An Antiapoptotic Neuroprotective Role for Neuroglobin

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
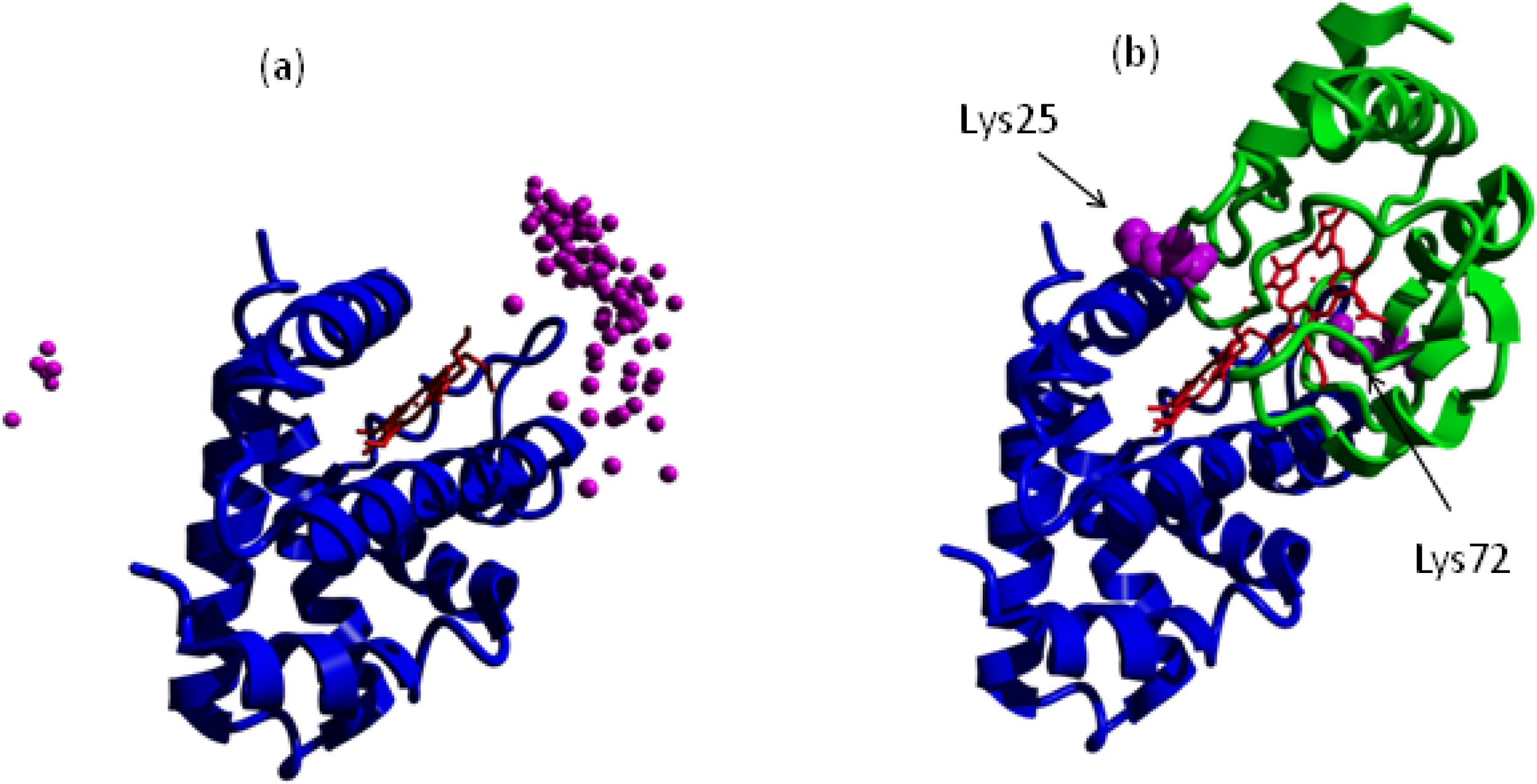{kind=link}
{kind=link}
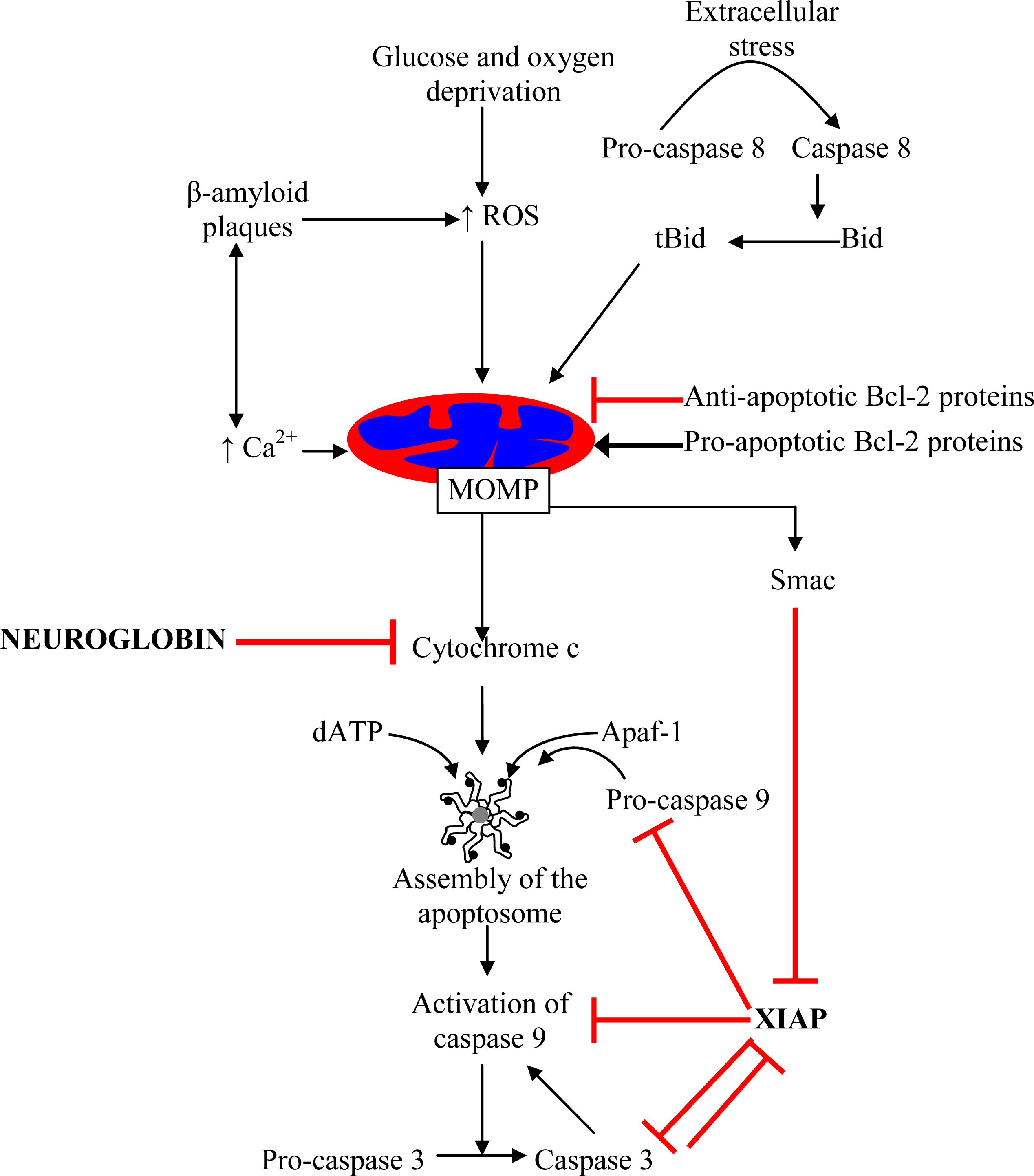
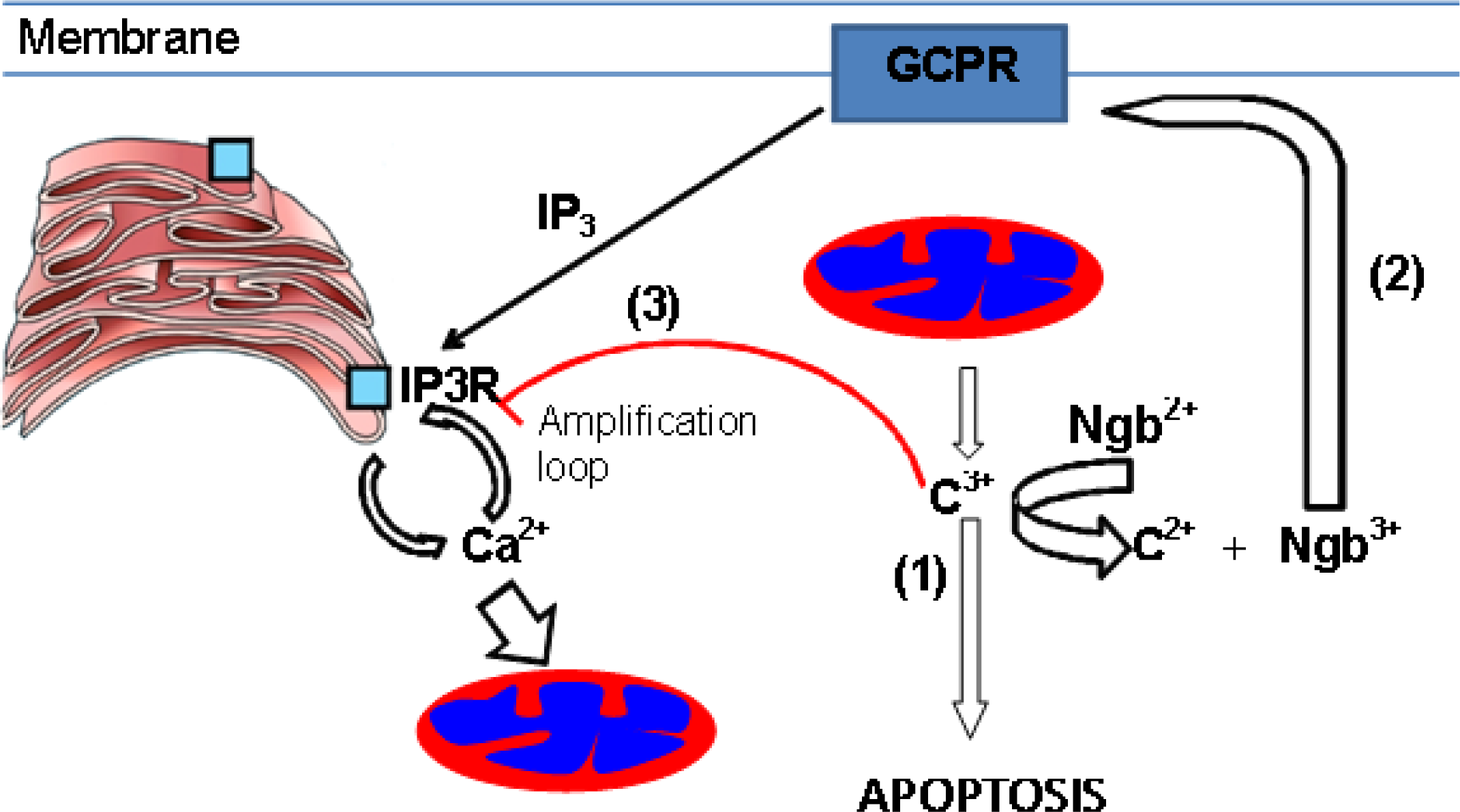
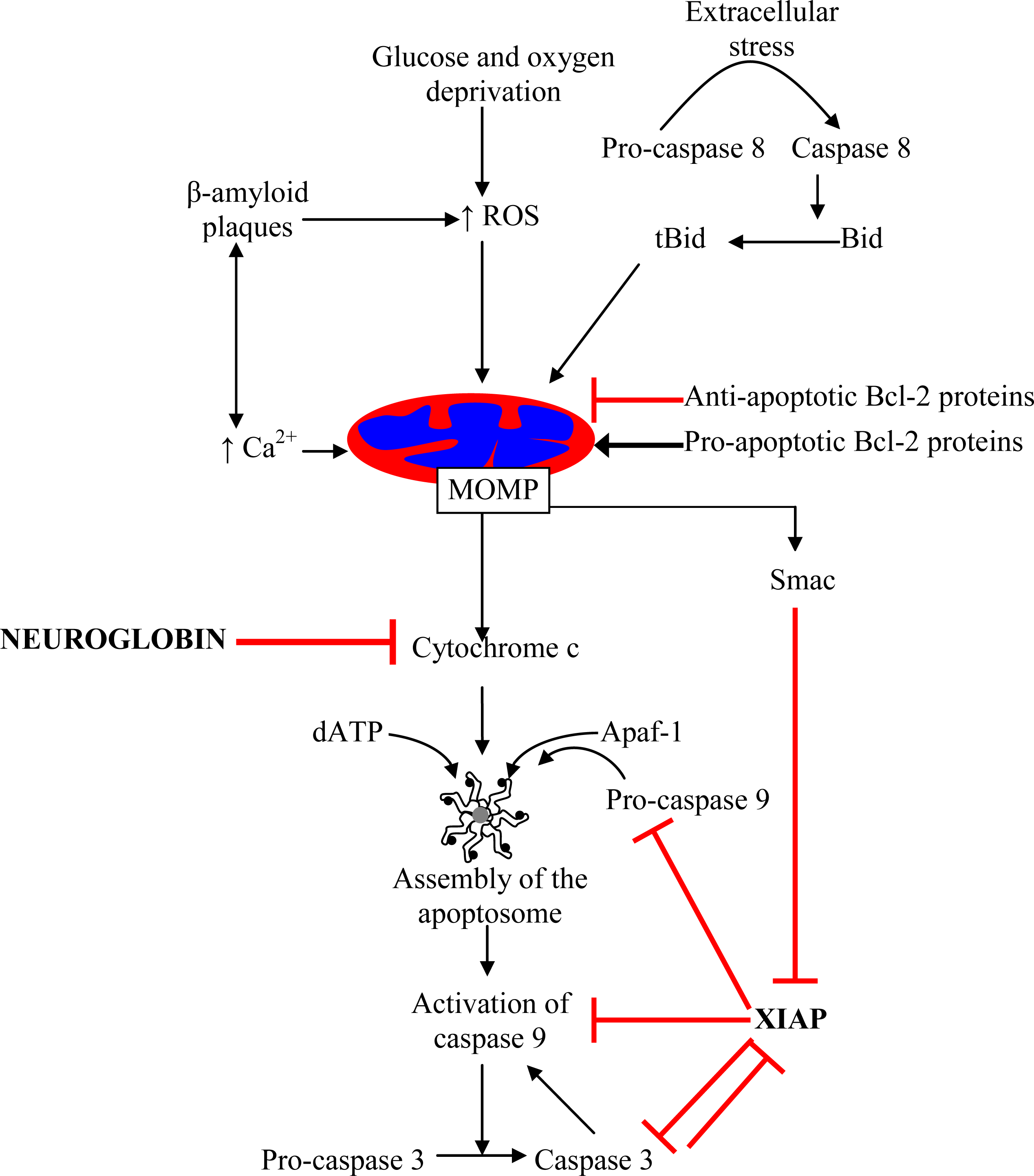
1. Mitochondrial Pathway of Cell Death–A Target for Neuroprotection
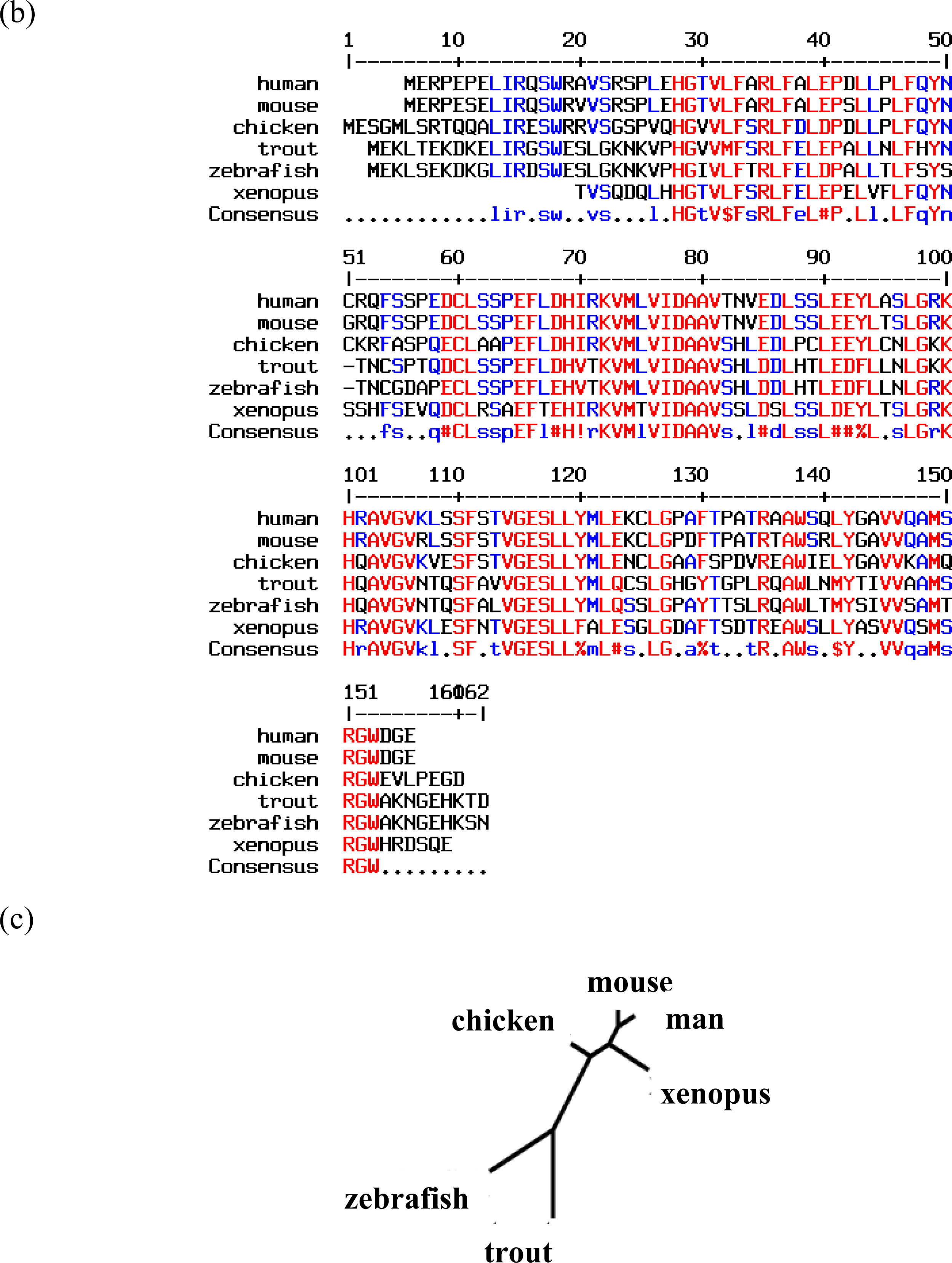
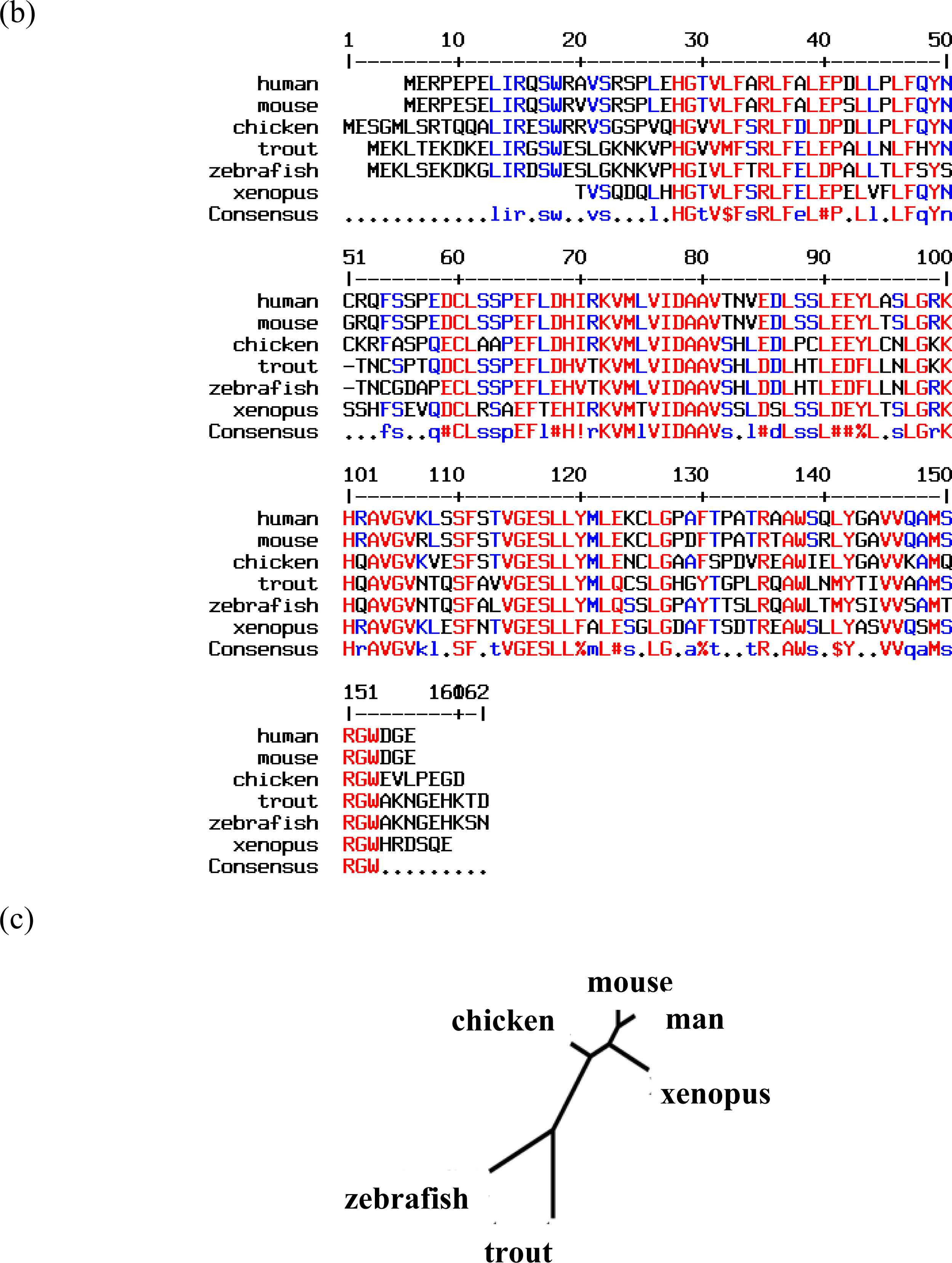
2. Neuroglobin Is an Evolutionarily Conserved Neuroprotective Protein
3. Proposed Mechanisms for Neuroglobin-Mediated Neuroprotection
4. Reactivity with Cytochrome C
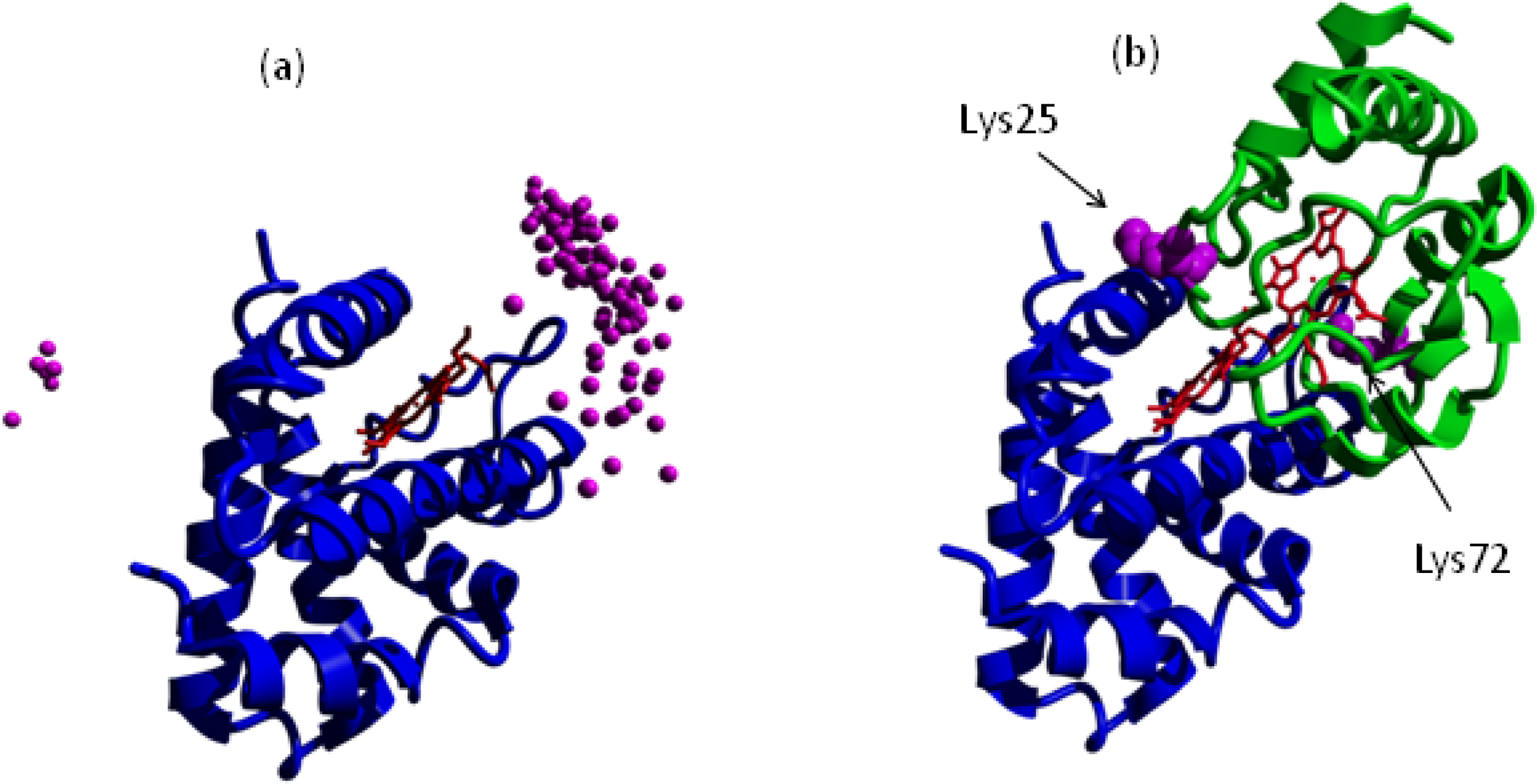
5. Experimental Evidence for Cytochrome c-Neuroglobin Interaction
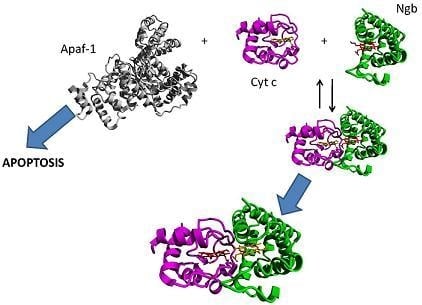
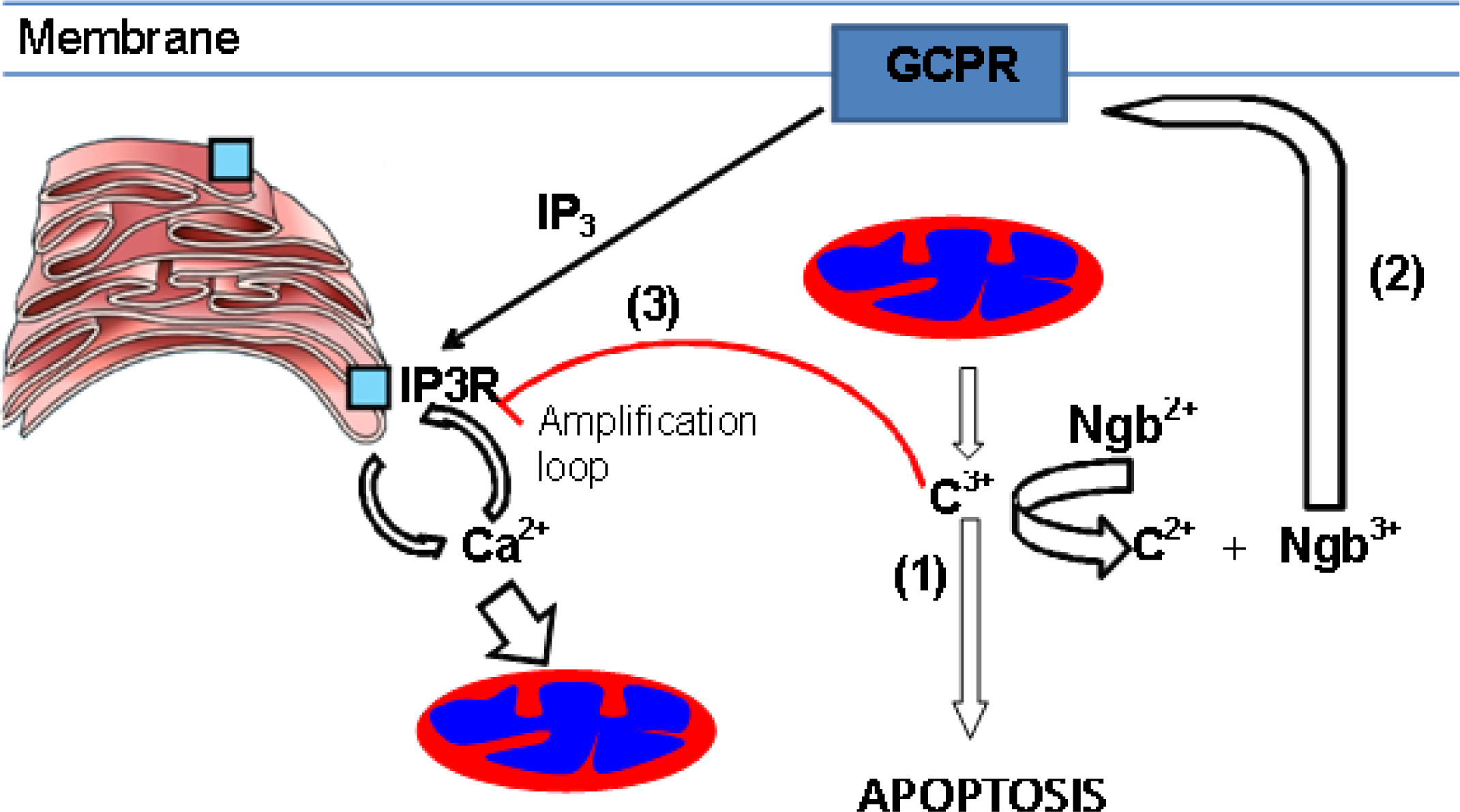
6. Hypothesis: A Co-Ordinating Role for Neuroglobin
7. Neuroglobin in Brain Tumours: Dr Jekyll or Mr Hyde?
8. Surviving the Release of Cytochrome c: Neuroglobin as a Drug Target
9. Conclusions
Acknowledgments
References
- Galluzzi, L; Blomgren, K; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci 2009, 10, 481–494. [Google Scholar]
- Lin, MT; Beal, MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar]
- Skommer, J; Wlodkowic, D; Deptala, A. Larger than life: Mitochondria and the Bcl-2 family. Leuk. Res 2007, 31, 277–286. [Google Scholar]
- Liston, P; Fong, WG; Korneluk, RG. The inhibitors of apoptosis: There is more to life than Bcl2. Oncogene 2003, 22, 8568–8580. [Google Scholar]
- Lankester, ER. A contribution to the knowledge of haemoglobion. Proc. Roy. Soc 1872, 21, 70–81. [Google Scholar]
- Burmester, T; Weich, B; Reinhardt, S; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar]
- Vallone, B; Nienhaus, K; Matthes, A; Brunori, M; Nienhaus, GU. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand binding. Proc. Natl. Acad. Sci. USA 2004, 101, 17351–17356. [Google Scholar]
