Functional Toxicogenomics: Mechanism-Centered Toxicology

Abstract
:1. Introduction
2. In Vitro Assessment of Toxicity Pathways
3. Toxicogenomics: Omic Tools Applied to Toxicology
4. Functional Toxicogenomics in Yeast
5. Functional Toxicogenomics in Higher Eukaryotes
6. Integrating Functional Data to Identify Toxicity Pathways Will Lead to Comprehensive Cellular Assays
Acknowledgments
References
- Judson, R; Richard, A; Dix, DJ; Houck, K; Martin, M; Kavlock, R; Dellarco, V; Henry, T; Holderman, T; Sayre, P; Tan, S; Carpenter, T; Smith, E. The toxicity data landscape for environmental chemicals. Environ. Health Perspect 2009, 117, 685–695. [Google Scholar]
- van Hummelen, P; Sasaki, J. State-of-the-art genomics approaches in toxicology. Mutat Res 2010. [Google Scholar] [CrossRef]
- Gad, SC. Recent developments in replacing, reducing, and refining animal use in toxicologic research and testing. Fundam. Appl. Toxicol 1990, 15, 8–16. [Google Scholar]
- Committee on Toxicity Testing and Assessment of Environmental Agents, National Research Council of the National Academies. Toxicity Testing in the 21st Century: A Vision and a Strategy; The National Academies Press: Washington, D.C., USA, 2007. [Google Scholar]
- Andersen, ME; Krewski, D. The Vision of Toxicity Testing in the 21st Century: Moving from discussion to action. Toxicol. Sci 2010, 117, 17–24. [Google Scholar]
- Schmidt, CW. TOX 21: New dimensions of toxicity testing. Environ. Health Perspect 2009, 117, A348–A353. [Google Scholar]
- Simmons, S; Fan, C; Ramabhadran, R. Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol. Sci 2009, 111, 202–225. [Google Scholar]
- Shukla, SJ; Huang, R; Austin, CP; Xia, M. The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov Today 2010. [Google Scholar] [CrossRef]
- Dix, DJ; Houck, KA; Martin, MT; Richard, AM; Setzer, RW; Kavlock, RJ. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci 2007, 95, 5–12. [Google Scholar]
- Judson, RS; Houck, KA; Kavlock, RJ; Knudsen, TB; Martin, MT; Mortensen, HM; Reif, DM; Rotroff, DM; Shah, I; Richard, AM; Dix, DJ. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ. Health Perspect 2010, 118, 485–492. [Google Scholar]
- Andersen, ME; Krewski, D. Toxicity testing in the 21st century: Bringing the vision to life. Toxicol. Sci 2009, 107, 324–330. [Google Scholar]
- Hamadeh, HK; Amin, RP; Paules, RS; Afshari, CA. An overview of toxicogenomics. Curr. Issues Mol. Biol 2002, 4, 45–56. [Google Scholar]
- Hayes, KR; Bradfield, CA. Advances in toxicogenomics. Chem. Res. Toxicol 2005, 18, 403–414. [Google Scholar]
- Gatzidou, ET; Zira, AN; Theocharis, SE. Toxicogenomics: A pivotal piece in the puzzle of toxicological research. J. Appl. Toxicol 2007, 27, 302–309. [Google Scholar]
- Nuwaysir, EF; Bittner, M; Trent, J; Barrett, JC; Afshari, CA. Microarrays and toxicology: The advent of toxicogenomics. Mol. Carcinog 1999, 24, 153–159. [Google Scholar]
- Aardema, MJ; Macgregor, JT. Toxicology and genetic toxicology in the new era of “toxicogenomics”: Impact of “-omics” technologies. Mutat. Res 2002, 499, 13–25. [Google Scholar]
