexation and oxidative products. J. Inorg. Biochem 2004, 98, 657–663. [Google Scholar]
- Fraga, CG; Oteiza, PI. Iron toxicity and antioxidant nutrients. Toxicology 2002, 180, 23–32. [Google Scholar]
- Hynes, MJ; Coinceanainn, MO. The kinetics and mechanisms of the reaction of iron(III) with gallic acid, gallic acid methyl ester and catechin. J. Inorg. Biochem 2001, 85, 131–142. [Google Scholar]
- Borsari, M; Gabbi, C; Ghelfi, F; Grandi, R; Saladini, M; Severi, S; Borella, F. Silybin, a new iron-chelating agent. J. Inorg. Biochem 2001, 85, 123–129. [Google Scholar]
- Kostyuk, VA; Potapovich, AI. Antiradical and chelating effects in flavonoid protection against silica-induced cell injury. Arch. Biochem. Biophys 1998, 355, 43–48. [Google Scholar]
- Schipper, HM. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med 2004, 37, 1995–2011. [Google Scholar]
- Abate, A; Yang, G; Wong, RJ; Schroder, H; Stevenson, DK; Dennery, PA. Apigenin decreases hemin-mediated heme oxygenase-1 induction. Free Radic. Biol. Med 2005, 39, 711–718. [Google Scholar]
- Kantengwa, S; Polla, BS. Flavonoids, but not protein kinase C inhibitors, prevent stress protein synthesis during erythrophagocytosis. Biochem. Biophys. Res. Commun 1991, 180, 308–314. [Google Scholar]
- Zatta, P; Lucchini, R; van Rensburg, SJ; Taylor, A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bullutin 2003, 62, 15–28. [Google Scholar]
- Schweizer, U; Bräuer, AU; Köhrle, J; Nitsch, R; Savaskan, NE. Selenium and brain function: a poorly recognized liaison. Brain Res. Brain Res. Rev 2004, 45, 164–178. [Google Scholar]
- Johnson, S. Is Parkinson’s disease the heterozygote form of Wilson’s disease: PD = 1/2 WD? Med Hypotheses 2001, 56, 171–173. [Google Scholar]
- Sziráki, I; Mohanakumar, KP; Rauhala, P; Kim, HG; Yeh, KJ; Chiueh, CC. Manganese: a transition metal protects nigrostriatal neurons from oxidative stress in the iron-induced animal model of Parkinsonism. Neuroscience 1998, 85, 1101–1111. [Google Scholar]
- Cuajungco, MP; Lees, GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol. Dis 1997, 4, 137–169. [Google Scholar]
- Bush, AI. Metals and neuroscience. Curr. Opin. Chem. Biol 2000, 4, 184–191. [Google Scholar]
- Zago, MP; Mackenzie, GG; Adamo, AM; Keen, CL; Oteiza, PI. Differential modulation of MAP kinases by zinc deficiency in IMR-32 cells: role of H2O2. Antioxid. Redox Signal 2005, 7, 1773–1782. [Google Scholar]
- Blonska, M; Bronikowska, J; Pietsz, G; Czuba, ZP; Scheller, S; Krol, W. Effects of ethanol extract of propolis (EEP) and its flavones on inducible gene expression in J774A.1 macrophages. J. Ethnopharmacol 2004, 91, 25–30. [Google Scholar]
- Cho, H; Yun, CW; Park, WK; Kong, JY; Kim, KS; Park, Y; Lee, S; Kim, BK. Modulation of the activity of pro-inflammatory enzymes, COX-2 and iNOS.; by chrysin derivatives. Pharmacol. Res 2004, 49, 37–43. [Google Scholar]