- Pesce, A; Dewide, S; Nardini, M; Moens, L; Ascenzi, P; Hankeln, T; Burmester, T; Bolognesi, M. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure 2003, 11, 1087–1095. [Google Scholar]
- Vallone, B; Nienhaus, K; Brunori, M; Nienhaus, GU. The structure of murine neuroglobin: Novel pathways for ligand migration and binding. Proteins 2004, 56, 85–92. [Google Scholar]
- Fago, A; Mathews, AJ; Dewilde, S; Moens, L; Brittain, T. The reactions of neuroglobin with CO: Evidence for two forms of the ferrous protein. J. Inorg. Biochem 2006, 100, 1339–1343. [Google Scholar]
- Dewilde, S; Kiger, L; Burmester, T; Hankeln, T; Baudin-Creuzal, V; Aerts, T; Marden, MC; Caubergs, R; Moens, L. Biochemical characterisation and ligand binding properties of neuroglobin, a novel member of the globin family. J. Biol. Chem 2001, 276, 38949–38955. [Google Scholar]
- Brunori, M; Giuffre, A; Nienhaus, K; Nienhaus, GU; Scandurra, FM; Vallone, B. Neuroglobin, Nitric Oxide and Oxygen: Functional pathways and conformational changes. Proc. Natl. Acad. Sci. USA 2005, 102, 8483–8488. [Google Scholar]
- Smagghe, BJ; Trent, JT; Hargrove, MS. NO dioxygenase activity is ubiquitous in vitro but limited by reduction in vivo. Plos One 2008, 3, e2039. [Google Scholar]
- Hankeln, T; Ebner, B; Fuchs, C; Gerlach, F; Haberkamp, M; Laufs, TL; Roesner, A; Schmidt, M; Weich, B; Wystub, S; Saaler-Reinhardt, S; Reuss, S; Bolognesi, M; De Sanctis, D; Marden, MC; Kiger, L; Moens, L; Dewilde, S; Nevo, E; Avivi, A; Weber, RE; Fago, A; Burmester, T. Neuroglobin and cytoglobin in search of their role in the vertebrate globin family. J. Inorg. Biochem 2005, 99, 110–119. [Google Scholar]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucl. Acids. Res 1988, 16, 10881–10890. [Google Scholar]
- Dereeper, A; Guignon, V; Blanc, G; Audic, S; Buffet, S; Chevenet, F; Dufayard, JF; Guindon, S; Lefort, V; Lescot, M; Claverie, JM; Gascuel, O. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucl. Acids Res 2008, 36, W465–W469. [Google Scholar]