- Jayapal, M; Bhattacharjee, RN; Melendez, AJ; Hande, MP. Environmental toxicogenomics: A post-genomic approach to analysing biological responses to environmental toxins. Int. J. Biochem. Cell Biol 2010, 42, 230–240. [Google Scholar]
- Uehara, T; Ono, A; Maruyama, T; Kato, I; Yamada, H; Ohno, Y; Urushidani, T. The Japanese toxicogenomics project: Application of toxicogenomics. Mol. Nutr. Food Res 2010, 54, 218–227. [Google Scholar]
- Mulrane, L; Rexhepaj, E; Smart, V; Callanan, JJ; Orhan, D; Eldem, T; Mally, A; Schroeder, S; Meyer, K; Wendt, M; O’Shea, D; Gallagher, WM. Creation of a digital slide and tissue microarray resource from a multi-institutional predictive toxicology study in the rat: An initial report from the PredTox group. Exp. Toxicol. Pathol 2008, 60, 235–245. [Google Scholar]
- McBurney, RN; Hines, WM; von Tungeln, LS; Schnackenberg, LK; Beger, RD; Moland, CL; Han, T; Fuscoe, JC; Chang, C-W; Chen, JJ; Su, Z; Fan, X-H; Tong, W; Booth, SA; Balasubramanian, R; Courchesne, PL; Campbell, JM; Graber, A; Guo, Y; Juhasz, PJ; Li, TY; Lynch, MD; Morel, NM; Plasterer, TN; Takach, EJ; Zeng, C; Beland, FA. The liver toxicity biomarker study: Phase I design and preliminary results. Toxicol. Pathol 2009, 37, 52–64. [Google Scholar]
- Vlaanderen, J; Moore, LE; Smith, MT; Lan, Q; Zhang, L; Skibola, CF; Rothman, N; Vermeulen, R. Application of OMICS technologies in occupational and environmental health research; current status and projections. Occup. Environ. Med 2010, 67, 136–143. [Google Scholar]
- Paules, R. Phenotypic anchoring: Linking cause and effect. Environ. Health Perspect 2003, 111, A338–A339. [Google Scholar]
- Waters, MD; Fostel, JM. Toxicogenomics and systems toxicology: Aims and prospects. Nat. Rev. Genet 2004, 5, 936–948. [Google Scholar]
- Hieter, P; Boguski, M. Functional genomics: It’s all how you read it. Science 1997, 278, 601–602. [Google Scholar]
- Burns, N; Grimwade, B; Ross-Macdonald, PB; Choi, EY; Finberg, K; Roeder, GS; Snyder, M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev 1994, 8, 1087–1105. [Google Scholar]
- Smith, V; Botstein, D; Brown, PO. Genetic footprinting: A genomic strategy for determining a gene’s function given its sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 6479–6483. [Google Scholar]
- Ross-MacDonald, P; Sheehan, A; Roeder, GS; Snyder, M. A multipurpose transposon system for analyzing protein production, localization, and function in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 190–195. [Google Scholar]
- Kumar, A; Snyder, M. Emerging technologies in yeast genomics. Nat. Rev. Genet 2001, 2, 302–312. [Google Scholar]
- Jin, Y; Dunlap, P; McBride, S; Al-Refai, H; Bushel, P; Freedman, J. Global transcriptome and deletome profiles of yeast exposed to transition metals. PLoS Genet 2008, 4, e1000053. [Google Scholar]
- Shoemaker, DD; Lashkari, DA; Morris, D; Mittmann, M; Davis, RW. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet 1996, 14, 450–456. [Google Scholar]
- Giaever, G; Shoemaker, DD; Jones, TW; Liang, H; Winzeler, EA; Astromoff, A; Davis, RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet 1999, 21, 278–283. [Google Scholar]
- Pierce, SE; Davis, RW; Nislow, C; Giaever, G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat. Protoc 2007, 2, 2958–2974. [Google Scholar]
