Binding Ligand Prediction for Proteins Using Partial Matching of Local Surface Patches



Abstract
:1. Introduction
2. Materials and Methods
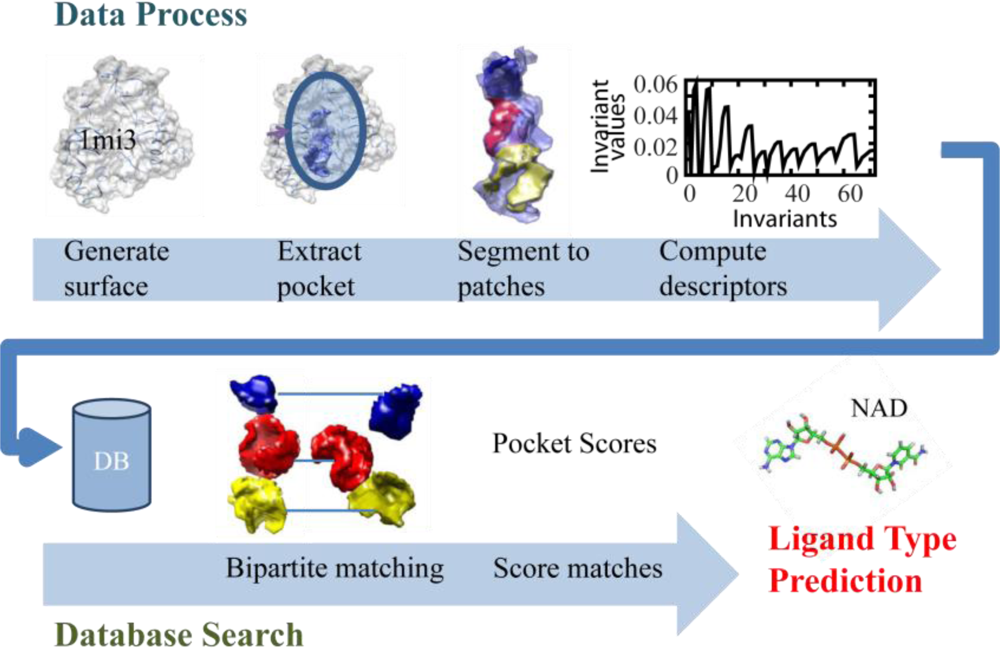
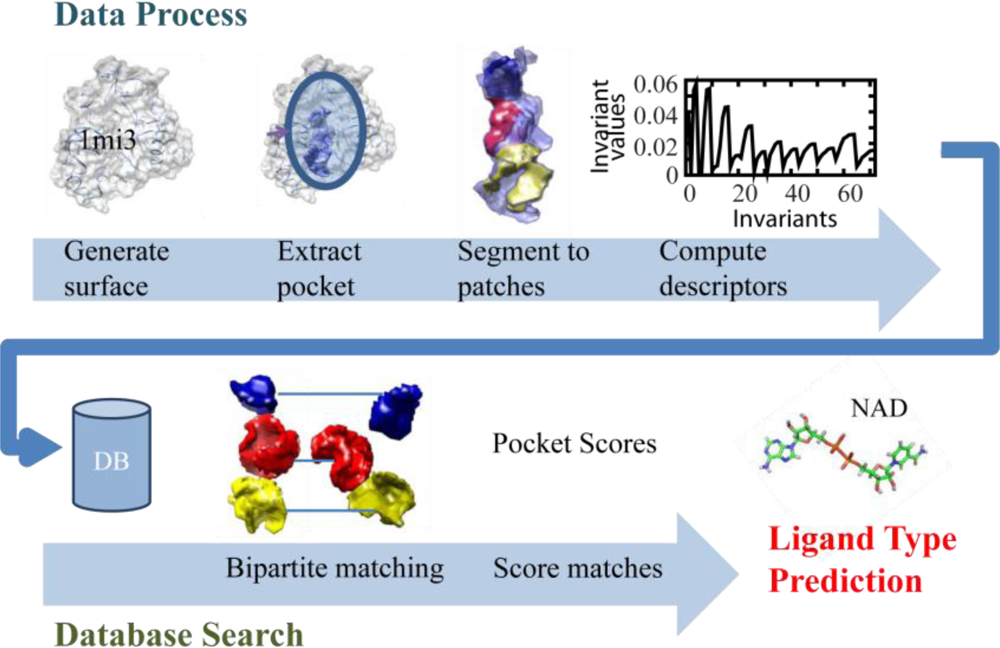
2.1. Overview of the Algorithm
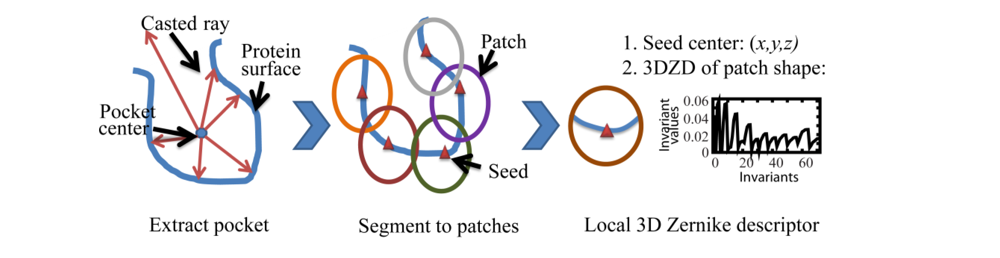
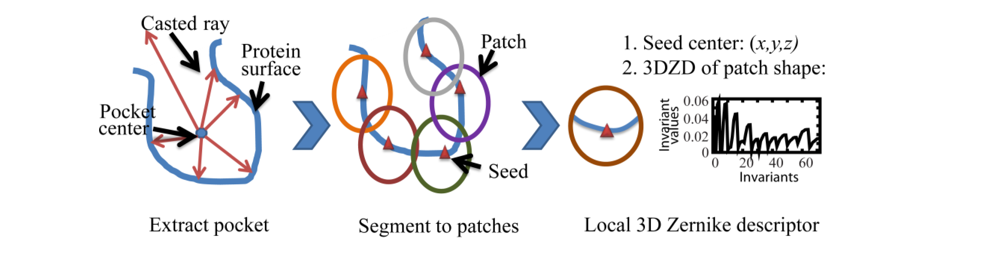
2.2. Local Surface Patch Extraction
2.3. Encoding Local Surface Patch Using the 3D Zernike Descriptor
2.4. Comparing Surface Patches of Pockets Using Partial Matching Algorithm
2.5. Scoring Pocket Distance and Binding Ligand Types
2.6. Dataset
2.7. Performance Evaluation
3. Results
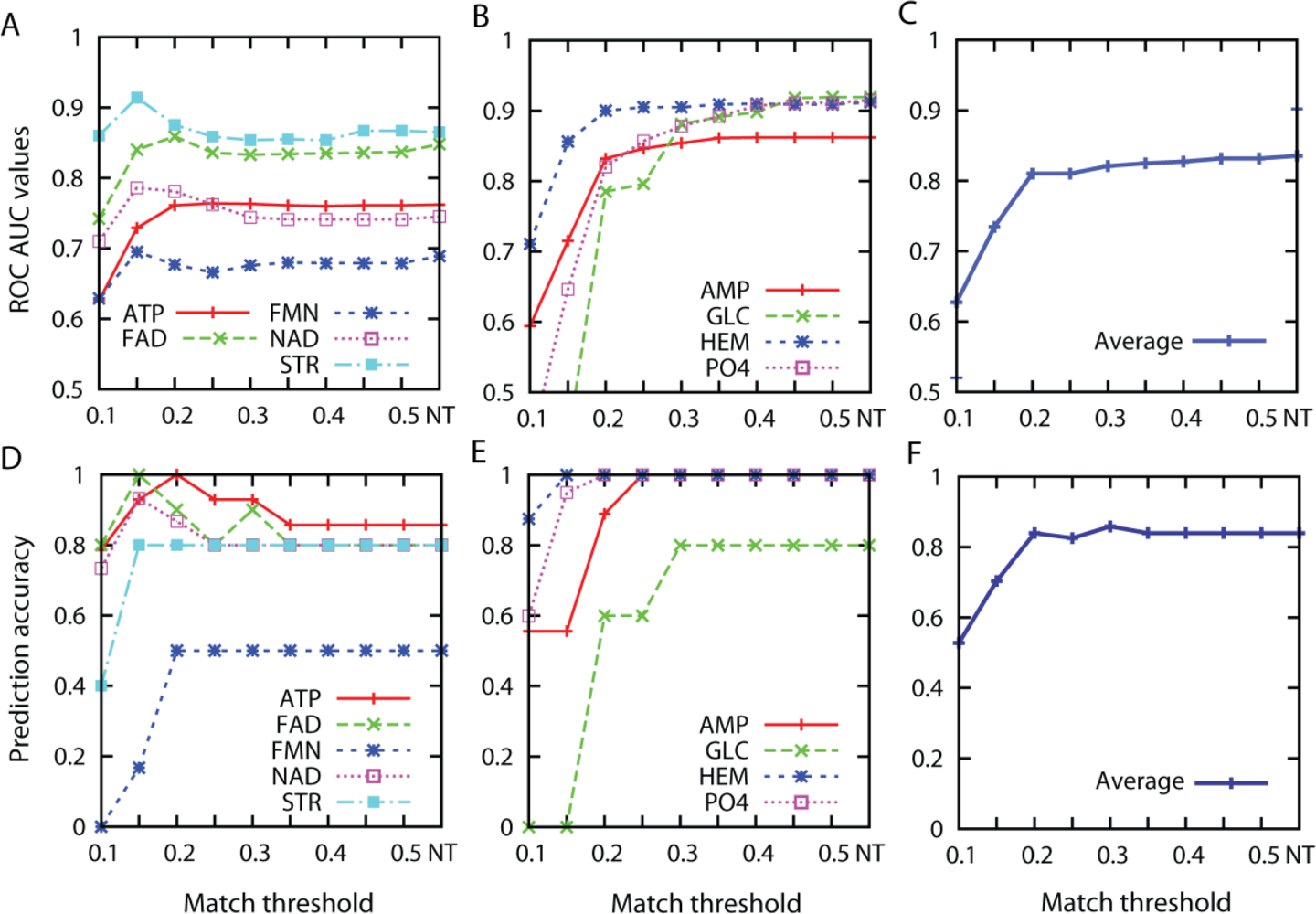
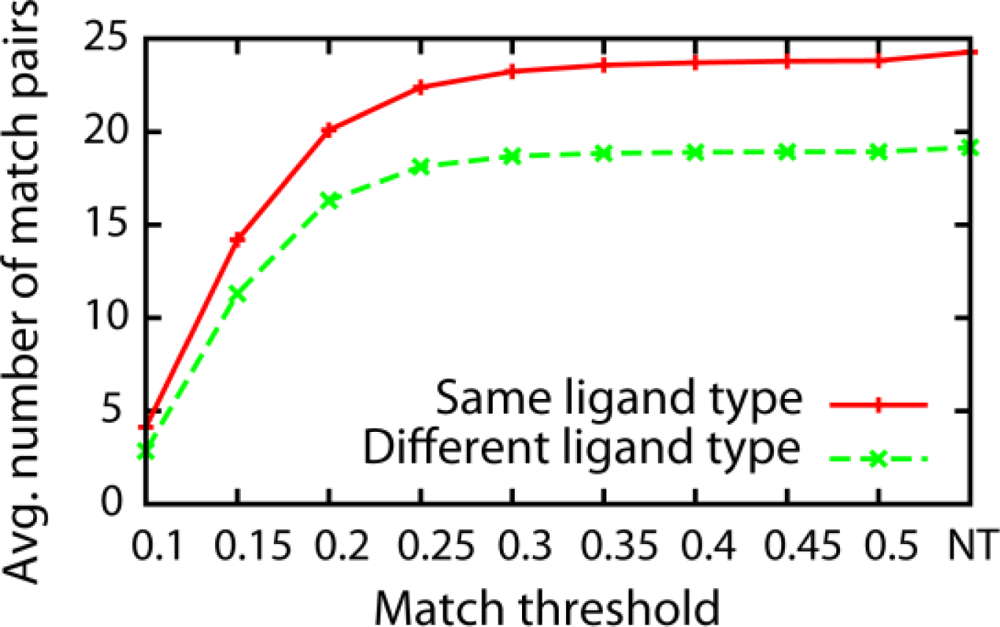
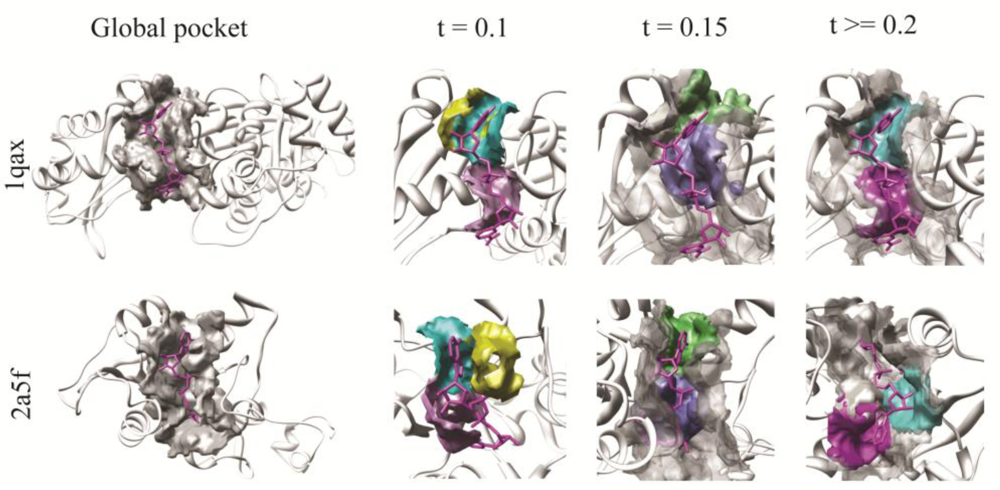
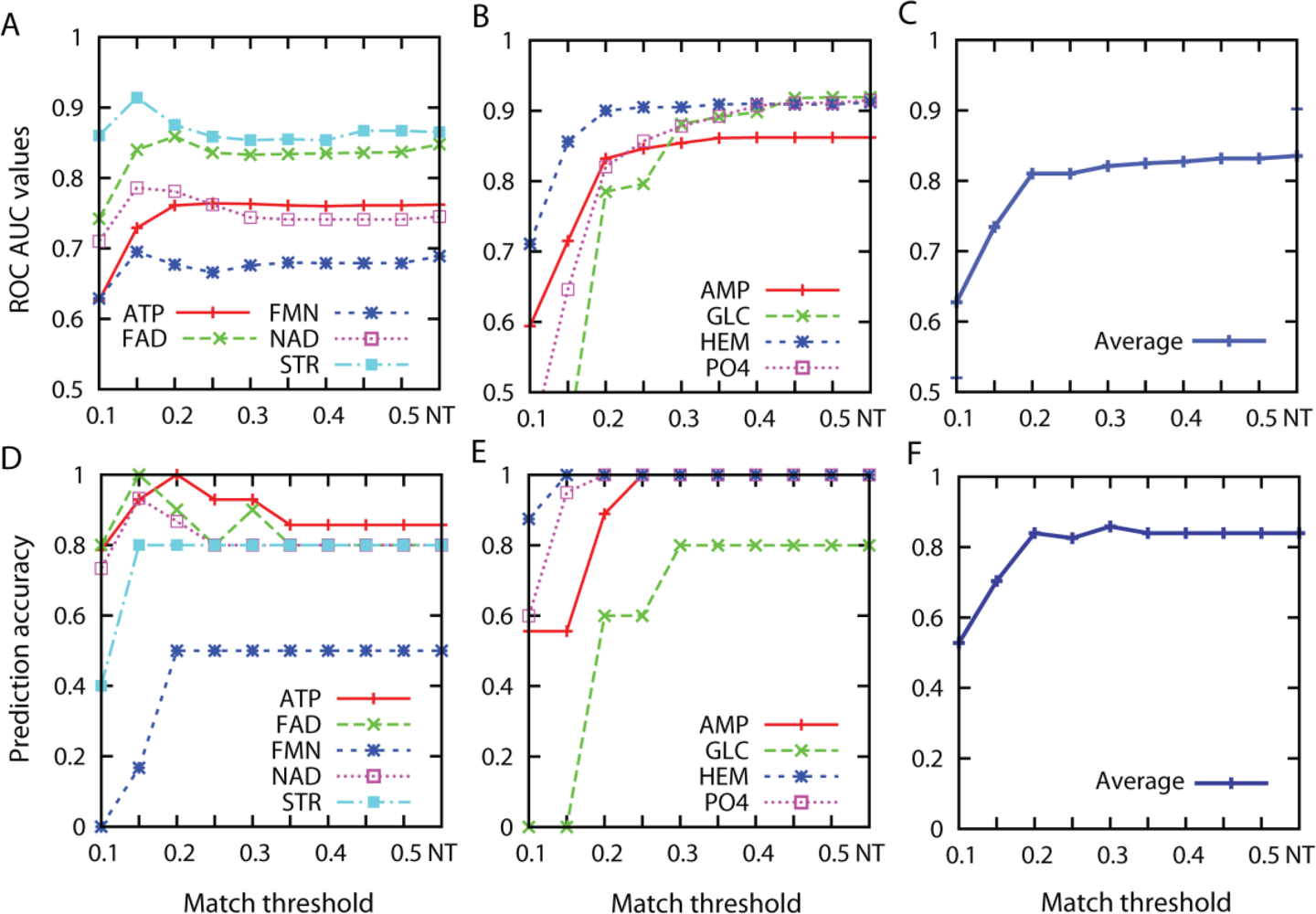
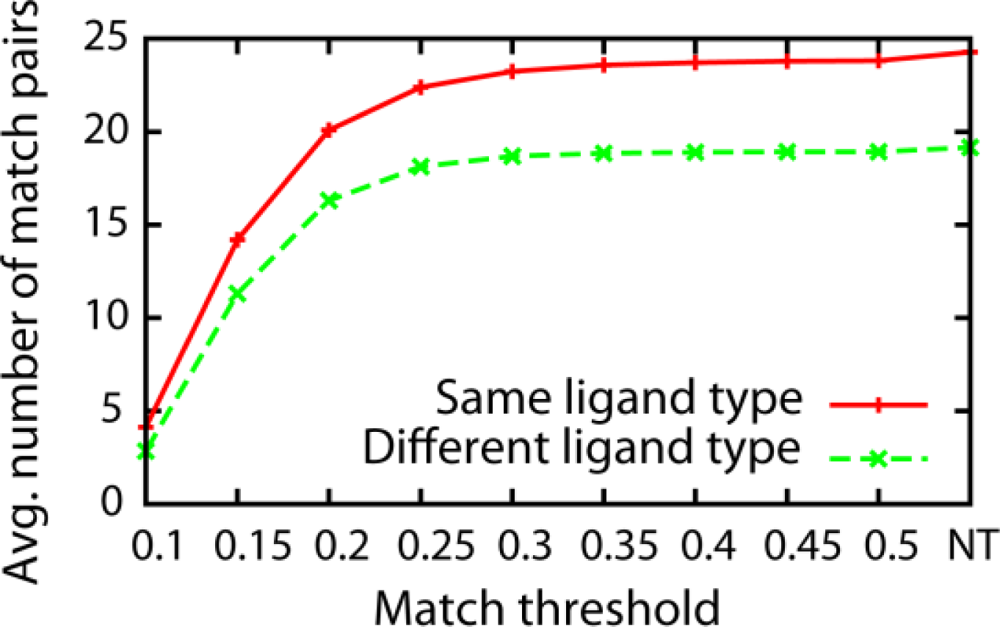
3.1. Effect of the Threshold Value for Patch Similarity
3.2. Prediction Performance
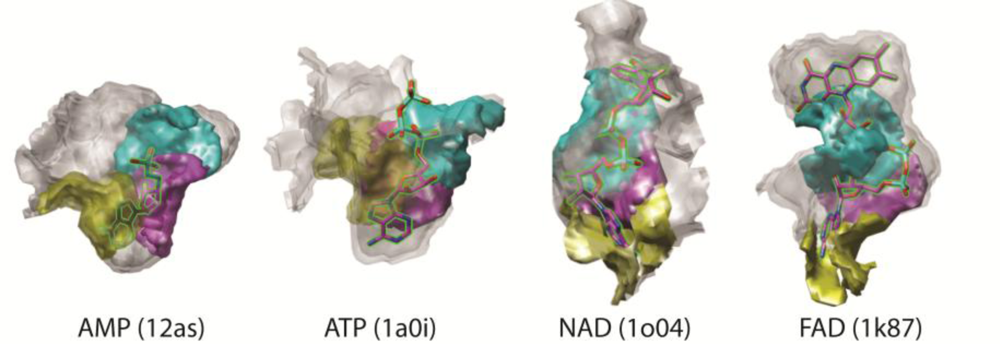
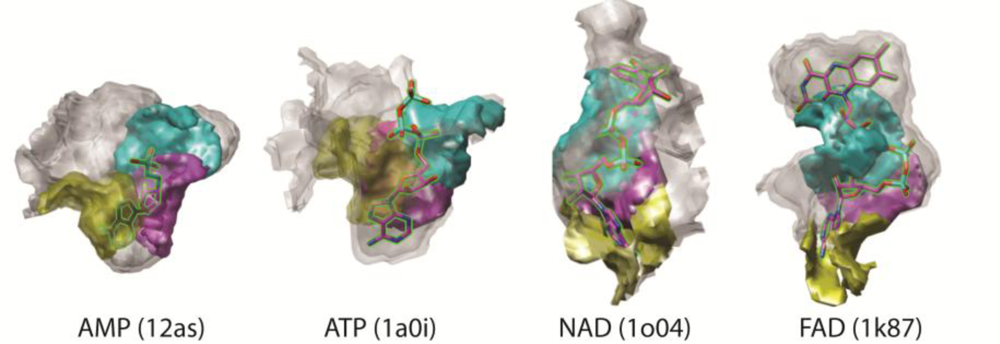
3.3. Examples of Matched Local Surface Patches
3.4. Computation Time