- Hundahl, CA; Allen, GC; Hannibal, J; Kjaer, K; Rehfeld, JF; Dewilde, S; Nyengaard, JR; Kelsen, J; Hay-Schmidt, A. Anatomical characterization of Cytoglobin and Neuroglobin mRNA and protein expression in the mouse brain. Brain Res, 2010; [Epub ahead of print]. [Google Scholar]
- Jin, K; Mao, Y; Mao, X; Xie, L; Greenberg, DA. Neuroglobin expression in ischemic stroke. Stroke 2010, 41, 557–559. [Google Scholar]
- Szymanski, M; Wang, R; Fallin, MD; Bassett, SS; Avramopoulos, D. Neuroglobin and Alzheimer's dementia: Genetic association and gene expression changes. Neurobiol Aging, 2008; doi:10.1016/j.neurobiolaging.2008.10.003. [Google Scholar]
- Chuang, PY; Conley, YP; Poloyac, SM; Okonkwo, DO; Ren, D; Sherwood, PR; Hravnak, M; Alexander, SA. Neuroglobin genetic polymorphisms and their relationship to functional outcomes following traumatic brain injury. J Neurotrauma, 2010; [Epub ahead of print]. [Google Scholar]
- Lin, Y; Fang, L; Xue, XH; Murong, SX; Wang, N; Wu, ZY. Association between Ngb polymorphisms and ischemic stroke in the Southern Chinese Han population. B.M.C. Med. Genet 2008, 9, 110. [Google Scholar]
- Sun, Y; Jin, K; Mao, XO; Zhu, Y; Greenberg, DA. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc. Natl. Acad. Sci. USA 2001, 98, 15306–15311. [Google Scholar]
- Fordel, E; Thijs, L; Martinet, W; Lenjou, M; Laufs, T; van Bockstaele, D; Moens, L; Dewilde, S. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci. Lett 2006, 410, 146–151. [Google Scholar]
- Li, RC; Pouranfar, F; Lee, SK; Morris, MW; Wang, Y; Gozal, D. Neuroglobin protects PC12 cells against beta-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar]
- Li, RC; Morris, MW; Lee, SK; Pouranfar, F; Wang, Y; Gozal, D. Neuroglobin protects PC12 cells against oxidative stress. Brain Res 2008, 1190, 159–166. [Google Scholar]
- Raychaudhuri, S; Skommer, J; Henty, K; Birch, N; Brittain, T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis 2010, 15, 401–411. [Google Scholar]