- Nislow, C; Giaever, G. Chemical genomic tools for understanding gene function and drug action. In Methods in Microbiology; Stansfield, I, Stark, M, Eds.; Academic Press: London, UK, 2007; Volume 36, pp. 387–414. [Google Scholar]
- Pierce, SE; Davis, RW; Nislow, C; Giaever, G. Chemogenomic approaches to elucidation of gene function and genetic pathways. Methods Mol. Biol 2009, 548, 115–143. [Google Scholar]
- Winzeler, E; Shoemaker, D; Astromoff, A; Liang, H; Anderson, K; Andre, B; Bangham, R; Benito, R; Boeke, J; Bussey, H; Chu, A; Connelly, C; Davis, K; Dietrich, F; Dow, S; El Bakkoury, M; Foury, F; Friend, S; Gentalen, E; Giaever, G; Hegemann, J; Jones, T; Laub, M; Liao, H; Liebundguth, N; Lockhart, D; Lucau-Danila, A; Lussier, M; M’Rabet, N; Menard, P; Mittmann, M; Pai, C; Rebischung, C; Revuelta, J; Riles, L; Roberts, C; Ross-MacDonald, P; Scherens, B; Snyder, M; Sookhai-Mahadeo, S; Storms, R; Veronneau, S; Voet, M; Volckaert, G; Ward, T; Wysocki, R; Yen, G; Yu, K; Zimmermann, K; Philippsen, P; Johnston, M; Davis, R. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar]
- Giaever, G; Chu, AM; Ni, L; Connelly, C; Riles, L; Véronneau, S; Dow, S; Lucau-Danila, A; Anderson, K; André, B; Arkin, AP; Astromoff, A; El-Bakkoury, M; Bangham, R; Benito, R; Brachat, S; Campanaro, S; Curtiss, M; Davis, K; Deutschbauer, A; Entian, K-D; Flaherty, P; Foury, F; Garfinkel, DJ; Gerstein, M; Gotte, D; Güldener, U; Hegemann, JH; Hempel, S; Herman, Z; Jaramillo, DF; Kelly, DE; Kelly, SL; Kötter, P; LaBonte, D; Lamb, DC; Lan, N; Liang, H; Liao, H; Liu, L; Luo, C; Lussier, M; Mao, R; Menard, P; Ooi, SL; Revuelta, JL; Roberts, CJ; Rose, M; Ross-Macdonald, P; Scherens, B; Schimmack, G; Shafer, B; Shoemaker, DD; Sookhai-Mahadeo, S; Storms, RK; Strathern, JN; Valle, G; Voet, M; Volckaert, G; Wang, C-Y; Ward, TR; Wilhelmy, J; Winzeler, EA; Yang, Y; Yen, G; Youngman, E; Yu, K; Bussey, H; Boeke, JD; Snyder, M; Philippsen, P; Davis, RW; Johnston, M. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar]
- Pierce, SE; Fung, EL; Jaramillo, DF; Chu, AM; Davis, RW; Nislow, C; Giaever, G. A unique and universal molecular barcode array. Nat. Meth 2006, 3, 601–603. [Google Scholar]
- Smith, AM; Ammar, R; Nislow, C; Giaever, G. A survey of yeast genomic assays for drug and target discovery. Pharmacol. Ther 2010, 127, 156–164. [Google Scholar]
- Ammar, R; Smith, AM; Heisler, LE; Giaever, G; Nislow, C. A comparative analysis of DNA barcode microarray feature size. BMC Genomics 2009, 10, 471. [Google Scholar]
- Smith, AM; Heisler, LE; Mellor, J; Kaper, F; Thompson, MJ; Chee, M; Roth, FP; Giaever, G; Nislow, C. Quantitative phenotyping via deep barcode sequencing. Genome Res 2009, 19, 1836–1842. [Google Scholar]
- Steinmetz, L; Scharfe, C; Deutschbauer, A; Mokranjac, D; Herman, Z; Jones, T; Chu, A; Giaever, G; Prokisch, H; Oefner, P; Davis, R. Systematic screen for human disease genes in yeast. Nat. Genet 2002, 31, 400–404. [Google Scholar]
- Giaever, G; Flaherty, P; Kumm, J; Proctor, M; Nislow, C; Jaramillo, DF; Chu, AM; Jordan, MI; Arkin, AP; Davis, RW. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. USA 2004, 101, 793–798. [Google Scholar]
