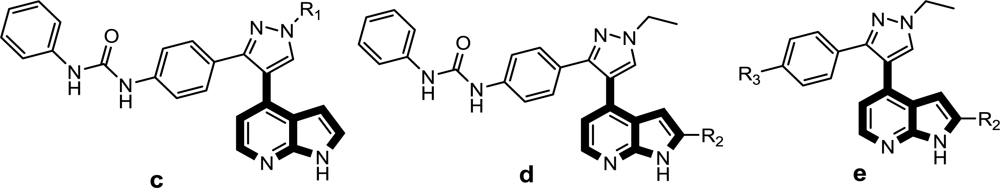
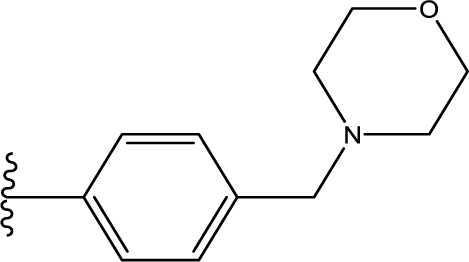
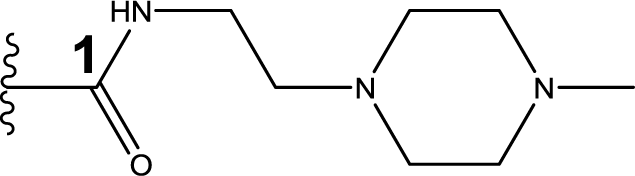
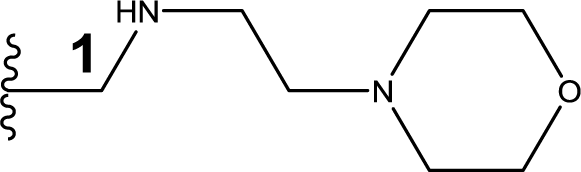
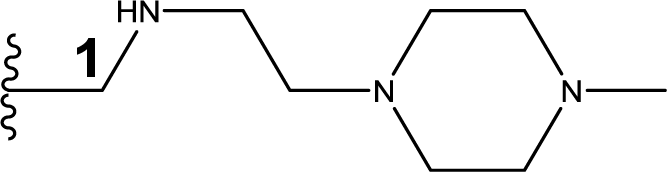
1. Introduction
The Aurora kinases are a family of three highly homologous serine-threonine protein kinases (Aurora A, B and C) that play a critical role in regulating many of the processes that are pivotal to mitosis [
1]. Since it was discovered that Aurora kinases are aberrantly over-expressed in various tumor cells [
2], there has been intense research in the area of identifying selective Aurora inhibitors as potential drugs; up to now more than 10 small molecules have entered clinical studies [
1]. In the last decades, compared with Aurora B, Aurora A has received most of the attention in terms of a link with human cancers in the field of drug development, since the inhibition of Aurora B could rapidly lead to a catastrophic mitosis and cell death, and the inhibition of Aurora B, rather than of Aurora A, is also more crucial for the inhibition of cell proliferation [
3].
Aurora B is involved in ensuring chromosome segregation and alignment as part of the chromosomal passenger protein complex (CPC), which plays a key role in regulating progression through and completion of mitosis [
4]. A number of studies have characterized the gross cellular effects of disrupting Aurora B in cells, including the expression of kinase dead protein, siRNA depletion of total protein, or microinjection of neutralizing antibodies [
1]. Some work also showed that the depression of Aurora B kinase activity by small inhibitors could lead to a failure in cytokinesis and abnormal exit from mitosis, resulting in the endoreduplication, accumulation of polyploidy cells and ultimately apoptosis [
5–
7].
Encouragingly, series of small molecules have been investigated and exhibited efficient inhibitory activities against Aurora B [
4,
8–
10]. MK-0457, the first Aurora inhibitor to enter clinical trials, can effectively disrupt mitosis and promote apoptosis in cycling cells while still leaving the non-cycling cells unaffected [
6]. It also possesses interesting characteristics in that this compound exhibits approximately equal potency to all three types of Aurora kinases, which definitely improves the efficiency of the molecule. GSK1070916 [
11], a kind of 7-azaindole derivative, is another potent and selective ATP-competitive inhibitor of both Aurora B and C with a >250-fold selectivity over Aurora A [
9]. Recently, this Aurora B inhibitor was also advanced as an agent for the treatment of cancer [
12,
13]. SNS-314, the third important pan-Aurora inhibitor based on a 4-aminothieno [3,2-d] pyrimidine scaffold, attracted much research interest not only due to its good affinity against all three isoforms of Aurora kinases [
1], but also because of its compelling preclinical profile; it has entered clinical trials in patients with solid tumors [
4,
10].