- Antao, ST; Duong, TT; Aran, R; Witting, PK. Neuroglobin over-expression in cultured human neuronal cells protects against hydrogen peroxide insult via activating phosphoinositide-3 kinase and opening the mitochondrial KATP channel. Antioxid Redox Signal, 2010; [Epub ahead of print]. [Google Scholar]
- Ye, SQ; Zhou, XY; Lai, XJ; Zheng, L; Chen, XQ. Silencing neuroglobin enhances neuronal vulnerability to oxidative injury by down-regulating 14-3-3gamma. Acta Pharmacol. Sin 2009, 30, 913–918. [Google Scholar]
- Hundahl, C; Kelsen, J; Ronn, LC; Weber, RE; Geuens, E; Hay-Schmidt, A; Nyengaard, JR. Does neuroglobin protect neurons from ischemic insult? A quantitative investigation of neuroglobin expression following transient MCAo in spontaneously hypertensive rats. Brains Res 2006, 1085, 19–27. [Google Scholar]
- Khan, AA; Wang, Y; Sun, Y; Mao, XO; Xie, L; Miles, E; Graboski, J; Chen, S; Ellerby, LM; Jin, K; Greenberg, DA. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc. Natl. Acad. Sci. USA 2006, 103, 17944–17948. [Google Scholar]
- Wang, X; Liu, J; Zhu, H; Tejima, E; Tsuji, K; Murata, Y; Atochin, DN; Huang, PL; Zhang, C; Lo, EH. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke 2008, 39, 1869–1874. [Google Scholar]
- Sun, Y; Jin, K; Peel, A; Mao, XO; Xie, L; Greenberg, DA. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 3497–3500. [Google Scholar]
- Garry, DJ; Mammen, PPA. Neuroprotection and the role of neuroglobin. Lancet 2003, 362, 342–343. [Google Scholar]
- Fago, A; Hundahl, C; Dewilde, S; Gilany, K; Moens, L; Weber, RE. Allosteric regulation and temperature dependence of oxygen binding in human neuroglobin and cytoglobin. J. Biol. Chem 2004, 279, 44417–444426. [Google Scholar]
- Fago, A; Hundahl, C; Malte, H; Weber, RE. Functional properties of neuroglobin and cytoglobin. Insights into the ancestral physiological roles of globins. I.U.B.M.B. Life 2004, 56, 689–696. [Google Scholar]