- Deutschbauer, AM; Jaramillo, DF; Proctor, M; Kumm, J; Hillenmeyer, ME; Davis, RW; Nislow, C; Giaever, G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 2005, 169, 1915–1925. [Google Scholar]
- Lee, W; St Onge, RP; Proctor, M; Flaherty, P; Jordan, MI; Arkin, AP; Davis, RW; Nislow, C; Giaever, G. Genome-wide requirements for resistance to functionally distinct DNA-damaging agents. PLoS Genet 2005, 1, e24. [Google Scholar]
- Chen, Y; Feldman, DE; Deng, C; Brown, JA; De Giacomo, AF; Gaw, AF; Shi, G; Le, QT; Brown, JM; Koong, AC. Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol. Cancer Res 2005, 3, 669–677. [Google Scholar]
- Holland, S; Lodwig, E; Sideri, T; Reader, T; Clarke, I; Gkargkas, K; Hoyle, D; Delneri, D; Oliver, S; Avery, S. Application of the comprehensive set of heterozygous yeast deletion mutants to elucidate the molecular basis of cellular chromium toxicity. Genome Biol 2007, 8, R268. [Google Scholar]
- Doostzadeh, J; Davis, RW; Giaever, GN; Nislow, C; Langston, JW. Chemical genomic profiling for identifying intracellular targets of toxicants producing Parkinson’s disease. Toxicol. Sci 2007, 95, 182–187. [Google Scholar]
- Hillenmeyer, M; Fung, E; Wildenhain, J; Pierce, S; Hoon, S; Lee, W; Proctor, M; St Onge, R; Tyers, M; Koller, D; Altman, R; Davis, R; Nislow, C; Giaever, G. The chemical genomic portrait of yeast: Uncovering a phenotype for all genes. Science 2008, 320, 362–365. [Google Scholar]
- Ericson, E; Gebbia, M; Heisler, LE; Wildenhain, J; Tyers, M; Giaever, G; Nislow, C; Snyder, M. Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet 2008, 4, e1000151. [Google Scholar]
- Jo, WJ; Loguinov, A; Chang, M; Wintz, H; Nislow, C; Arkin, AP; Giaever, G; Vulpe, CD. Identification of genes involved in the toxic response of Saccharomyces cerevisiae against iron and copper overload by parallel analysis of deletion mutants. Toxicol. Sci 2008, 101, 140–151. [Google Scholar]
- Yu, L; Lopez, A; Anaflous, A; El Bali, B; Hamal, A; Ericson, E; Heisler, LE; Mcquibban, A; Giaever, G; Nislow, C; Boone, C; Brown, GW; Bellaoui, M; Snyder, M. Chemical–genetic profiling of imidazo[1,2-a]pyridines and -pyrimidines reveals target pathways conserved between yeast and human cells. PLoS Genet 2008, 4, e1000284. [Google Scholar]
- Jo, WJ; Kim, JH; Oh, E; Jaramillo, D; Holman, P; Loguinov, AV; Arkin, AP; Nislow, C; Giaever, G; Vulpe, CD. Novel insights into iron metabolism by integrating deletome and transcriptome analysis in an iron deficiency model of the yeast Saccharomyces cerevisiae. BMC Genomics 2009, 10, 130. [Google Scholar]
- Jo, W; Loguinov, A; Wintz, H; Chang, M; Smith, A; Kalman, D; Zhang, L; Smith, M; Vulpe, C. Comparative functional genomic analysis identifies distinct and overlapping sets of genes required for resistance to monomethylarsonous acid (MMAIII) and arsenite (AsIII) in yeast. Toxicol. Sci 2009, 111, 424–436. [Google Scholar]
- Smith, AM; Heisler, LE; St Onge, RP; Farias-Hesson, E; Wallace, IM; Bodeau, J; Harris, AN; Perry, KM; Giaever, G; Pourmand, N; Nislow, C. Highly-multiplexed barcode sequencing: An efficient method for parallel analysis of pooled samples. Nucleic Acids Res 2010, 38, e142. [Google Scholar]