Structure-activity analysis is the foundation for understanding the structural features of both the inhibitors and the target receptors involved in a particular biological process and thus helps to design more effective inhibitors [
14]. Therefore, this method has encouraged its wide use as a rational way to gain insight into the influence of various interactive fields on the activity and thus to aid in the design and forecasting of the inhibitory activity of novel inhibitors [
15–
21]. In this work, the most widely used computational tools, comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods [
22,
23], were used to derive 3D-QSAR models for the above three different chemical series of Aurora B inhibitors. Meanwhile, molecular docking was also performed to combine with the 3D-QSAR method, presenting more informative data for the drug design.
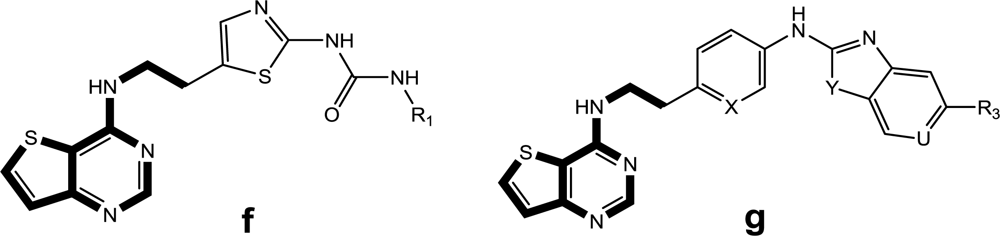
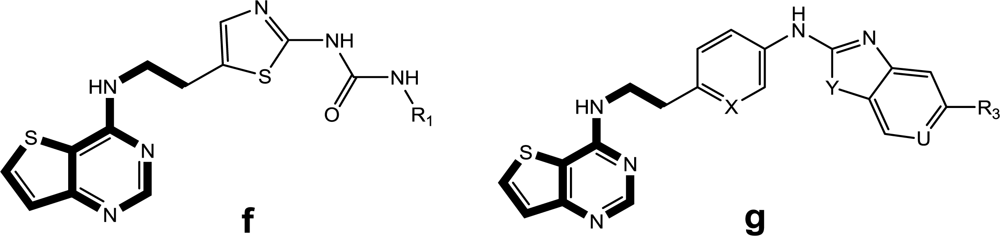
To date, a number of Aurora B small molecule inhibitors, from structurally diverse chemical series, have already been reported or reviewed elsewhere [
1,
4,
8–
10]. However, very few series of Aurora B inhibitors have so far received much attention from a theoretical perspective. More recently, an elegant 3D-QSAR work concerning the quinazoline derivatives of AZD1152 and ZM447439 classes combined with molecular docking was reported [
24]. The authors found the highly active ligands could be designed by varying positively charged, bulky, hydrophobic substitutes at the quinazoline ring, and bulky and hydrophobic groups around the thiazole ring were desirable for higher activity [
24]. More recently, several other series of compounds, such as MK-0457 [
8], GSK1070916 [
9] and SNS-314 [
4] derivatives, have been reported as promising Aurora B inhibitors. However, no comprehensive features of the ligand-receptor interactions or detailed structural determinants at the atomic level were obtained for these inhibitors since the X-ray crystallographic structure for the human Aurora B kinase has not been reported to date. Therefore, in the present study, we mainly focus on the study of the above three classes of inhibitors with an attempt to disclose the structural features of anticancer Aurora B inhibitors using an integrated computational method including 3D-QSAR, homology modeling and molecular docking simulations. A comparison was also performed to identify similarities and differences in the binding modes for each class, and thus a set of vital amino acid residues were found to play a critical role in stabilizing the ligand-receptor interactions of Aurora B kinase. To our knowledge, this work presents the first 3D-QSAR study for these series of compounds, which will provide a platform for the screening and design of novel Aurora B inhibitors as important weapons in the fight against tumors.