- Chen, JC; Ho, FM; Pei-Dawn, LC; Chen, CP; Jeng, KC; Hsu, HB; Lee, ST; Wen, TW; Lin, WW. Inhibition of iNOS gene expression by quercetin is mediated by the inhibition of IkappaB kinase, nuclear factor-kappa B and STAT1, and depends on heme oxygenase-1 induction in mouse BV-2 microglia. Eur. J. Pharmacol 2005, 521, 9–20. [Google Scholar]
- Martínez-Flórez, S; Gutiérrez-Fernández, B; Sánchez-Campos, S; González-Gallego, J; Tuñón, MJ. Quercetin attenuates nuclear factor-kappaB activation and nitric oxide production in interleukin-1beta-activated rat hepatocytes. J. Nutr 2005, 135, 1359–1365. [Google Scholar]
- Jung, WJ; Sung, MK. Effects of major dietary antioxidants on inflammatory markers of RAW 264.7 macrophages. Biofactors 2004, 21, 113–117. [Google Scholar]
- Cho, SY; Park, SJ; Kwon, MJ; Jeong, TS; Bok, SH; Choi, WY; Jeong, WI; Ryu, SY; Do, SH; Lee, CS; Song, JC; Jeong, KS. Quercetin suppresses proinflammatory cytokines production through MAP kinases andNF-kappaB pathway in lipopolysaccharide-stimulated macrophage. Mol. Cell Biochem 2003, 243, 153–160. [Google Scholar]
- van Meeteren, ME; Hendriks, JJ; Dijkstra, CD; van Tol, EA. Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease. Biochem. Pharmacol 2004, 67, 967–975. [Google Scholar]
- Scuro, LS; Simioni, PU; Grabriel, DL; Saviani, EE; Modolo, LV; Tamashiro, WM; Salgado, I. Suppression of nitric oxide production in mouse macrophages by soybean flavonoids accumulated in response to nitroprusside and fungal elicitation. BMC Biochem 2004, 21, 5. [Google Scholar]
- Hu, C; Kitts, DD. Luteolin and luteolin-7-O-glucoside from dandelion flower suppress iNOS and COX-2 in RAW264.7 cells. Mol. Cell Biochem 2004, 265, 107–113. [Google Scholar]
- Kim, SJ; Park, H; Kim, HP. Inhibition of nitric oxide production from lipopolysaccharidetreated RAW 264.7 cells by synthetic flavones: structure-activity relationship and action mechanism. Arch. Pharm. Res 2004, 27, 937–943. [Google Scholar]
- Matsuda, H; Morikawa, T; Ando, S; Toguchida, I; Yoshikawa, M. Structural requirements of flavonoids for nitric oxide production inhibitory activity and mechanism of action. Bioorg. Med. Chem 2003, 11, 1995–2000. [Google Scholar]
- Rojas, J; Payá, M; Devesa, I; Dominguez, JN; Ferrándiz, ML. Therapeutic administration of 3,4,5-trimethoxy-4′-fluorochalcone, a selective inhibitor of iNOS expression, attenuates the development of adjuvant-induced arthritis in rats. Naunyn Schmiedebergs Arch. Pharmacol 2003, 368, 225–233. [Google Scholar]
- Ko, HH; Tsao, LT; Yu, KL; Liu, CT; Wang, JP; Lin, CN. Structure-activity relationship studies on chalcone derivatives. The potent inhibition of chemical mediators release. Bioorg. Med. Chem 2003, 11, 105–111. [Google Scholar]