- Weiss, A; Delproposto, J; Giroux, CN. High-throughput phenotypic profiling of gene-environment interactions by quantitative growth curve analysis in Saccharomyces cerevisiae. Anal. Biochem 2004, 327, 23–34. [Google Scholar]
- Birrell, GW; Giaever, G; Chu, AM; Davis, RW; Brown, JM. A genome-wide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc. Natl. Acad. Sci. USA 2001, 98, 12608–12613. [Google Scholar]
- Bassett, DE; Boguski, MS; Spencer, F; Reeves, R; Kim, S; Weaver, T; Hieter, P. Genome cross-referencing and XREFdb: Implications for the identification and analysis of genes mutated in human disease. Nat. Genet 1997, 15, 339–344. [Google Scholar]
- Foury, F. Human genetic diseases: A cross-talk between man and yeast. Gene 1997, 195, 1–10. [Google Scholar]
- Jo, W; Ren, X; Chu, F; Aleshin, M; Wintz, H; Burlingame, A; Smith, M; Vulpe, C; Zhang, L. Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicol. Appl. Pharmacol 2009, 241, 294–302. [Google Scholar]
- Shannon, P; Markiel, A; Ozier, O; Baliga, NS; Wang, JT; Ramage, D; Amin, N; Schwikowski, B; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar]
- Ideker, T; Ozier, O; Schwikowski, B; Siegel, AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 2002, 18, S233–S240. [Google Scholar]
- Robinson, M; Grigull, J; Mohammad, N; Hughes, T. FunSpec: A web-based cluster interpreter for yeast. BMC Bioinf 2002, 3, 35. [Google Scholar]
- Penkett, CJ; Morris, JA; Wood, V; Bähler, J. YOGY: A web-based, integrated database to retrieve protein orthologs and associated Gene Ontology terms. Nucleic Acids Res 2006, 34, W330–334. [Google Scholar]
- Lehár, J; Stockwell, BR; Giaever, G; Nislow, C. Combination chemical genetics. Nat. Chem. Biol 2008, 4, 674–681. [Google Scholar]
- Brenner, C. Chemical genomics in yeast. Genome Biol 2004, 5, 240. [Google Scholar]
- Hoon, S; St Onge, R; Giaever, G; Nislow, C. Yeast chemical genomics and drug discovery: An update. Trends Pharmacol. Sci 2008, 29, 499–504. [Google Scholar]
- Breslow, DK; Cameron, DM; Collins, SR; Schuldiner, M; Stewart-Ornstein, J; Newman, HW; Braun, S; Madhani, HD; Krogan, NJ; Weissman, JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar]
- Yan, Z; Costanzo, M; Heisler, LE; Paw, J; Kaper, F; Andrews, BJ; Boone, C; Giaever, G; Nislow, C. Yeast Barcoders: A chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat. Methods 2008, 5, 719–725. [Google Scholar]
- Yan, Z; Berbenetz, NM; Giaever, G; Nislow, C. Precise gene-dose alleles for chemical genetics. Genetics 2009, 182, 623–626. [Google Scholar]
- Ho, CH; Magtanong, L; Barker, SL; Gresham, D; Nishimura, S; Natarajan, P; Koh, JLY; Porter, J; Gray, CA; Andersen, RJ; Giaever, G; Nislow, C; Andrews, B; Botstein, D; Graham, TR; Yoshida, M; Boone, C. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotechnol 2009, 27, 369–377. [Google Scholar]
- Hoon, S; Smith, AM; Wallace, IM; Suresh, S; Miranda, M; Fung, E; Proctor, M; Shokat, KM; Zhang, C; Davis, RW; Giaever, G; St Onge, RP; Nislow, C. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol 2008, 4, 498–506. [Google Scholar]