4. Discussion
Acknowledgments
References
- Hawkins, T; Kihara, D. Function prediction of uncharacterized proteins. J. Bioinf. Comput. Biol 2007, 5, 1–30. [Google Scholar]
- Hawkins, T; Chitale, M; Kihara, D. New paradigm in protein function prediction for large scale omics analysis. Mol. BioSyst 2008, 4, 223–231. [Google Scholar]
- Watson, JD; Laskowski, RA; Thornton, JM. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol 2005, 15, 275–284. [Google Scholar]
- Valencia, A. Automatic annotation of protein function. Curr. Opin. Struct. Biol 2005, 15, 267–74. [Google Scholar]
- Berman, HM; Westbrook, J; Feng, Z; Gilliland, G; Bhat, TN; Weissig, H; Shindyalov, IN; Bourne, PE. The protein data bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Chandonia, J; Brenner, SE. The impact of structural genomics: Expectations and outcomes. Science 2006, 311, 347–351. [Google Scholar]
- Skolnick, J; Brylinski, M. FINDSITE: A combined evolution/structure-based approach to protein function prediction. Brief. Bioinform 2009, 10, 378. [Google Scholar]
- Kihara, D; Skolnick, J. Microbial genomes have over 72% structure assignment by the threading algorithm PROSPECTOR_Q. Proteins: Struct. Funct. Bioinf 2004, 55, 464–473. [Google Scholar]
- Pal, D; Eisenberg, D. Inference of protein function from protein structure. Structure 2005, 13, 121–130. [Google Scholar]
- Orengo, CA; Thornton, JM. Protein families and their evolution—A structural perspective. Biochemistry 2005, 74, 867–900. [Google Scholar]
- Orengo, CA; Jones, DT; Thornton, JM. Protein superfamilies and domain superfolds. Nature 1994, 372, 631–634. [Google Scholar]
- Ausiello, G; Peluso, D; Via, A; Helmer-Citterish, M. Local comparison of protein structures highlights cases of convergent evolution in analogous functional sites. BMC Bioinformatics 2007, 8, S24. [Google Scholar]
- Chikhi, R; Sael, L; Kihara, D. Real-time ligand binding pocket database search using local surface descriptors. Proteins: Struct. Funct. Bioinf 2010, 78, 2007–2028. [Google Scholar]