- Chiu, FL; Lin, JK. HPLC analysis of naturally occurring methylated catechins, 3″- and 4″-methyl-epigallocatechin gallate, in various fresh tea leaves and commercial teas and their potent inhibitory effects on inducible nitric oxide synthase in macrophages. J. Agric. Food Chem 2005, 53, 7035–7042. [Google Scholar]
- Sutherland, BA; Shaw, OM; Clarkson, AN; Jackson, DN; Sammut, IA; Appleton, I. Neuroprotective effects of (−)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: novel mechanisms of action. FASEB J 2005, 19, 258–260. [Google Scholar]
- Singh, R; Ahmed, S; Islam, N; Goldberg, VM; Haqqi, TM. Epigallocatechin-3-gallate inhibits interleukin-1beta-induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: suppression of nuclear factor kappaB activation by degradation of the inhibitor of nuclear factor kappaB. Arthritis Rheum 2002, 46, 2079–2086. [Google Scholar]
- Lee, SH; Seo, GS; Sohn, DH. Inhibition of lipopolysaccharide-induced expression of inducible nitric oxide synthase by butein in RAW 264.7 cells. Biochem. Biophys. Res. Commun 2004, 323, 125–132. [Google Scholar]
- Sautebin, L; Rossi, A; Serraino, I; Dugo, P; Di Paola, R; Mondello, L; Genovese, T; Britti, D; Peli, A; Dugo, G; Caputi, AP; Cuzzocrea, S. Effect of anthocyanins contained in a blackberry extract on the circulatory failure and multiple organ dysfunction caused by endotoxin in the rat. Planta Med 2004, 70, 745–752. [Google Scholar]
- Liu, H; Yang, X; Tang, R; Liu, J; Xu, H. Effect of scutellarin on nitric oxide production in early stages of neuron damage induced by hydrogen peroxide. Pharmacol. Res 2005, 51, 205–210. [Google Scholar]
- Lin, CM; Huang, ST; Liang, YC; Lin, MS; Shih, CM; Chang, YC; Chen, TY; Chen, CT. Isovitexin suppresses lipopolysaccharide-mediated inducible nitric oxide synthase through inhibition of NF-kappa B in mouse macrophages. Planta Med 2005, 71, 748–753. [Google Scholar]
- Sakata, K; Hirose, Y; Qiao, Z; Tanaka, T; Mori, H. Inhibition of inducible isoforms of cyclooxygenase and nitric oxide synthase by flavonoid hesperidin in mouse macrophage cell line. Cancer Lett 2003, 199, 139–145. [Google Scholar]
- Kanno, S; Shouji, A; Tomizawa, A; Hiura, T; Osanai, Y; Ujibe, M; Obara, Y; Nakahata, N; Ishikawa, M. Inhibitory effect of naringin on lipopolysaccharide (LPS)-induced endotoxin shock in mice and nitric oxide production in RAW 264.7 macrophages. Life Sci 2006, 78, 673–681. [Google Scholar]
- Lin, HY; Shen, SC; Chen, YC. Anti-inflammatory effect of heme oxygenase 1: glycosylation and nitric oxide inhibition in macrophages. J. Cell Physiol 2005, 202, 579–590. [Google Scholar]
- Chen, CJ; Raung, SL; Liao, SL; Chen, SY. Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem. Pharmacol 2004, 67, 957–965. [Google Scholar]