- Brunori, M; Vallone, B. A globin for the brain. FASEB. J 2006, 20, 2192–2197. [Google Scholar]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev 1999, 79, 1431–1568. [Google Scholar]
- Zhu, C; Wang, X; Qiu, L; Peeters-Scholte, C; Hagberg, H; Blomgren, K. Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor in neonatal rat cerebral hypoxia-ischaemia. J. Neurochem 2004, 90, 462–471. [Google Scholar]
- Petersen, MG; Dewilde, S; Fago, A. Reactions of ferrous neuroglobin and cytoglobin with nitrite under anaerobic conditions. J. Inorg. Biochem 2008, 102, 1777–1782. [Google Scholar]
- Herold, S; Fago, A; Weber, RE; Dewilde, S; Moens, L. Reactivity studies of the Fe(III) and Fe(II) NO forms of human neuroglobin reveal a potential role against oxidative stress. J. Biol. Chem 2004, 279, 22841–22847. [Google Scholar]
- Moschetti, T; Guiffre, A; Ardiccino, C; Vallone, B; Modjtahedi, N; Kroemer, G; Brunori, M. Failure of apoptosis-inducing factor to act as a neuroglobin reductase. Biochem. Biophys. Res. Comm 2008, 390, 121–124. [Google Scholar]
- Trandafir, F; Hoogewijs, D; Altieri, F; Rivetti de val Cervo, P; Ramser, K; van Doorslaer, S; Vanfleteren, JR; Moens, L; Dewilde, S. Neuroglobin and cytoglobin as potential enzymes or substrates. Gene 2007, 398, 103–113. [Google Scholar]
- Guiffre, A; Moschetti, T; Vallone, B; Brunori, M. Neuroglobin: Enzymatic reduction and oxygen affinity. Biochem. Biophys. Res. Comm 2008, 367, 893–898. [Google Scholar]
- Khan, AA; Mao, XO; Banwait, S; DerMardirossian, CM; Bokoch, GM; Jin, K; Greenberg, DA. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J 2008, 22, 1737–1747. [Google Scholar]
- Khan, AA; Mao, XO; Banwait, S; Jin, K; Greenberg, DA. Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19114–19119. [Google Scholar]
- Pan, LP; Hibdon, S; Liu, RQ; Durham, B; Millet, F. Intracomplex electron transfer between ruthenium-cytochrome c derivatives and cytochrome c oxidase. Biochemistry 1993, 32, 8492–8498. [Google Scholar]
