Relative Stabilities of Conserved and Non-Conserved Structures in the OB-Fold Superfamily

Abstract
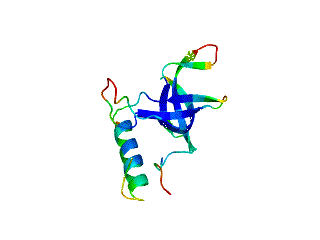
:
1. Introduction
2. Results
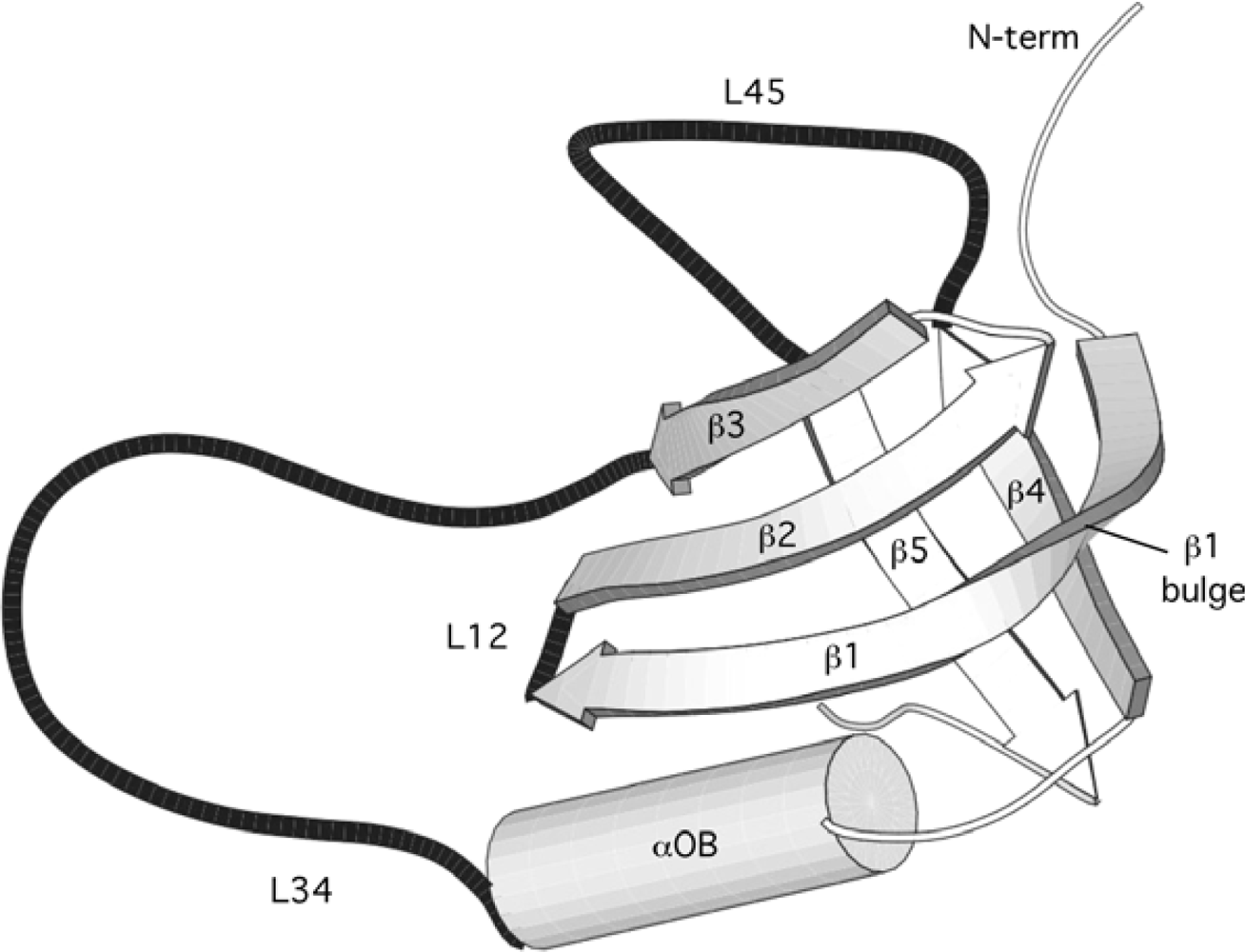
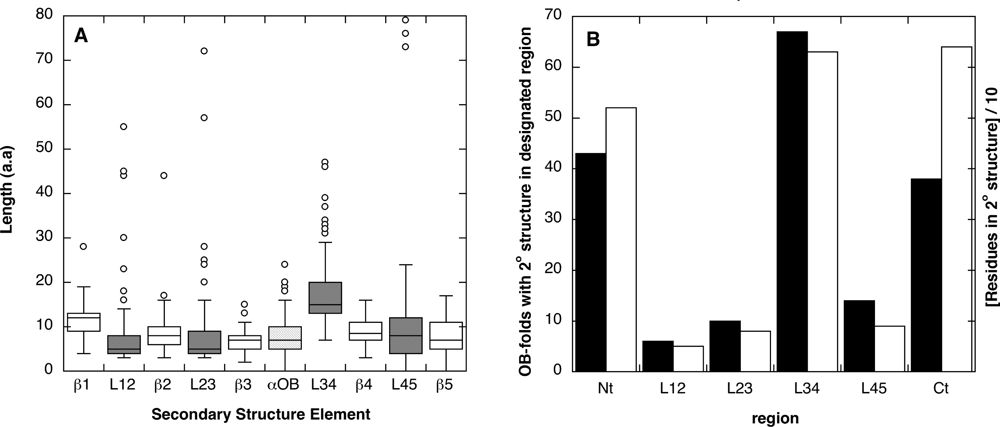
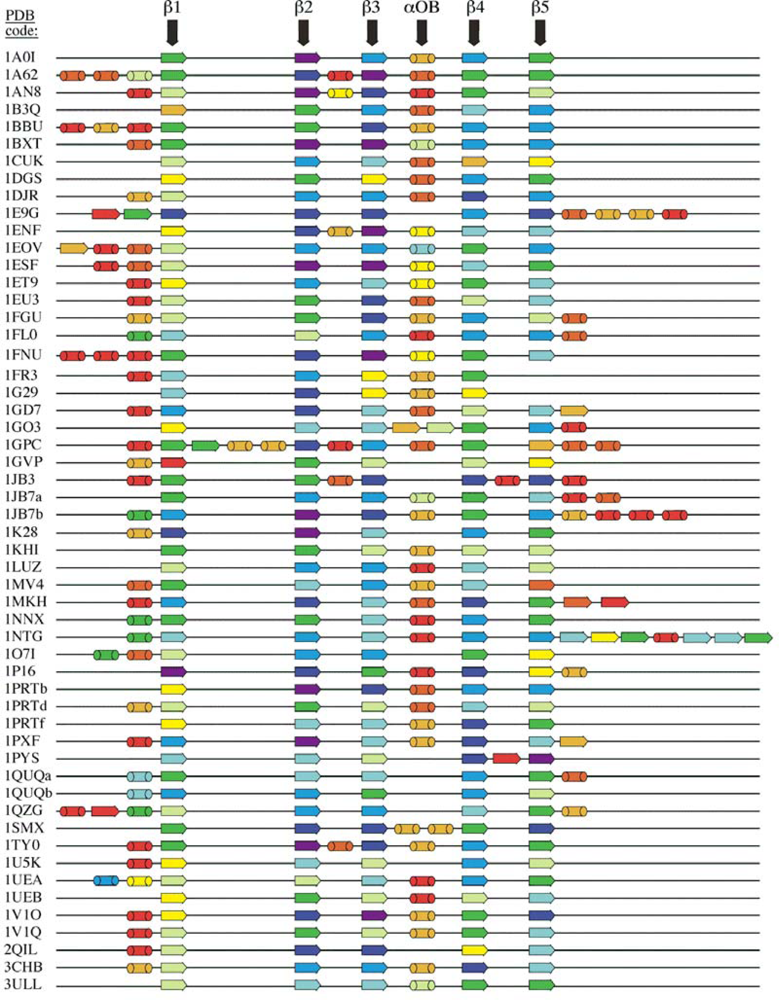
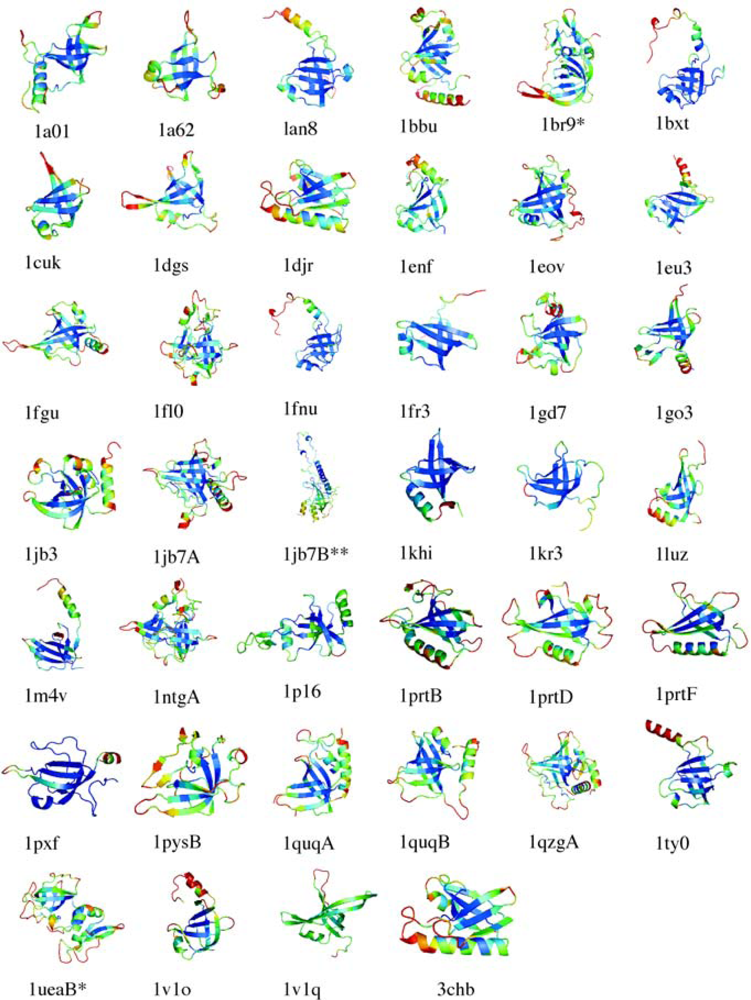
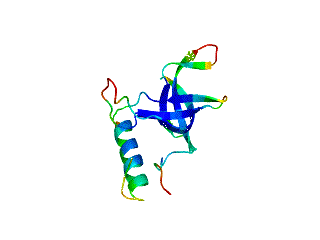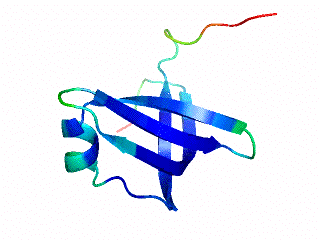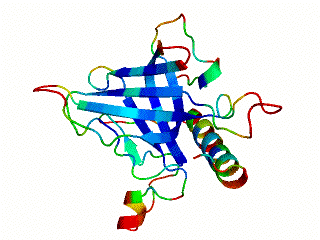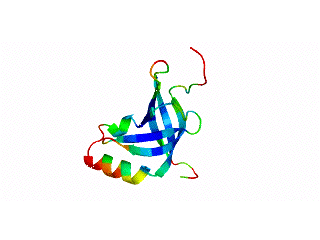
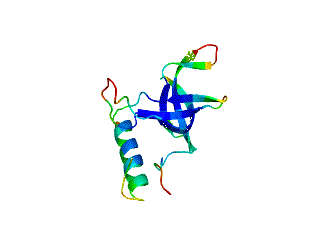
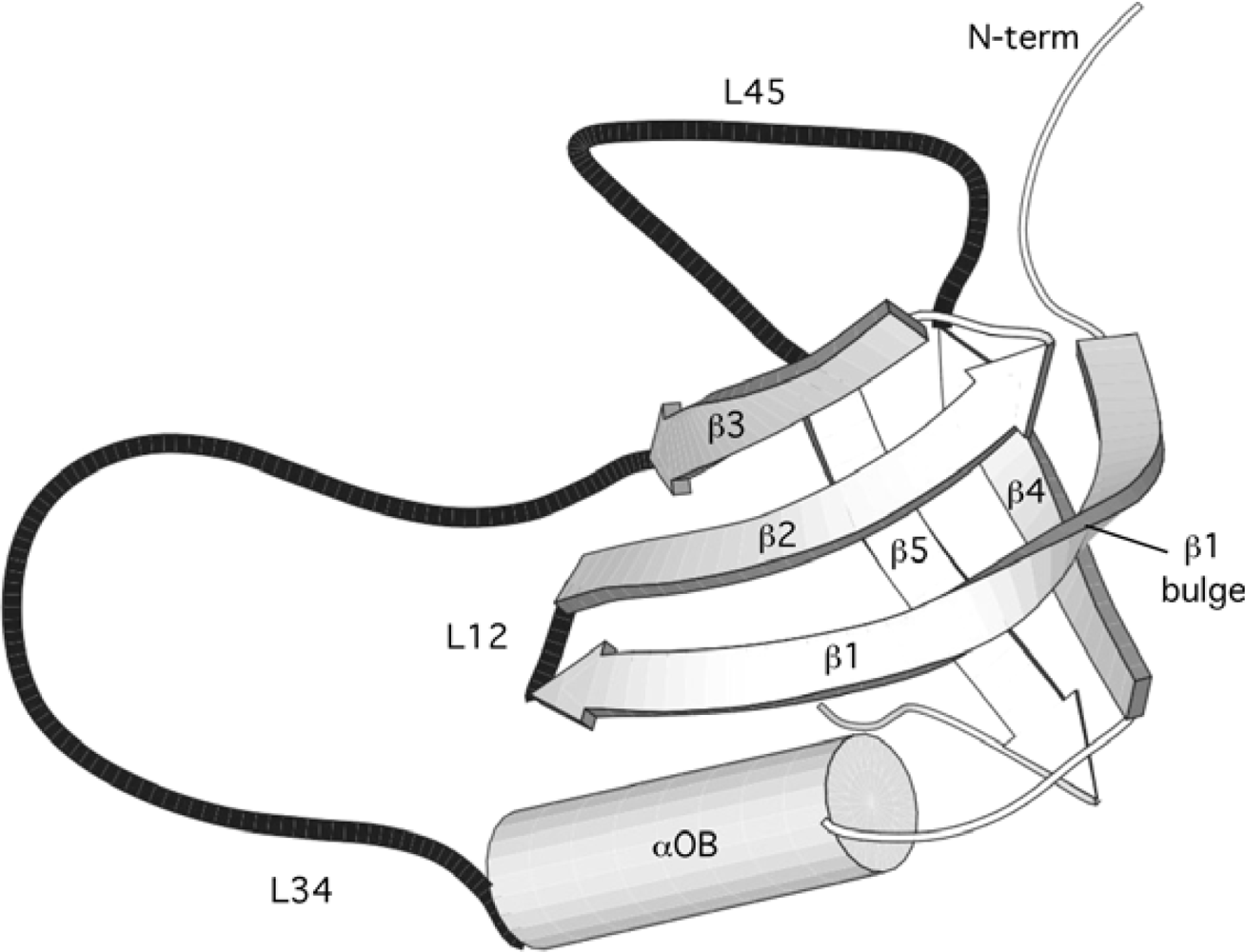
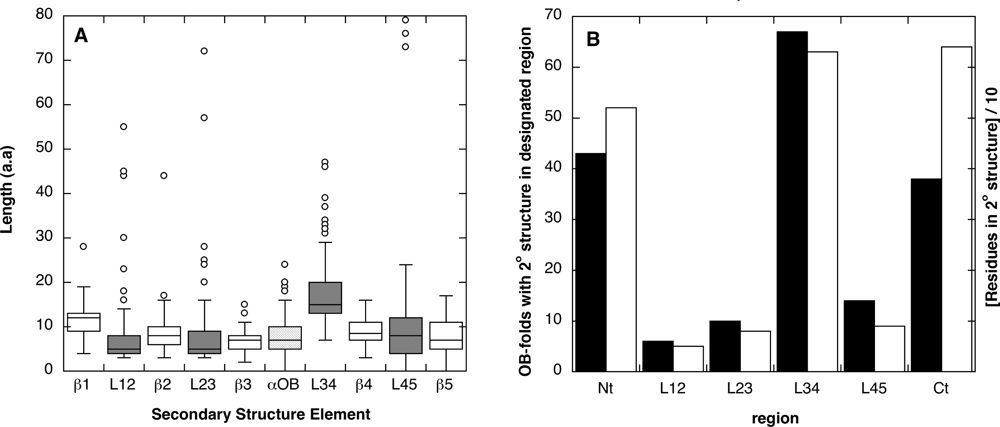
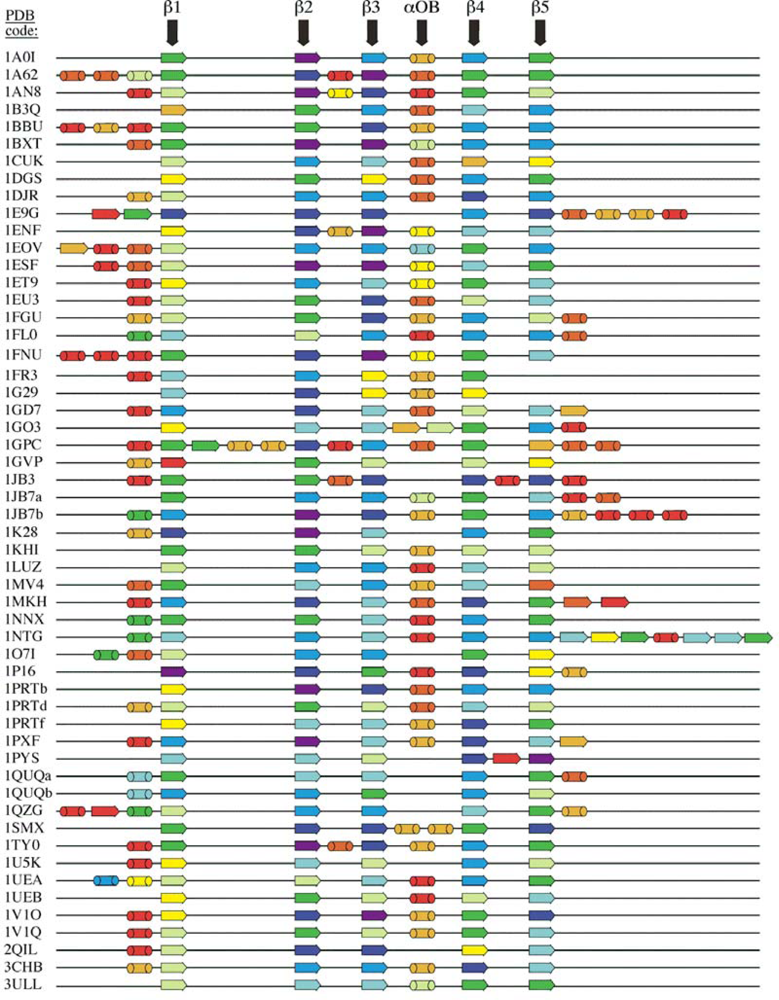
2.1. Length distribution of OB-fold secondary structures
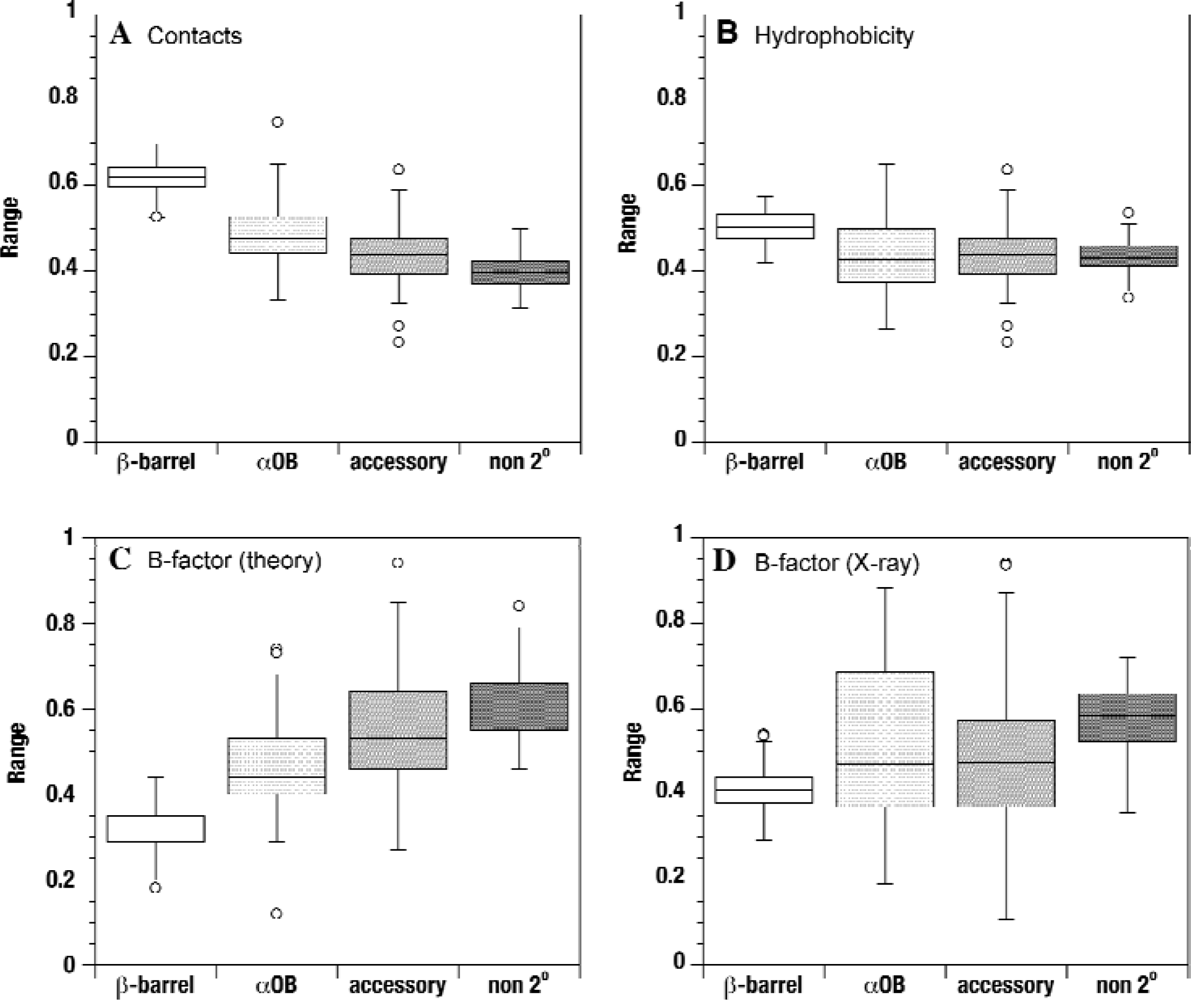
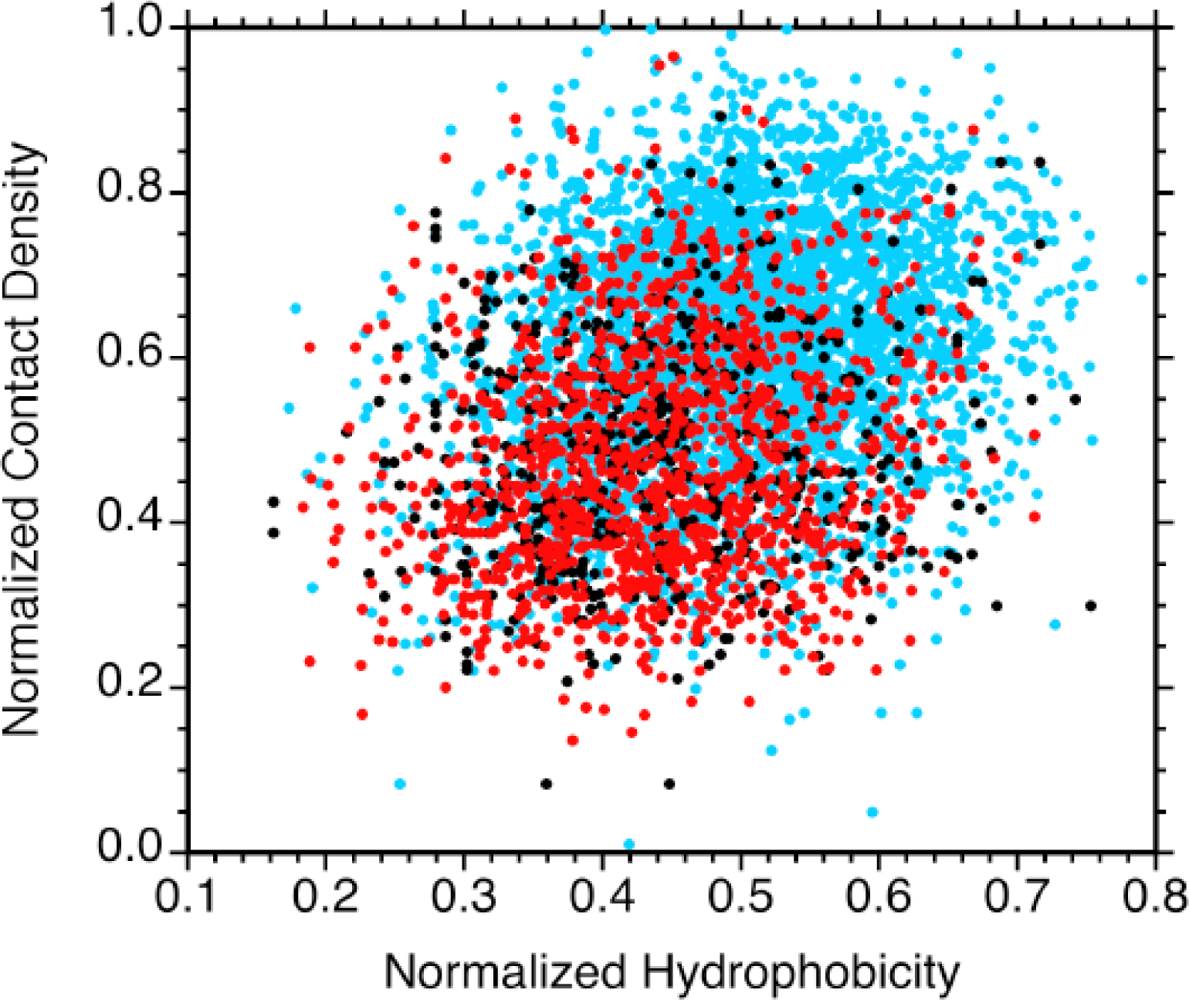
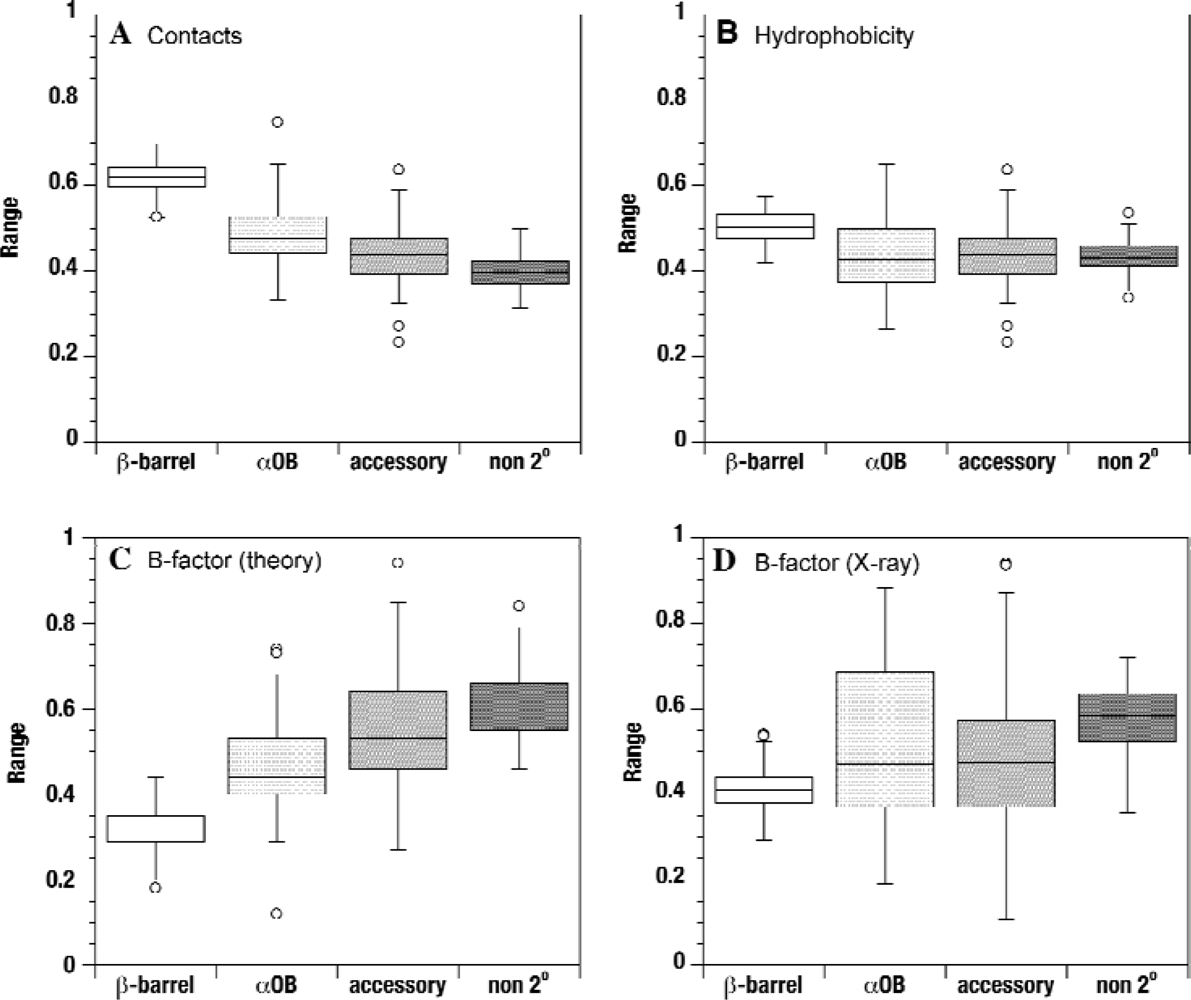
2.2. Contact densities distinguish conserved from non-conserved structures
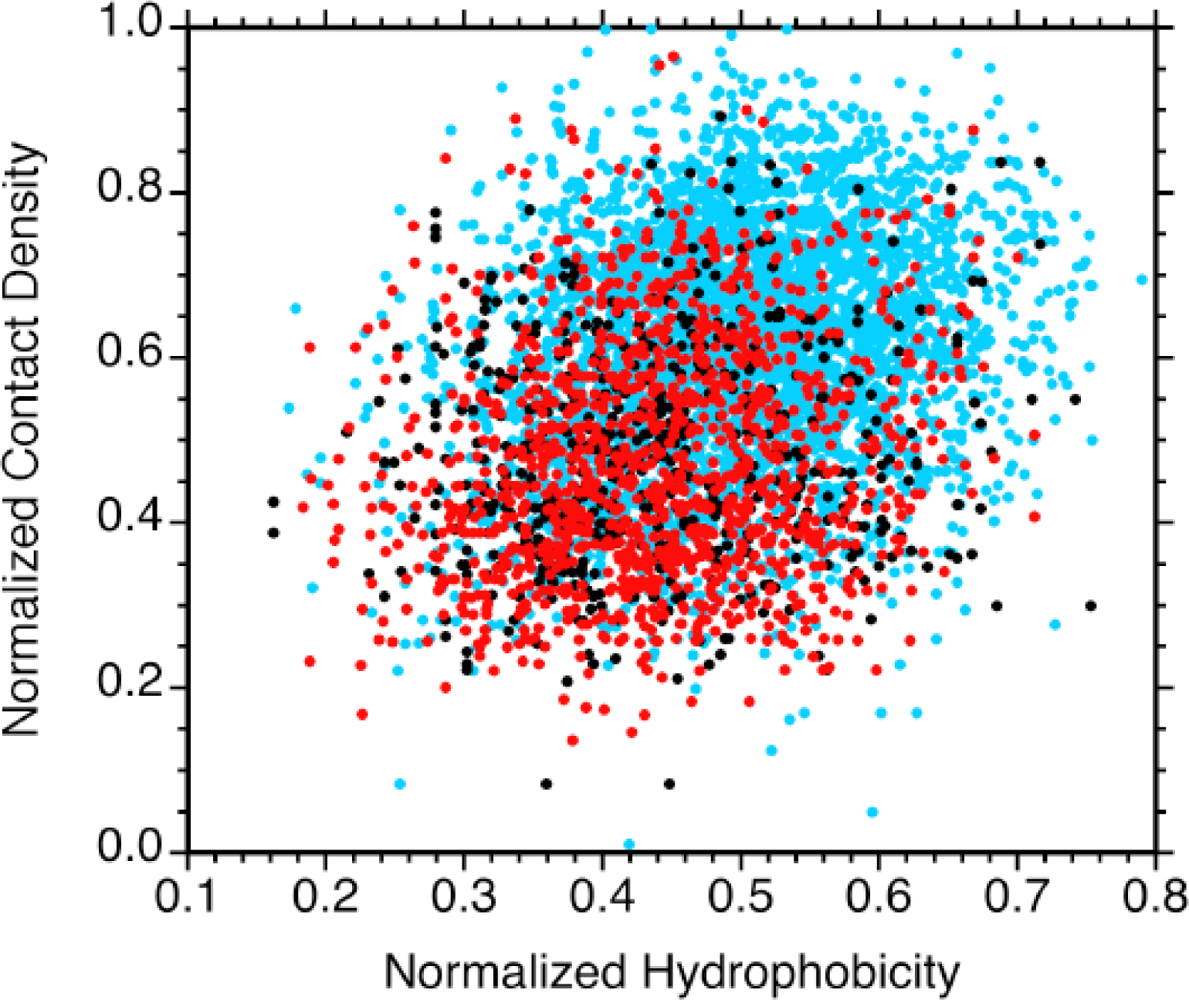