- Lin, HY; Juan, SH; Shen, SC; Hsu, FL; Chen, YC. Inhibition of lipopolysaccharideinduced nitric oxide production by flavonoids in RAW264.7 macrophages involves heme oxygenase-1. Biochem. Pharmacol 2003, 66, 1821–1832. [Google Scholar]
- Schümann, J; Prockl, J; Kiemer, AK; Vollmar, AM; Bang, R; Tiegs, G. Silibinin protects mice from T cell-dependent liver injury. J. Hepatol 2003, 39, 333–340. [Google Scholar]
- Kang, JS; Jeon, YJ; Kim, HM; Han, SH; Yang, KH. Inhibition of inducible nitric-oxide synthase expression by silymarin in lipopolysaccharide-stimulated macrophages. J. Pharmacol. Exp. Ther 2002, 302, 138–144. [Google Scholar]
- Banerjee, T; van der Vliet, A; Ziboh, VA. Downregulation of COX-2 and iNOS by amentoflavone and quercetin in A549 human lung adenocarcinoma cell line. Prostaglandins Leukot. Essent Fatty Acids 2002, 66, 485–492. [Google Scholar]
- Takahashi, T; Takasuka, N; Iigo, M; Baba, M; Nishino, H; Tsuda, H; Okuyama, T. Isoliquiritigenin, a flavonoid from licorice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci 2004, 95, 448–453. [Google Scholar]
- Lee, H; Kim, YO; Kim, H; Kim, SY; Noh, HS; Kang, SS; Cho, GJ; Choi, WS; Suk, K. lavonoid wogonin from medicinal herb is neuroprotective by inhibiting inflammatory activation of microglia. FASEB J 2003, 17, 1943–1944. [Google Scholar]
- Markovic, M; Miljkovic, Dj; Trajkovic, V. Regulation of inducible nitric oxide synthase by cAMP-elevating phospho-diesterase inhibitors. Curr. Drug Targets Inflamm Allergy 2003, 2, 63–79. [Google Scholar]
- Hulley, P; Hartikka, J; Abdel’Al, S; Engels, P; Buerki, HR; Wiederhold, KH; Müller, T; Kelly, P; Lowe, D; Lübbert, H. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur. J. Neurosci 1995, 7, 2431–2440. [Google Scholar]
- Yu, SM; Cheng, ZJ; Kuo, SC. Endothelium-dependent relaxation of rat aorta by butein, a novel cyclic AMP-specific phosphodiesterase inhibitor. Eur. J. Pharmacol 1995, 280, 69–77. [Google Scholar]
- Girotti, C; Ginet, M; Demarne, FC; Lagarde, M; Géloën, A. Lipolytic activity of cirsimarin extracted from Microtea debilis. Planta Med 2005, 71, 1170–1172. [Google Scholar]
- Dell’agli, M; Bellosta, S; Rizzi, L; Galli, GV; Canavesi, M; Rota, F; Parente, R; Bosisio, E; Romeo, S. A structure-activity study for the inhibition of metalloproteinase-9 activity and gene expression by analogues of gallocatechin-3-gallate. Cell Mol. Life Sci 2005, 62, 2896–2903. [Google Scholar]
- Ko, WC; Shih, CM; Lai, YH; Chen, JH; Huang, HL. Inhibitory effects of flavonoids on phosphodiesterase isozymes from guinea pig and their structure-activity relationships. Biochem. Pharmacol 2004, 68, 2087–2094. [Google Scholar]
- Paliyath, G; Poovaiah, BW. Identification of naturally occurring calmodulin inhibitors in plants and their effects on calcium- and calmodulin-promoted protein phosphorylation. Plant Cell Physiol 1985, 26, 201–209. [Google Scholar]