- Willie, A; Stayton, PS; Sligar, SG; Durham, B; Millet, F. Genetic engineering of redox donor sites: Measurement of intracomplex electron transfer between ruthenium-65-cytochrome b5 and cytochrome c. Biochemistry 1992, 31, 7237–7242. [Google Scholar]
- Naito, NR; Huang, H; Sturgess, AW; Nocek, JM; Hoffman, BM. Binding and electron transfer between cytochrome b5 and the hemoglobin α and β subunits through use of [Zn,Fe] hybrids. J. Am. Chem. Soc 1998, 120, 11256–11262. [Google Scholar]
- Fago, A; Mathews, AJ; Moens, L; Dewilde, S; Brittain, T. Reactivity of neuroglobin with the potential redox protein partners cytochrome b5 and cytochrome c. FEBS. Lett 2006, 580, 4884–4888. [Google Scholar]
- Bonding, SH; Henty, K; Dingley, A; Brittain, T. The binding of cytochrome c to neuroglobin: A docking and Surface Plasmon Resonance study. Int.J. Biol. Macromol 2008, 43, 295–299. [Google Scholar]
- Fago, A; Mathews, AJ; Brittain, T. A role for neuroglobin: Resetting the trigger level for apoptosis in neuronal and retinal cells. IUBMB. Life 2008, 60, 398–401. [Google Scholar]
- Yu, T; Wang, X; Purring-Koch, C; Weil, Y; McLendon, GL. A mutational epitope for cytochrome c binding to the apoptosis protease activation factor-1. J. Biol. Chem 2001, 276, 13034–13038. [Google Scholar]
- Abdullaev, ZK; Bodrova, ME; Chernyak, BV; Dolgihk, DA; Kluck, RM; Pereverzev, MO; Aseniev, AS; Efremov, RG; Kirpichnikov, MP; Mokhova, EN; Newmeyer, DD; Roder, H; Skulachev, VP. A cytochrome c mutant with high electron transfer and antioxidant activities but devoid of apoptogenic effect. Biochem J 2002, 362, 749–754. [Google Scholar]
- Kluck, RM; Ellerby, LM; Ellerby, HM; Naiem, S; Yaffe, MP; Margoliash, E; Bredesen, D; Mauk, AG; Sherman, F; Newmaeyer, DD. Determinants of cytochrome c pro-apoptotic activity. J. Biol. Chem 2000, 275, 16127–16133. [Google Scholar]