2.3. Sequence hydrophobicity
2.4. Theoretical B-factors
2.5. Experimental B-factors
2.6. Differences between conserved and non-conserved structure from NMR data
3. Discussion
4. Conclusions
5. Experimental Section
5.1. OB-fold database selection
5.2. Secondary structure assignment
5.3. Analysis of sequence and structure parameters
Acknowledgments
References and Notes
- Efimov, AV. Structural trees for protein superfamilies. Proteins 1997, 28, 241–260. [Google Scholar]
- Murzin, AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J 1993, 12, 861–867. [Google Scholar]
- Murzin, AG; Brenner, SE; Hubbard, T; Chothia, C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol 1995, 247, 536–540. [Google Scholar]
- Orengo, CA; Jones, DT; Thornton, JM. Protein superfamilies and domain superfolds. Nature 1994, 372, 631–634. [Google Scholar]
- Taylor, WR. A ‘periodic table’ for protein structures. Nature 2002, 416, 657–660. [Google Scholar]
- Alexandrescu, AT; Gittis, AG; Abeygunawardana, C; Shortle, D. NMR structure of a stable ‘OB-fold’ sub-domain isolated from staphylococcal nuclease. J. Mol. Biol 1995, 250, 134–143. [Google Scholar]
- Watson, E; Matousek, WM; Irimies, EL; Alexandrescu, AT. Partially folded states of staphylococcal nuclease highlight the conserved structural hierarchy of OB-fold proteins. Biochemistry 2007, 46, 9484–9494. [Google Scholar]
- Murzin, AG. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol 1998, 8, 380–387. [Google Scholar]
- Bai, Y; Sosnick, TR; Mayne, L; Englander, SW. Protein folding intermediates: native-state hydrogen exchange. Science 1995, 269, 192–197. [Google Scholar]
- Jeng, MF; Englander, SW. Stable submolecular folding units in a non-compact form of cytochrome c. J. Mol. Biol 1991, 221, 1045–1061. [Google Scholar]
- Lindorff-Larsen, K; Rogen, P; Paci, E; Vendruscolo, M; Dobson, CM. Protein folding and the organization of the protein topology universe. Trends Biochem. Sci 2005, 30, 13–19. [Google Scholar]