- Campos-Toimil, M; Lugnier, C; Droy-Lefaix, MT; Takeda, K. Inhibition of type 4 phosphodiesterase by rolipram and Ginkgo biloba extract (EGb 761) decreases agonist-induced rises in internal calcium in human endothelial cells. Arterioscler. Thromb. Vasc. Biol 2000, 20, E34–E40. [Google Scholar]
- Nichols, MR; Morimoto, BH. Differential inhibition of multiple cAMP phosphodiesterase isozymes by isoflavones and tyrphostins. Mol. Pharmacol 2000, 57, 738–745. [Google Scholar]
- Satake, N; Imanishi, M; Shibata, S. Increased nitroglycerin-induced relaxation by genistein in rat aortic rings. Eur. J. Pharmacol 1999, 377, 193–197. [Google Scholar]
- Saponara, R; Bosisio, E. Inhibition of cAMP-phosphodiesterase by biflavones of Ginkgo biloba in rat adipose tissue. J. Nat. Prod 1998, 61, 1386–1387. [Google Scholar]
- Kuppusamy, UR; Das, NP. Effects of flavonoids on cyclic AMP phosphodiesterase and lipid mobilization in rat adipocytes. Biochem. Pharmacol 1992, 44, 1307–1315. [Google Scholar]
- Orallo, F; Camiña, M; Alvarez, E; Basaran, H; Lugnier, C. Implication of cyclic nucleotide phosphodiesterase inhibition in the vasorelaxant activity of the citrus-fruits flavonoid (+/−)-naringenin. Planta Med 2005, 71, 99–107. [Google Scholar]
- Kumar, S; Sarkar, A; Sundar, D. Controlling aggregation propensity in A53T mutant of alphasynuclein causing Parkinson’s disease. Biochem. Biophys. Res. Commun 2009, 387, 305–309. [Google Scholar]
- McCormack, AL; Mak, SK; Shenasa, M; Langston, WJ; Forno, LS; Di Monte, DA. Pathologic modifications of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated squirrel monkeys. J. Neuropathol. Exp. Neurol 2008, 67, 793–802. [Google Scholar]
- Ulusoy, A; Febbraro, F; Jensen, PH; Kirik, D; Romero-Ramos, M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci 2010, 32, 409–422. [Google Scholar]
- Paxinou, E; Chen, Q; Weisse, M; Giasson, BI; Norris, EH; Rueter, SM; Trojanowski, JQ; Lee, VM; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci 2001, 21, 8053–8061. [Google Scholar]
- Santner, A; Uversky, VN. Metalloproteomics and metal toxicology of α-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar]
- Perez, RG; Hastings, TG. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J. Neurochem 2004, 89, 1318–1324. [Google Scholar]
- Volles, MJ; Lansbury, PT, Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar]
- Parihar, MS; Parihar, A; Fujita, M; Hashimoto, M; Ghafourifar, P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J. Biochem. Cell Biol 2009, 41, 2015–2024. [Google Scholar]
- Reindl, W; Yuan, J; Krämer, A; Strebhardt, K; Berg, T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008, 15, 459–466. [Google Scholar]