4. Conclusions
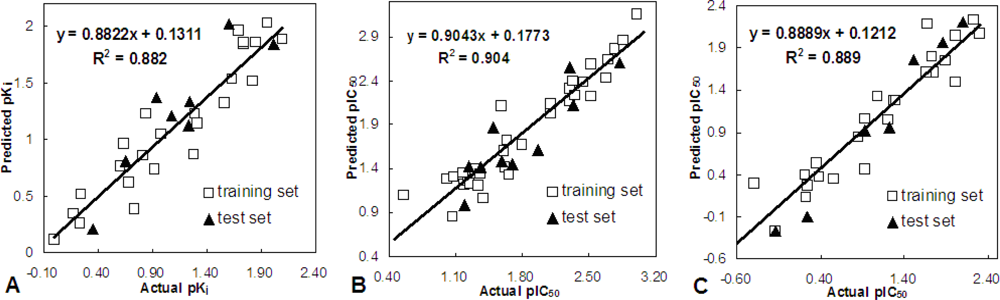
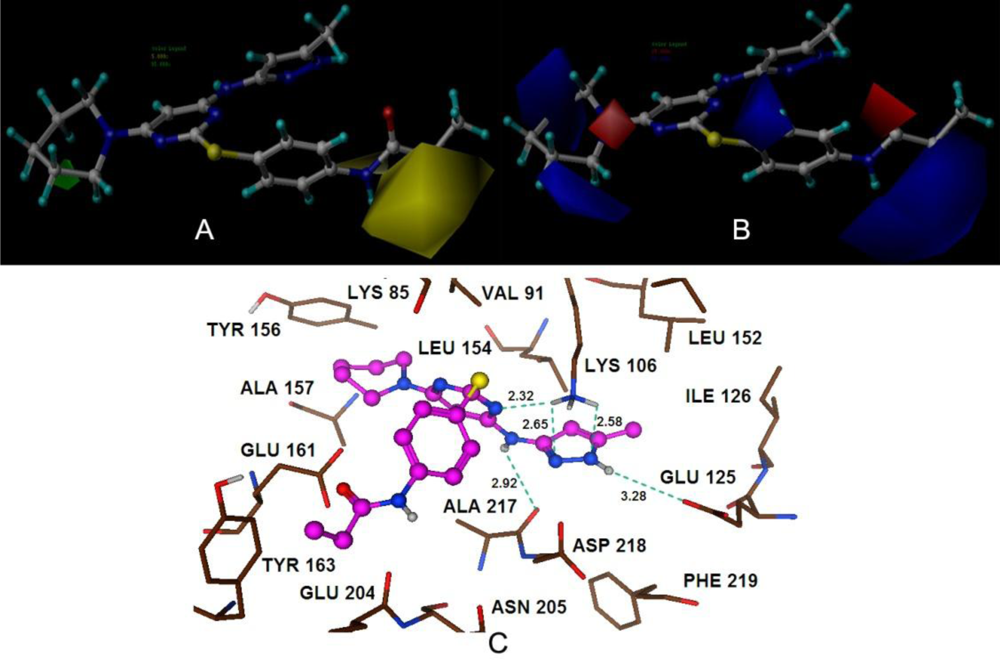
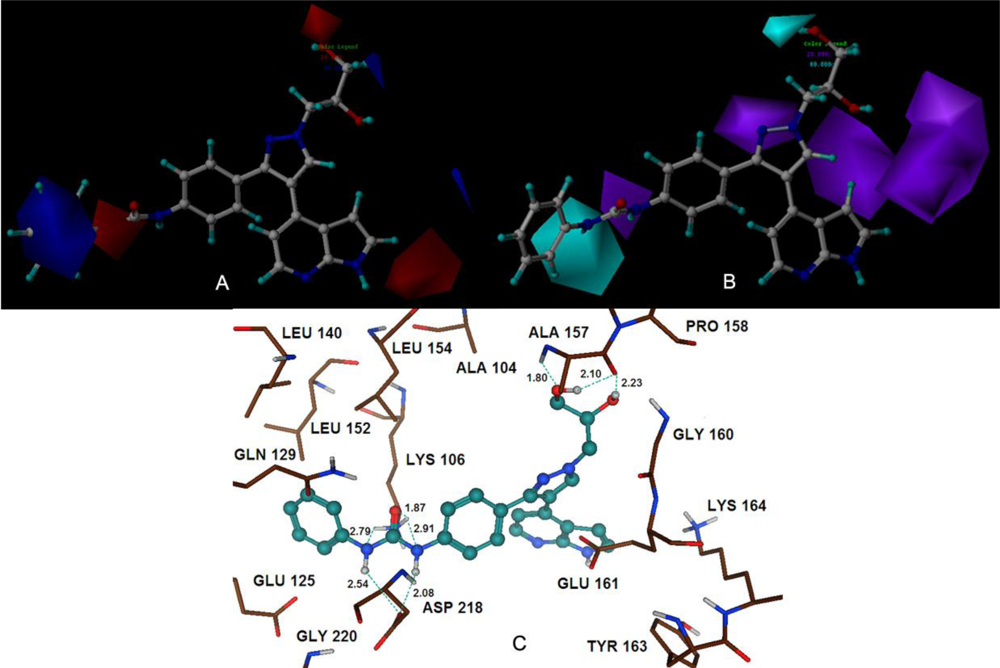
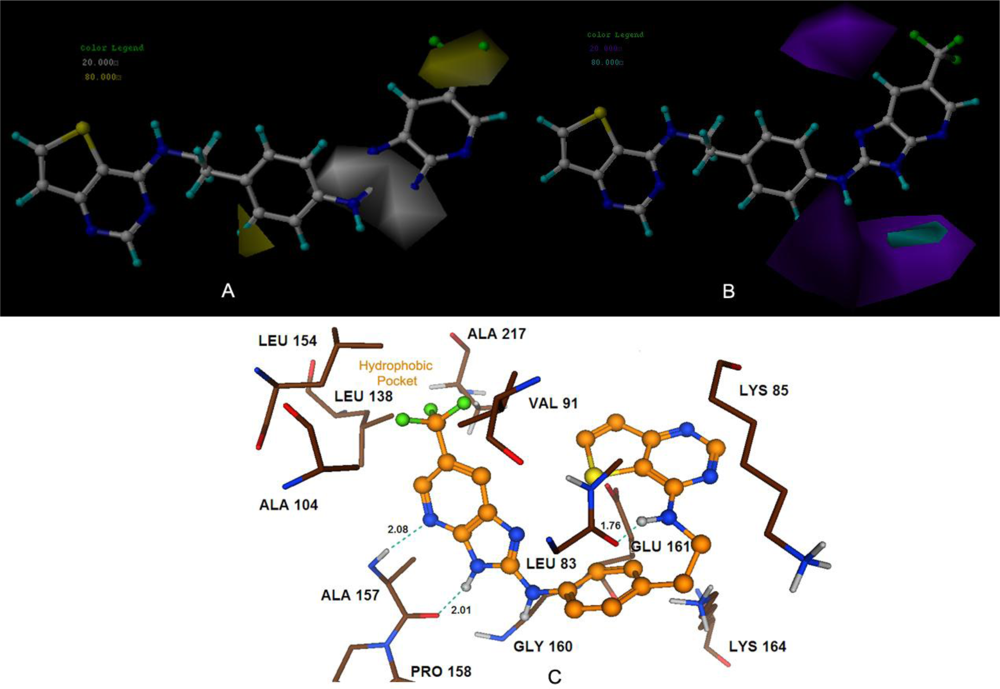
The 3D-QSAR studies yielded stable and statistically significant predictive models with relative high cross-correlation coefficients for predicting the activities of new Aurora B inhibitors. A high LOOCV r2 value and a small standard deviation indicate the existence of a similar relationship in all compounds of the series used in the study. The overall study for the optimal model from the MK-0457 class implies the crucial roles of steric and electrostatic field effects, while the GSK1070916 model revealed the importance of electrostatic and hydrogen-bond donor fields. In addition, for SNS-314, hydrophobic and hydrogen-bond donor fields were found to be more important than the other descriptors.