- Kahraman, A; Morris, RJ; Laskowski, RA; Thornton, JM. Shape variation in protein binding pockets and their ligands. J. Mol. Biol 2007, 368, 283–301. [Google Scholar]
- Laskowski, RA. SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graphics 1995, 13, 323–330. [Google Scholar]
- Levitt, DG; Banaszak, LJ. POCKET: A computer graphics method for identifying and displaying protein cavities and their surrounding amino acids. J. Mol. Graphics 1992, 10, 229–234. [Google Scholar]
- Kawabata, T; Go, N. Detection of pockets on protein surfaces using small and large probe spheres to find putative ligand binding sites. Proteins: Struct. Funct. Bioinf 2007, 68, 516–529. [Google Scholar]
- Weisel, M; Proschak, E; Schneider, G. PocketPicker: Analysis of ligand binding-sites with shape descriptors. Chem. Cent. J 2007, 1, 1–17. [Google Scholar]
- Li, B; Turuvekere, S; Agrawal, M; La, D; Ramani, K; Kihara, D. Characterization of local geometry of protein surfaces with the visibility criterion. Proteins: Struct. Funct. Bioinf 2008, 71, 670–683. [Google Scholar]
- Kalidas, Y; Chandra, N. PocketDepth: A new depth based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol 2008, 161, 31–42. [Google Scholar]
- Liang, J; Edelsbrunner, H; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci 1998, 7, 1884–1897. [Google Scholar]
- Huang, B; Schroeder, M. LIGSITEcsc: Predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol 2006, 6, 19. [Google Scholar]
- Tseng, YY; Dundas, J; Liang, J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J. Mol. Biol 2009, 387, 451–464. [Google Scholar]
- Elcock, AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J. Mol. Biol 2001, 312, 885–896. [Google Scholar]
- Laurie, AT; Jackson, RM. Q-SiteFinder: An energy-based method for the prediction of protein– ligand binding sites. Bioinformatics 2005, 21, 1908. [Google Scholar]
- An, J; Totrov, M; Abagyan, R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell. Proteomics 2005, 4, 752. [Google Scholar]
- Sael, L; Kihara, D. Protein surface representation and comparison: New approaches in structural proteomics. In Biological Data Mining; Chen, JY, Lonardi, S, Eds.; Chapman & Hall/CRC Press: Boca Raton, FL, USA, 2009aa; pp. 89–109. [Google Scholar]