- Mbefo, MK; Paleologou, KE; Boucharaba, A; Oueslati, A; Schell, H; Fournier, M; Olschewski, D; Yin, G; Zweckstetter, M; Masliah, E; Kahle, PJ; Hirling, H; Lashuel, HA. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem 2010, 285, 2807–2822. [Google Scholar]
- Cozza, G; Bonvini, P; Zorzi, E; Poletto, G; Pagano, MA; Sarno, S; Donella-Deana, A; Zagotto, G; Rosolen, A; Pinna, LA; Meggio, F; Moro, S. Identification of ellagic acid as potent inhibitor of protein kinase CK2: a successful example of a virtual screening application. J. Med. Chem 2006, 49, 2363–2366. [Google Scholar]
- Chen, L; Feany, MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci 2005, 8, 657–663. [Google Scholar]
- Chinta, SJ; Mallajosyula, JK; Rane, A; Andersen, JK. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett 2010, 486, 235–239. [Google Scholar]
- Mandel, SA; Fishman-Jacob, T; Youdim, MB. Modeling sporadic Parkinson’s disease by silencing the ubiquitin E3 ligase component, SKP1A. Parkinsonism Relat. Disord 2009, 15, S148–S151. [Google Scholar]
- Yasuda, T; Mochizuki, H. The regulatory role of α-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis 2010, 15, 1312–1321. [Google Scholar]
- Xie, W; Li, X; Li, C; Zhu, W; Jankovic, J; Le, W. Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. J. Neurochem 2010, 115, 188–199. [Google Scholar]
- Hyun, DH; Lee, M; Halliwell, B; Jenner, P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem 2003, 86, 363–373. [Google Scholar]
- Zhu, W; Xie, W; Pan, T; Xu, P; Fridkin, M; Zheng, H; Jankovic, J; Youdim, MB; Le, W. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J 2007, 21, 3835–3844. [Google Scholar]
- Zhou, ZD; Lim, TM. Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic. Res 2009, 43, 417–430. [Google Scholar]
- McNaught, KS; Jnobaptiste, R; Jackson, T; Jengelley, TA. The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson’s disease. Synapse 2010, 64, 241–250. [Google Scholar]
- Olanow, CW. The pathogenesis of cell death in Parkinson’s disease—2007. Mov. Disord 2007, 22, S335–S342. [Google Scholar]
- Chang, CR; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci 2010, 1201, 34–39. [Google Scholar]
- Lee, JY; Nagano, Y; Taylor, JP; Lim, KL; Yao, TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol 2010, 189, 671–679. [Google Scholar]
- Renna, M; Jimenez-Sanchez, M; Sarkar, S; Rubinsztein, DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem 2010, 285, 11061–11067. [Google Scholar]
- Cherra, SJ, III; Kulich, SM; Uechi, G; Balasubramani, M; Mountzouris, J; Day, BW; Chu, CT. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol 2010, 190, 533–539. [Google Scholar]
- Sevlever, D; Jiang, P; Yen, SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar]