- Costanzo, M; Baryshnikova, A; Bellay, J; Kim, Y; Spear, ED; Sevier, CS; Ding, H; Koh, JLY; Toufighi, K; Mostafavi, S; Prinz, J; St Onge, RP; VanderSluis, B; Makhnevych, T; Vizeacoumar, FJ; Alizadeh, S; Bahr, S; Brost, RL; Chen, Y; Cokol, M; Deshpande, R; Li, Z; Lin, Z-Y; Liang, W; Marback, M; Paw, J; San Luis, B-J; Shuteriqi, E; Tong, AHY; van Dyk, N; Wallace, IM; Whitney, JA; Weirauch, MT; Zhong, G; Zhu, H; Houry, WA; Brudno, M; Ragibizadeh, S; Papp, B; Pál, C; Roth, FP; Giaever, G; Nislow, C; Troyanskaya, OG; Bussey, H; Bader, GD; Gingras, A-C; Morris, QD; Kim, PM; Kaiser, CA; Myers, CL; Andrews, BJ; Boone, C. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar]
- Ridpath, JR; Nakamura, A; Tano, K; Luke, AM; Sonoda, E; Arakawa, H; Buerstedde, J-M; Gillespie, DAF; Sale, JE; Yamazoe, M; Bishop, DK; Takata, M; Takeda, S; Watanabe, M; Swenberg, JA; Nakamura, J. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res 2007, 67, 11117–11122. [Google Scholar]
- Fire, A; Xu, S; Montgomery, MK; Kostas, SA; Driver, SE; Mello, CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Kiefer, J; Yin, HH; Que, QQ; Mousses, S. High-throughput siRNA screening as a method of perturbation of biological systems and identification of targeted pathways coupled with compound screening. Methods Mol. Biol 2009, 563, 275–287. [Google Scholar]
- Mullenders, J; Bernards, R. Loss-of-function genetic screens as a tool to improve the diagnosis and treatment of cancer. Oncogene 2009, 28, 4409–4420. [Google Scholar]
- Root, DE; Hacohen, N; Hahn, WC; Lander, ES; Sabatini, DM. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat. Methods 2006, 3, 715–719. [Google Scholar]
- Luo, B; Cheung, HW; Subramanian, A; Sharifnia, T; Okamoto, M; Yang, X; Hinkle, G; Boehm, JS; Beroukhim, R; Weir, BA; Mermel, C; Barbie, DA; Awad, T; Zhou, X; Nguyen, T; Piqani, B; Li, C; Golub, TR; Meyerson, M; Hacohen, N; Hahn, WC; Lander, ES; Sabatini, DM; Root, DE. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20380–20385. [Google Scholar]
- Kimura, J; Nguyen, ST; Liu, H; Taira, N; Miki, Y; Yoshida, K. A functional genome-wide RNAi screen identifies TAF1 as a regulator for apoptosis in response to genotoxic stress. Nucleic Acids Res 2008, 36, 5250–5259. [Google Scholar]
- Wolters, NM; MacKeigan, JP. From sequence to function: Using RNAi to elucidate mechanisms of human disease. Cell Death Differ 2008, 15, 809–819. [Google Scholar]
- Maine, EM. Studying gene function in Caenorhabditis elegans using RNA-mediated interference. Brief Funct. Genomic. Proteomic 2008, 7, 184–194. [Google Scholar]
- Menzel, R; Yeo, HL; Rienau, S; Li, S; Steinberg, CEW; Stürzenbaum, SR. Cytochrome P450s and short-chain dehydrogenases mediate the toxicogenomic response of PCB52 in the nematode Caenorhabditis elegans. J. Mol. Biol 2007, 370, 1–13. [Google Scholar]
- Mohr, S; Bakal, C; Perrimon, N. Genomic screening with RNAi: Results and challenges. Annu. Rev. Biochem 2010, 79, 37–64. [Google Scholar]
- Aleström, P; Holter, JL; Nourizadeh-Lillabadi, R. Zebrafish in functional genomics and aquatic biomedicine. Trends Biotechnol 2006, 24, 15–21. [Google Scholar]
- Carette, JE; Guimaraes, CP; Varadarajan, M; Park, AS; Wuethrich, I; Godarova, A; Kotecki, M; Cochran, BH; Spooner, E; Ploegh, HL; Brummelkamp, TR. haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar]