- Chertkova, RV; Sharanov, GV; Feofanov, AV; Bocharova, OV; Latypov, RF; Chernyak, BV; Arseniev, AS; Dolgikh, DA; Kirpichnikov, MP. Porapototic activity of cytochrome c in living cells: Effect of K72 substitution and species differences. Mol. Cell. Biochem 2008, 314, 85–93. [Google Scholar]
- Raychaudhuri, S; Willgohs, E; Nguyen, TN; Khan, EM; Goldkorn, T. Monte Carlo simulation of cell death signaling predicts large cell-to-cell stochastic fluctuations through the type 2 pathway of apoptosis. Biophys. J 2008, 95, 3559–3562. [Google Scholar]
- Spencer, SL; Gaudet, S; Albeck, JG; Burke, JM; Sorger, PK. Non-genetic origins of cell-to-cell variability in tRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar]
- Borutaite, V; Brown, GC. Mitochondrial regulation of caspase activation by cytochrome oxidase and tetramethylphenylenediamine via cytosolic cytochrome c redox state. J. Biol. Chem 2007, 282, 31124–31130. [Google Scholar]
- Brown, GC; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta 2008, 1777, 877–881. [Google Scholar]
- Suto, D; Sato, K; Ohba, Y; Yoshimura, T; Fujii, J. Suppression of the pro-apoptotic function of cytochrome c by singlet oxygen via a haem redox state-independent mechanism. Biochem. J 2006, 392, 399–406. [Google Scholar]
- Boehning, D; Patterson, RL; Sedaghat, L; Glebova, NO; Kurosaki, T; Snyder, SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell. Biol 2003, 5, 1051–1061. [Google Scholar]
- Duong, TTH; Witting, PK; Antao, ST; Parry, SN; Kennerson, M; Lai, B; Vogt, S; Lay, PA; Harris, HM. Multiple protective activities of neuroglobin in cultured neuronal cells exposed to hypoxia re-oxygenation injury. J. Neurochem 2009, 108, 1143–1154. [Google Scholar]
- Liu, J; Yu, Z; Guo, S; Lee, S; Xing, C; Zhang, C; Gao, Y; Nicholls, DG; Lo, EH; Wang, X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J. Neurosci. Res 2009, 87, 164–170. [Google Scholar]
- Wakasugi, K; Nakano, T; Morishima, I. Oxidised human neuroglobin acts as a heterotrimeric Gα protein guanine nucleotide dissocation inhibitor. J. Biol. Chem 2003, 278, 36505–36512. [Google Scholar]