- Wang, Y; Alexandrescu, AT; Shortle, D. Initial studies of the equilibrium folding pathway of staphylococcal nuclease. Philos. Trans. R. Soc. Lond. B Biol. Sci 1995, 348, 27–34. [Google Scholar]
- Agrawal, V; Kishan, KV. OB-fold: growing bigger with functional consistency. Curr. Protein Pept. Sci 2003, 4, 195–206. [Google Scholar]
- Arcus, V. OB-fold domains: a snapshot of the evolution of sequence, structure and function. Curr. Opin. Struct. Biol 2002, 12, 794–801. [Google Scholar]
- Theobald, DL; Mitton-Fry, RM; Wuttke, DS. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct 2003, 32, 115–133. [Google Scholar]
- Alexandrescu, AT; Jaravine, VA; Dames, SA; Lamour, FP. NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J. Mol. Biol 1999, 289, 1041–1054. [Google Scholar]
- Jaravine, VA; Rathgeb-Szabo, K; Alexandrescu, AT. Microscopic stability of cold shock protein A examined by NMR native state hydrogen exchange as a function of urea and trimethylamine N-oxide. Protein Sci 2000, 9, 290–301. [Google Scholar]
- Bahar, I; Wallqvist, A; Covell, DG; Jernigan, RL. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry 1998, 37, 1067–1075. [Google Scholar]
- Shortle, D; Stites, WE; Meeker, AK. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry 1990, 29, 8033–8041. [Google Scholar]
- Tartaglia, GG; Cavalli, A; Vendruscolo, M. Prediction of local structural stabilities of proteins from their amino acid sequences. Structure 2007, 15, 139–143. [Google Scholar]
- Andreeva, A; Howorth, D; Chandonia, JM; Brenner, SE; Hubbard, TJ; Chothia, C; Murzin, AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 2008, 36, D419–D425. [Google Scholar]
- Yang, LW; Liu, X; Jursa, CJ; Holliman, M; Rader, AJ; Karimi, HA; Bahar, I. iGNM: a database of protein functional motions based on Gaussian Network Model. Bioinformatics 2005, 21, 2978–2987. [Google Scholar]