- Berns, K; Hijmans, EM; Mullenders, J; Brummelkamp, TR; Velds, A; Heimerikx, M; Kerkhoven, RM; Madiredjo, M; Nijkamp, W; Weigelt, B; Agami, R; Ge, W; Cavet, G; Linsley, PS; Beijersbergen, RL; Bernards, R. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004, 428, 431–437. [Google Scholar]
- Brummelkamp, TR; Fabius, AWM; Mullenders, J; Madiredjo, M; Velds, A; Kerkhoven, RM; Bernards, R; Beijersbergen, RL. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat. Chem. Biol 2006, 2, 202–206. [Google Scholar]
- Berns, K; Horlings, HM; Hennessy, BT; Madiredjo, M; Hijmans, EM; Beelen, K; Linn, SC; Gonzalez-Angulo, AM; Stemke-Hale, K; Hauptmann, M; Beijersbergen, RL; Mills, GB; van de Vijver, MJ; Bernards, R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar]
- Whitehurst, AW; Bodemann, BO; Cardenas, J; Ferguson, D; Girard, L; Peyton, M; Minna, JD; Michnoff, C; Hao, W; Roth, MG; Xie, X-J; White, MA. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar]
- Sudo, H; Tsuji, AB; Sugyo, A; Imai, T; Saga, T; Harada, Y-N. A loss of function screen identifies nine new radiation susceptibility genes. Biochem. Biophys. Res. Commun 2007, 364, 695–701. [Google Scholar]
- Ohn, T; Kedersha, N; Hickman, T; Tisdale, S; Anderson, P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nature 2008, 10, 1224–1231. [Google Scholar]
- Schlabach, MR; Luo, J; Solimini, NL; Hu, G; Xu, Q; Li, MZ; Zhao, Z; Smogorzewska, A; Sowa, ME; Ang, XL; Westbrook, TF; Liang, AC; Chang, K; Hackett, JA; Harper, JW; Hannon, GJ; Elledge, SJ. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar]
- O’Connor, MN; Salles, II; Cvejic, A; Watkins, NA; Walker, A; Garner, SF; Jones, CI; Macaulay, IC; Steward, M; Zwaginga, J-J; Bray, SL; Dudbridge, F; de Bono, B; Goodall, AH; Deckmyn, H; Stemple, DL; Ouwehand, WH; Consortium, B. Functional genomics in zebrafish permits rapid characterization of novel platelet membrane proteins. Blood 2009, 113, 4754–4762. [Google Scholar]
- Zhao, L; Pan, Y; Gang, Y; Wang, H; Jin, H; Tie, J; Xia, L; Zhang, Y; He, L; Yao, L; Qiao, T; Li, T; Liu, Z; Fan, D. Identification of GAS1 as an epirubicin resistance-related gene in human gastric cancer cells with a partially randomized small interfering RNA library. J. Biol. Chem 2009, 284, 26273–26285. [Google Scholar]
- Zhang, S; Binari, R; Zhou, R; Perrimon, N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics 2010, 184, 1165–1179. [Google Scholar]
- Venancio, TM; Balaji, S; Aravind, L. High-confidence mapping of chemical compounds and protein complexes reveals novel aspects of chemical stress response in yeast. Mol. BioSyst 2010, 6, 165–171. [Google Scholar]
- Zhang, L; McHale, CM; Rothman, N; Li, G; Ji, Z; Vermeulen, R; Hubbard, AE; Ren, X; Shen, M; Rappaport, SM; North, M; Skibola, CF; Yin, S; Vulpe, C; Chanock, SJ; Smith, MT; Lan, Q. Systems biology of human benzene exposure. Chem. Biol. Interact 2010, 184, 86–93. [Google Scholar]
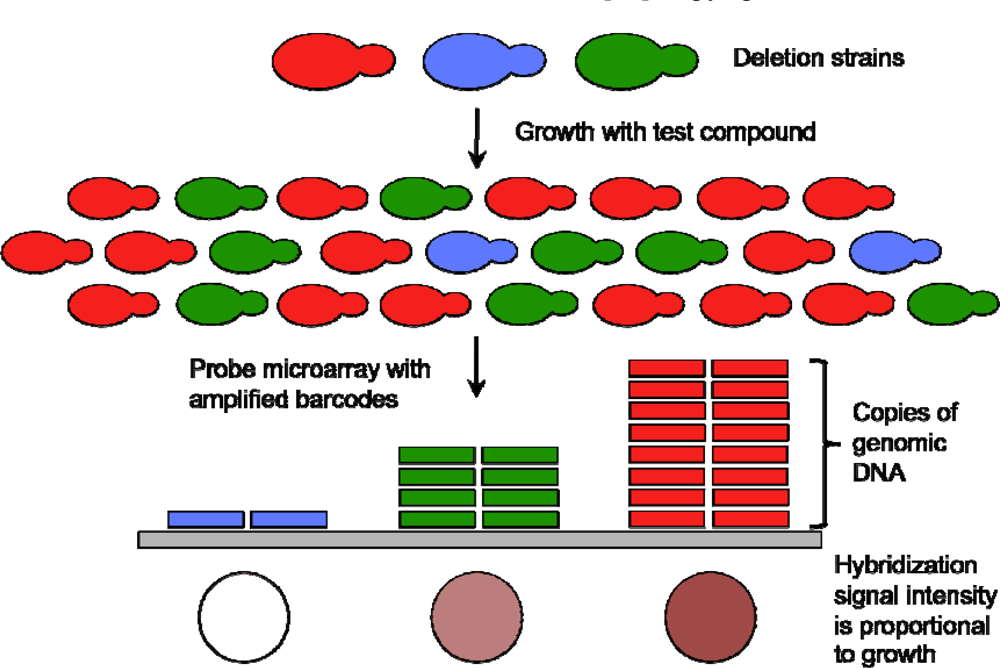

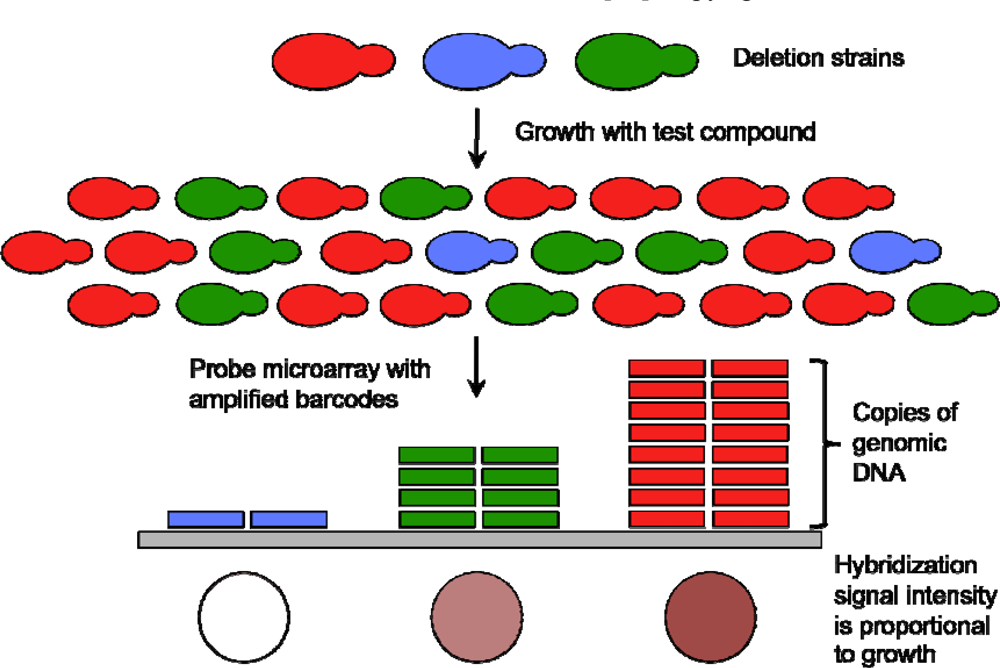{kind=link}
| Array Platform | Compound(s) Tested | YKO Collection Used | Reference |
|---|---|---|---|
| TAG3 | High salt; sorbitol; galactose; pH8; minimal medium; nystatin | Homozygous | [36] |
| TAG3 | Glycerol; ethanol; lactate; dinitrophenol | Homozygous | [41] |