- Porter, CT; Bartlett, GJ; Thornton, JM. The Catalytic Site Atlas: A resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 2004, 32, D129–D133. [Google Scholar]
- Arakaki, AK; Zhang, Y; Skolnick, J. Large-scale assessment of the utility of low-resolution protein structures for biochemical function assignment. Bioinformatics 2004, 20, 1087–1096. [Google Scholar]
- Ferrè, F; Ausiello, G; Zanzoni, A; Helmer-Citterich, M. SURFACE: A database of protein surface regions for functional annotation. Nucleic Acids Res 2004, 32, D240–D244. [Google Scholar]
- Gold, ND; Jackson, RM. Fold independent structural comparisons of protein-ligand binding sites for exploring functional relationships. J. Mol. Biol 2006, 355, 1112–1124. [Google Scholar]
- Kinoshita, K; Murakami, Y; Nakamura, H. eF-seek: Prediction of the functional sites of proteins by searching for similar electrostatic potential and molecular surface shape. Nucleic Acids Res 2007, 35, W398–W402. [Google Scholar]
- Morris, RJ; Najmanovich, RJ; Kahraman, A; Thornton, JM. Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons. Bioinformatics 2005, 21, 2347–2355. [Google Scholar]
- Hoffmann, B; Zaslavskiy, M; Vert, J; Stoven, V. A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: Application to ligand prediction. BMC Bioinformatics 2010, 11, 99. [Google Scholar]
- Canterakis, N. 3D Zernike moments and zernike affine invariants for 3D image analysis and recognition. 11th Scandinavian Conference on Image Analysis, Kangerlussuaq, Greenland, 7–11 June 1999.
- Baker, N; Holst, M; Wang, F. Adaptive multilevel finite element solution of the Poisson– Boltzmann equation II. Refinement at solvent-accessible surfaces in biomolecular systems. J. Comput. Chem 2000, 21, 1343–1352. [Google Scholar]
- Novotni, M; Klein, R. Proceedings of the eighth ACM symposium on solid modeling and applications. Proceedings of the Eighth ACM Symposium on Solid Modeling and Applications, Seattle, Washington, DC, USA, 16–20 June 2003; pp. 216–225.