- Murgas Torrazza, R; Suryawan, A; Gazzaneo, MC; Orellana, RA; Frank, JW; Nguyen, HV; Fiorotto, ML; El-Kadi, S; Davis, TA. Leucine supplementation of a low-protein meal increases skeletal muscle and visceral tissue protein synthesis in neonatal pigs by stimulating mTOR-dependent translation initiation. J. Nutr 2010, 140, 2145–5212. [Google Scholar]
- Bauchart-Thevret, C; Cui, L; Wu, G; Burrin, DG. Arginine-induced stimulation of protein synthesis and survival in IPEC-J2 cells is mediated by mTOR but not nitric oxide. Am. J. Physiol. Endocrinol. Metab 2010, 299, E899–E909. [Google Scholar]
- Kim, E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr. Res. Pract 2009, 3, 64–71. [Google Scholar]
- Machiya, Y; Hara, S; Arawaka, S; Fukushima, S; Sato, H; Sakamoto, M; Koyama, S; Kato, T. Phosphorylated {alpha}-Synuclein at Ser-129 Is Targeted to the Proteasome Pathway in a Ubiquitin-independent Manner. Biol. Chem 2010, 285, 40732–40744. [Google Scholar]
- Crews, L; Spencer, B; Desplats, P; Patrick, C; Paulino, A; Rockenstein, E; Hansen, L; Adame, A; Galasko, D; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5, e9313. [Google Scholar]
- Tang, FY; Chiang, EP; Pai, MH. Consumption of S-Allylcysteine Inhibits the Growth of Human Non-Small-Cell Lung Carcinoma in a Mouse Xenograft Model. J Agric Food Chem 2010, in press. [Google Scholar]
- Petrovski, G; Das, DK. Does autophagy take a front seat in lifespan extension? J. Cell Mol. Med 2010, 14, 2543–2551. [Google Scholar]
- Zhou, H; Luo, Y; Huang, S. Updates of mTOR inhibitors. Anticancer Agents Med. Chem 2010, 10, 571–581. [Google Scholar]
- Lee, YK; Lee, WS; Kim, GS; Park, OJ. Anthocyanins are novel AMPKα1 stimulators that suppress tumor growth by inhibiting mTOR phosphorylation. Oncol. Rep 2010, 24, 1471–1477. [Google Scholar]
- Lee, YK; Park, SY; Kim, YM; Kim, DC; Lee, WS; Surh, YJ; Park, OJ. Suppression of mTOR via Akt-dependent and -independent mechanisms in selenium-treated colon cancer cells: involvement of AMPKalpha1. Carcinogenesis 2010, 31, 1092–1099. [Google Scholar]
- Tang, FY; Cho, HJ; Pai, MH; Chen, YH. Concomitant supplementation of lycopene and eicosapentaenoic acid inhibits the proliferation of human colon cancer cells. J. Nutr. Biochem 2009, 20, 426–434. [Google Scholar]
- Balgi, AD; Fonseca, BD; Donohue, E; Tsang, TC; Lajoie, P; Proud, CG; Nabi, IR; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009, 4, e7124. [Google Scholar]
- Bruno, P; Calastretti, A; Priulla, M; Asnaghi, L; Scarlatti, F; Nicolin, A; Canti, G. Cell survival under nutrient stress is dependent on metabolic conditions regulated by Akt and not by autophagic vacuoles. Cell Signal 2007, 19, 2118–2126. [Google Scholar]
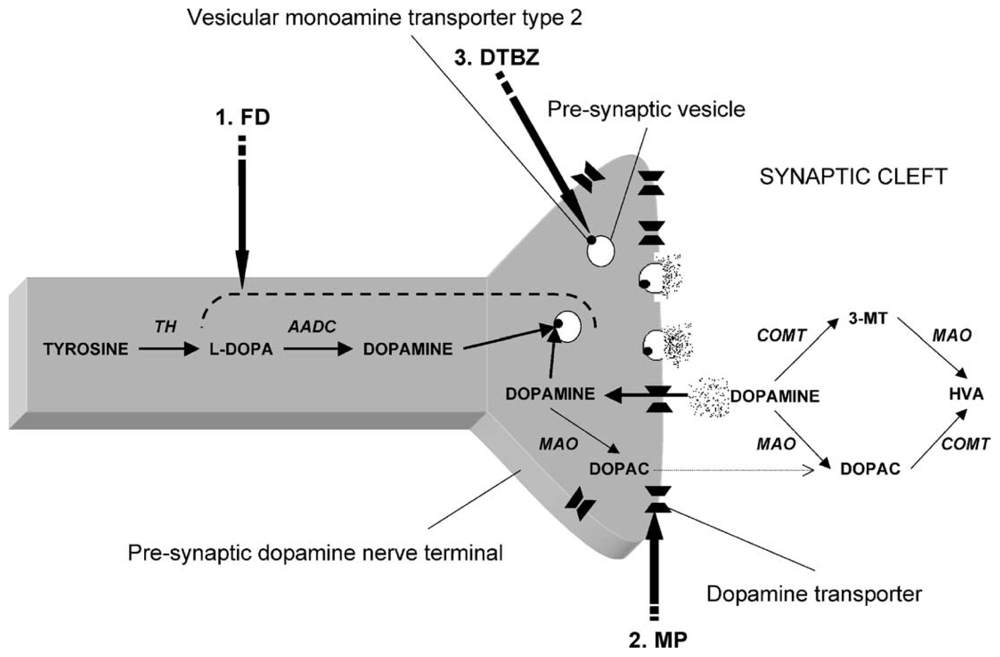
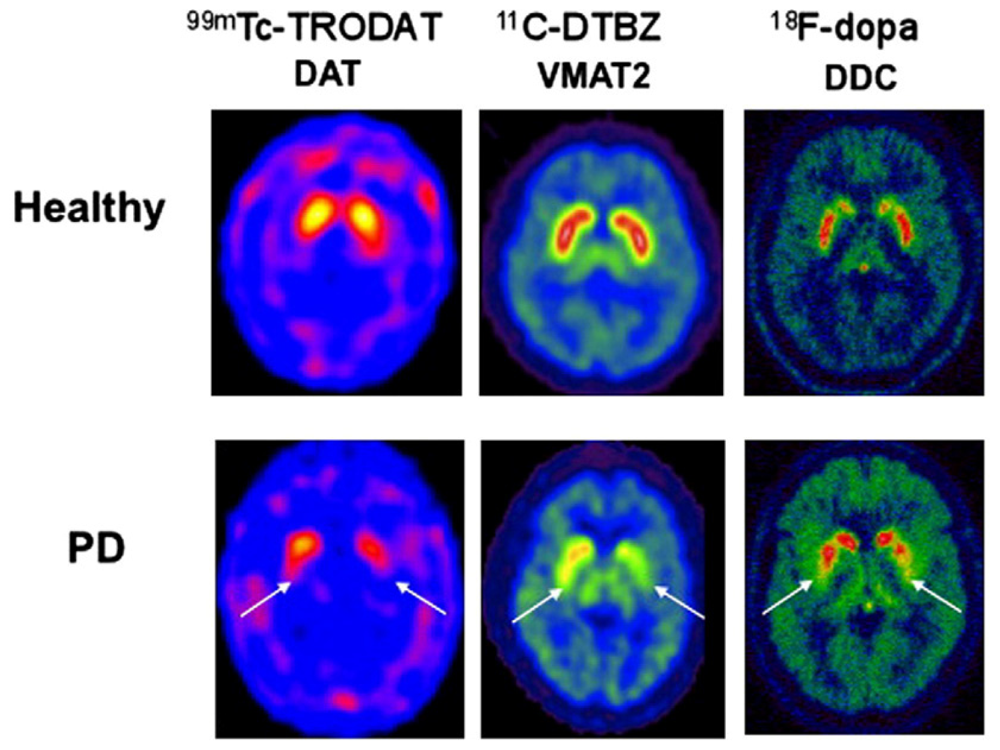

© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).