| TAG3 | Alverine citrate; atorvastatin; methotrexate; 5-fluorouracil (5-FU); miconazole; amphotericin B; lovastatin; cisplatin; itraconazole; fluconazole; dyclonine; fenpropimorph | Heterozygous | [42] |
| TAG3 | None (haploinsufficiency profiling) | Heterozygous | [43] |
| TAG3 | DNA damaging agents | Homozygous | [44] |
| TAG3 | ER stressors (tunicamycin; β-mercaptoethanol) | Homozygous | [45] |
| TAG3 | Chromium | Heterozygous | [46] |
| TAG3 | Neurotoxicants | Homozygous | [47] |
| TAG3 | 316 compounds | Homozygous; heterozygous | [48] |
| TAG3/4* | 214 psychoactive drugs | Homozygous; heterozygous | [49] |
| TAG3 | Iron and copper overload | Homozygous | [50] |
| TAG4 | Imidazo[1,2-a]pyridines and –Pyrimidines | Homozygous; heterozygous; haploid | [51] |
| TAG3 | Iron deficiency | Homozygous | [52] |
| TAG4 | Sodium arsenite and monomethylarsonous acid (MMA3) | Homozygous | [53] |
| TAG4 | Methyl methanesulfonate (MMS); cisplatin; compound 1561-0023 | Homozygous; heterozygous | [54] |
| System (organism) | Outcome | Reference |
|---|---|---|
| shRNA bar code (human) | Identification of genes whose suppression confers resistance to p53-induced growth arrest | [86] |
| shRNA bar code (human) | Identification of genes whose suppression confers resistance to p53-induced growth arrest | [87] |
| shRNA bar code (human) | Identification of genes whose suppression confers resistance to Herceptin | [88] |
| siRNA (human) | Identification of genes required for paclitaxel tolerance | [89] |
| RNAi (C. elegans) | Identification of genes involved in the toxicogenesis of a polychlorinated biphenyl (PCB) | [82] |
| shRNA (human) | Identification of radiation susceptibility genes | [90] |
| siRNA (human) | Identification of genes required for stress resistance | [91] |
| shRNA bar code (human) | Identification of genes required for cancer proliferation | [92] |
| shRNA (human) | Identification of genes required for induction of apoptosis | [79] |
| RNAi (zebrafish) | Characterization of novel human platelet proteins | [93] |
| siRNA (human) | Identification of genes whose suppression confers resistance to epirubicin | [94] |
| Mutagenized haploid cells (human) | Identification of diptheria toxin pathway genes | [85] |
| RNAi (D. melanogaster) | Identification of modifiers of mutant Huntingtin aggregate formation | [95] |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
North, M.; Vulpe, C.D. Functional Toxicogenomics: Mechanism-Centered Toxicology. Int. J. Mol. Sci. 2010, 11, 4796-4813. https://doi.org/10.3390/ijms11124796
North M, Vulpe CD. Functional Toxicogenomics: Mechanism-Centered Toxicology. International Journal of Molecular Sciences. 2010; 11(12):4796-4813. https://doi.org/10.3390/ijms11124796
Chicago/Turabian StyleNorth, Matthew, and Chris D. Vulpe. 2010. "Functional Toxicogenomics: Mechanism-Centered Toxicology" International Journal of Molecular Sciences 11, no. 12: 4796-4813. https://doi.org/10.3390/ijms11124796
APA StyleNorth, M., & Vulpe, C. D. (2010). Functional Toxicogenomics: Mechanism-Centered Toxicology. International Journal of Molecular Sciences, 11(12), 4796-4813. https://doi.org/10.3390/ijms11124796