- Venkatraman, V; Sael, L; Kihara, D. Potential for protein surface shape analysis using spherical harmonics and 3D Zernike descriptors. Cell Biochem. Biophys 2009, 54, 23–32. [Google Scholar]
- Kihara, D; Sael, L; Chikhi, R; Esquivel-Rodriguez, J. Molecular surface representation using 3D Zernike descriptors for protein shape comparison and docking. Curr Protein Peptide Sci, 2010; accepted. [Google Scholar]
- Sael, L; Li, B; La, D; Fang, Y; Ramani, K; Rustamov, R; Kihara, D. Fast protein tertiary structure retrieval based on global surface shape similarity. Proteins: Struct. Funct. Bioinf 2008, 72, 1259–1273. [Google Scholar]
- La, D; Esquivel-Rodríguez, J; Venkatraman, V; Li, B; Sael, L; Ueng, S; Ahrendt, S; Kihara, D. 3D-SURFER: Software for high-throughput protein surface comparison and analysis. Bioinformatics 2009, 25, 2843–2844. [Google Scholar]
- Sael, L; La, D; Li, B; Rustamov, R; Kihara, D. Rapid comparison of properties on protein surface. Proteins: Struct. Funct. Bioinf 2008, 73, 1–10. [Google Scholar]
- Venkatraman, V; Chakravarthy, PR; Kihara, D. Application of 3D Zernike descriptors to shape-based ligand similarity searching. J. Cheminformatics 2009, 1, 19. [Google Scholar]
- Venkatraman, V; Yang, YD; Sael, L; Kihara, D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinformatics 2009, 10, 407. [Google Scholar]
- Sael, L; Kihara, D. Protein surface representation for application to comparing low-resolution protein structure data. BMC Bioinformatics (GIW 2010 issue), 2010; in press. [Google Scholar]
- Demange, G; Gale, D; Sotomayor, M. Multi-item auctions. J. Polit. Economy 1986, 94, 863–872. [Google Scholar]
- Sael, L; Kihara, D. Characterization and classification of local protein surfaces using self-organizing map. Int. J. Knowl. Discov. Bioinformatics 2010, 1, 32–47. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand type | Average Number of Seed Points | Molecular mass (g/mol) a |
|---|---|---|
| AMP | 23.7 | 347.22 |
| ATP | 29.5 | 507.18 |
| FAD | 44.1 | 785.55 |
| FMN | 27.7 | 456.34 |
| GLC | 15.2 | 180.16 |
| HEM | 36.9 | 616.49 |
| NAD | 36.8 | 663.43 |
| PO4 | 9.7 | 94.97 |
| STR | 22.2 | 278.8 |
| 2D Pseudo-Zernike a) | 2D Zernikea) | Spherical Harmonics b) | Global 3DZD a) | Patch 3DZD | |
|---|---|---|---|---|---|
| shape only | 0.66 | 0.66 | 0.64 | 0.66 | 0.76 |
| shape + pocket size | 0.79 | 0.78 | 0.77 | 0.81 | 0.82 |
| Descriptor type | Rank | AMP | ATP | FAD | FMN | GLC | HEM | NAD | PO4 | STR | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Shape | AUC | 0.72 | 0.74 | 0.80 | 0.57 | 0.72 | 0.92 | 0.69 | 0.83 | 0.85 | 0.76 |
| Top1 | 0.11 | 0.14 | 0.40 | 0.00 | 0.00 | 1.00 | 0.00 | 0.90 | 0.00 | 0.28 | |
| Top3 | 0.67 | 0.93 | 0.90 | 0.00 | 0.40 | 1.00 | 0.60 | 1.00 | 0.80 | 0.70 | |
| Shape + size | AUC | 0.85 | 0.76 | 0.83 | 0.68 | 0.88 | 0.91 | 0.74 | 0.88 | 0.85 | 0.82 |
| Top1 | 0.67 | 0.43 | 0.60 | 0.00 | 0.40 | 0.94 | 0.00 | 1.00 | 0.00 | 0.45 | |
| Top3 | 1.00 | 0.93 | 0.90 | 0.50 | 0.80 | 1.00 | 0.80 | 1.00 | 0.80 | 0.86 | |
| Pocket Size a | Top1 | 0.22 | 0.07 | 0.50 | 0.00 | 0.00 | 0.00 | 0.27 | 1.00 | 0.00 | 0.23 |
| Top3 | 0.56 | 0.79 | 0.80 | 0.00 | 0.00 | 0.81 | 0.60 | 1.00 | 0.00 | 0.51 | |
| Random a | Top 1 | 0.10 | 0.13 | 0.10 | 0.06 | 0.05 | 0.15 | 0.14 | 0.19 | 0.06 | 0.11 |
| Top 3 | 0.28 | 0.40 | 0.31 | 0.21 | 0.17 | 0.45 | 0.42 | 0.55 | 0.19 | 0.33 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sael, L.; Kihara, D. Binding Ligand Prediction for Proteins Using Partial Matching of Local Surface Patches. Int. J. Mol. Sci. 2010, 11, 5009-5026. https://doi.org/10.3390/ijms11125009
Sael L, Kihara D. Binding Ligand Prediction for Proteins Using Partial Matching of Local Surface Patches. International Journal of Molecular Sciences. 2010; 11(12):5009-5026. https://doi.org/10.3390/ijms11125009
Chicago/Turabian StyleSael, Lee, and Daisuke Kihara. 2010. "Binding Ligand Prediction for Proteins Using Partial Matching of Local Surface Patches" International Journal of Molecular Sciences 11, no. 12: 5009-5026. https://doi.org/10.3390/ijms11125009