- Xia, JX; Fan, SY; Yan, J; Chen, F; Li, Y; Yu, ZP; Hu, ZA. Orexin A-induced extracellular calcium influx in prefrontal cortex neurons involves L-type calcium channels. J. Physiol Biochem 2009, 65, 125–36. [Google Scholar]
- Hanahan, D; Weinberg, RA. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Emara, M; Salloum, N; Allalunis-Turner, J. Expression and hypoxic up-regulation of neuroglobin in human glioblastoma cells. Mol. Oncol 2009, 3, 45–53. [Google Scholar]
- Johnson, CE; Huang, YY; Parrish, AB; Smith, MI; Vaughn, AE; Zhang, Q; Wright, KM; van Dyke, T; Wechsler-Reya, RJ; Kornbluth, S; Deshmukh, M. Differential Apaf-1 levels allow cytochrome c to induce apoptosis in brain tumors but not in normal neural tissues. Proc. Natl. Acad. Sci. USA 2007, 104, 20820–20825. [Google Scholar] [Green Version]
- Sun, L; Hui, AM; Su, Q; Vortmeyer, A; Kotliarov, Y; Pastorino, S; Passaniti, A; Menon, J; Walling, J; Bailey, R; Rosenblum, M; Mikkelsen, T; Fine, HA. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar]
- Kanungo, AK; Hao, Z; Elia, AJ; Mak, TW; Henderson, JT. Inhibition of apoptosome activation protects injured motor neurons from cell death. J. Biol. Chem 2008, 283, 22105–22112. [Google Scholar]

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brittain, T.; Skommer, J.; Raychaudhuri, S.; Birch, N. An Antiapoptotic Neuroprotective Role for Neuroglobin. Int. J. Mol. Sci. 2010, 11, 2306-2321. https://doi.org/10.3390/ijms11062306
Brittain T, Skommer J, Raychaudhuri S, Birch N. An Antiapoptotic Neuroprotective Role for Neuroglobin. International Journal of Molecular Sciences. 2010; 11(6):2306-2321. https://doi.org/10.3390/ijms11062306
Chicago/Turabian StyleBrittain, Thomas, Joanna Skommer, Subadhip Raychaudhuri, and Nigel Birch. 2010. "An Antiapoptotic Neuroprotective Role for Neuroglobin" International Journal of Molecular Sciences 11, no. 6: 2306-2321. https://doi.org/10.3390/ijms11062306