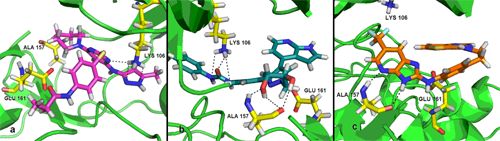
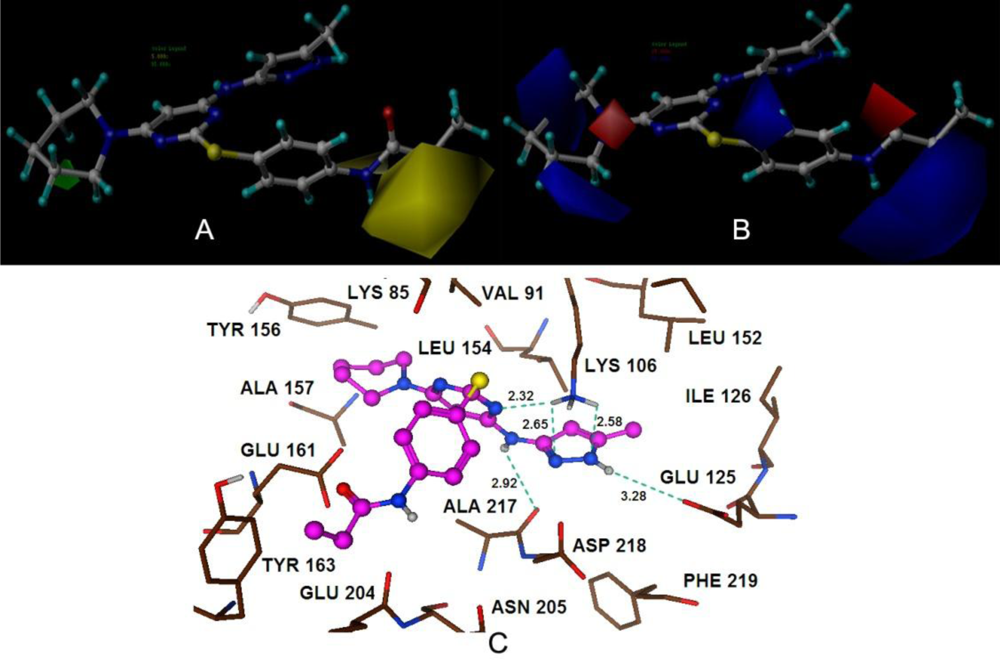
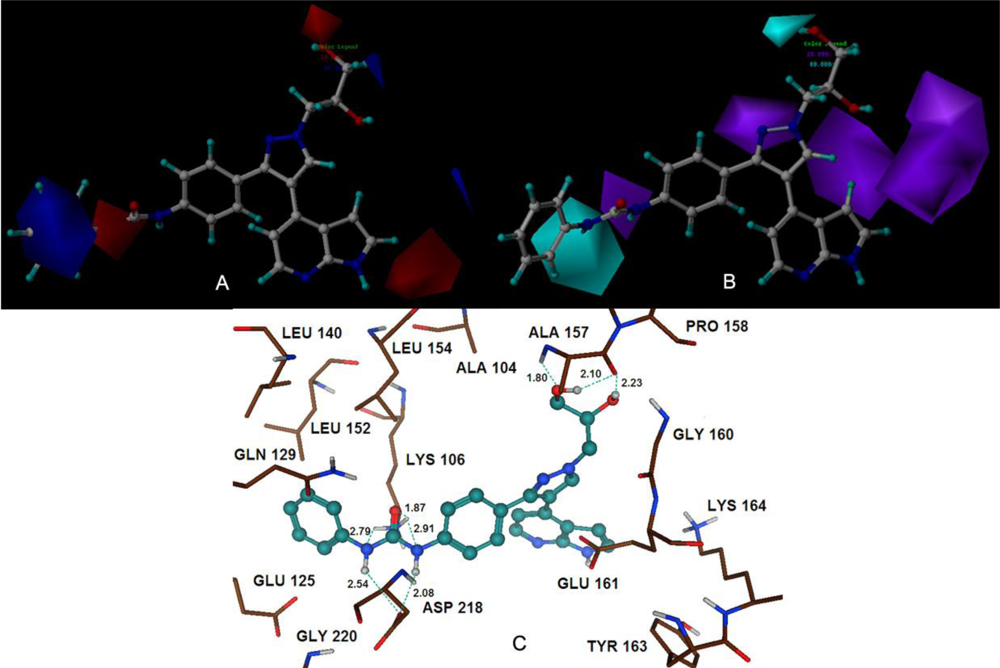
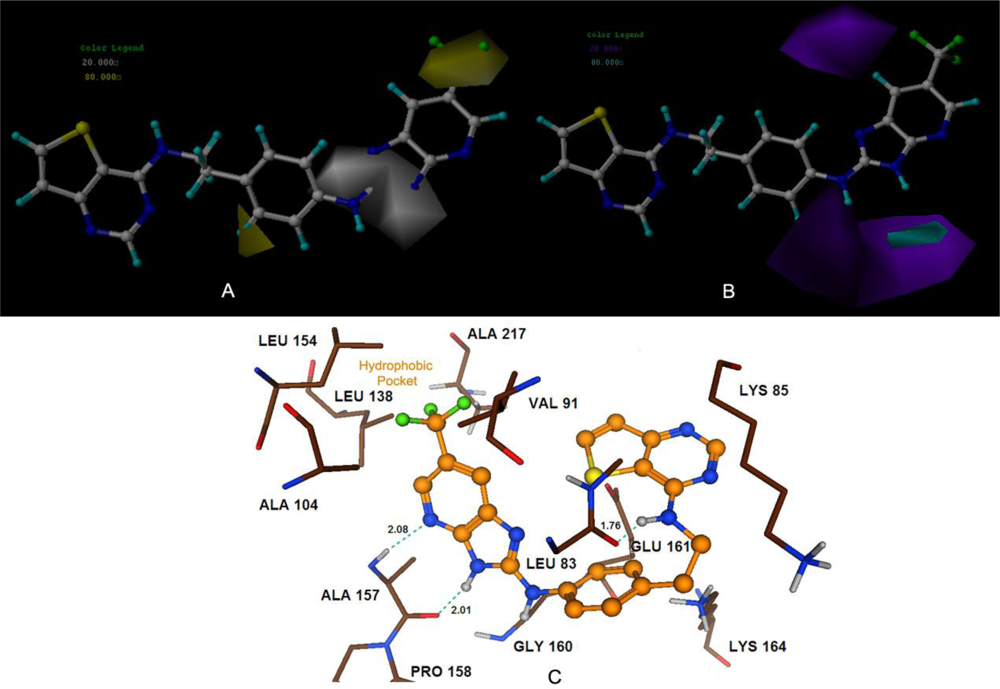
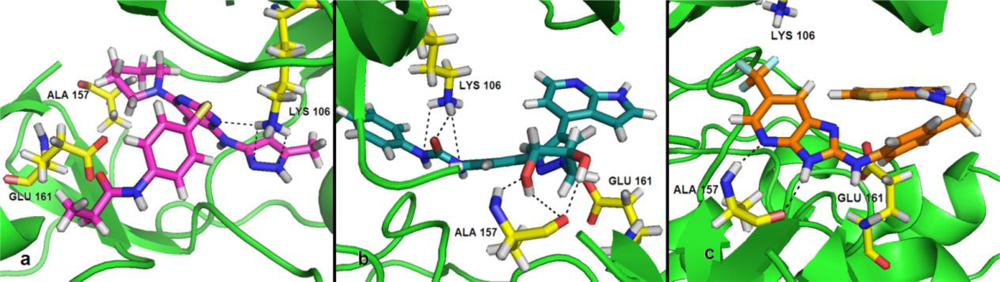
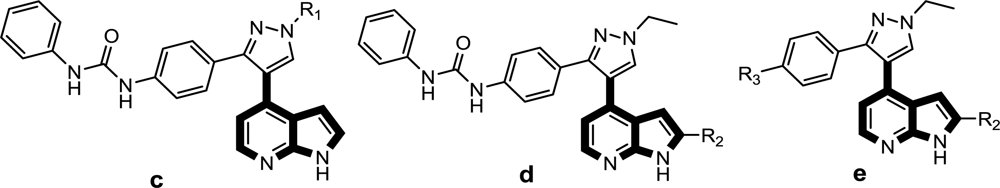
Satisfyingly, a good correlation was attained between the 3D-QSAR contour maps and the corresponding predictive binding mode. For the MK-0457 model, the bulky substituent of –R2 group plays a main contribution toward the inhibitory activity, which is consistent with the existence of a wide steric gorge enclosing this group. In addition, the carbonyl group at 1-position is critical for the increase in the inhibitory activity. For GSK1070916 compounds, the preference for electronegative groups with hydrogen bond donating capacity at 2-position and –R1 group shows a great impact on the overall inhibitory activities. The model for SNS-314 revealed the hydrophobic favorable property at the –R3 group, which is consistent with the docking results. And the docking analysis demonstrated the importance of Glu161, Ala157 and Lys106 in facilitating Aurora B recognition of its inhibitors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}