- Yang, LW; Rader, AJ; Liu, X; Jursa, CJ; Chen, SC; Karimi, HA; Bahar, I. oGNM: online computation of structural dynamics using the Gaussian Network Model. Nucleic Acids Res 2006, 34, W24–W31. [Google Scholar]
- Bahar, I; Atilgan, AR; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des 1997, 2, 173–181. [Google Scholar]
- Kabsch, W; Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Alexandrescu, AT; Rathgeb-Szabo, K. An NMR investigation of solution aggregation reactions preceding the misassembly of acid-denatured cold shock protein A into fibrils. J. Mol. Biol 1999, 291, 1191–1206. [Google Scholar]
- Max, KE; Zeeb, M; Bienert, R; Balbach, J; Heinemann, U. Common mode of DNA binding to cold shock domains. Crystal structure of hexathymidine bound to the domain-swapped form of a major cold shock protein from Bacillus caldolyticus. FEBS J 2007, 274, 1265–1279. [Google Scholar]
- Reichmann, L; Winter, G. Novel folded protein domains generated by combinatorial shuffling of polypeptide segments. Proc. Natl. Acad. Sci. USA 2000, 97, 10068–10073. [Google Scholar]
- Wang, Y; Shortle, D. A dynamic bundle of four adjacent hydrophobic segments in the denatured state of staphylococcal nuclease. Protein Sci 1996, 5, 1898–1906. [Google Scholar]
- Gu, Z; Zitzewitz, JA; Matthews, CR. Mapping the Structure of Folding Cores in TIM Barrel Proteins by Hydrogen Exchange Mass Spectrometry: The Roles of Motif and Sequence for the Indole-3-glycerol Phosphate Synthase from Sulfolobus solfataricus. J. Mol. Biol 2007, 368, 582–594. [Google Scholar]
- Yuan, Z; Bailey, TL; Teasdale, RD. Prediction of protein B-factor profiles. Proteins 2005, 58, 905–912. [Google Scholar]
- Yuan, Z; Zhao, J; Wang, ZX. Flexibility analysis of enzyme active sites by crystallographic temperature factors. Protein Eng 2003, 16, 109–114. [Google Scholar]
- Kyte, J; Doolittle, RF. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 1982, 157, 105–132. [Google Scholar]
- Finkelstein, AV; Ptitsyn, OB. Protein Physics: A Course of Lectures; Academic Press: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Berman, HM; Westbrook, J; Feng, Z; Gilliland, G; Bhat, TN; Weissig, H; Shindyalov, IN; Bourne, PE. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Kuriyan, J; Weis, WI. Rigid protein motion as a model for crystallographic temperature factors. Proc. Natl. Acad. Sci. USA 1991, 88, 2773–2777. [Google Scholar]
- Alexandrescu, AT; Jahnke, W; Wiltscheck, R; Blommers, MJ. Accretion of structure in staphylococcal nuclease: an 15N NMR relaxation study. J. Mol. Biol 1996, 260, 570–587. [Google Scholar]
- Feng, W; Tejero, R; Zimmerman, DE; Inouye, M; Montelione, GT. Solution NMR structure and backbone dynamics of the major cold-shock protein (CspA) from Escherichia coli: evidence for conformational dynamics in the single-stranded RNA-binding site. Biochemistry 1998, 37, 10881–10896. [Google Scholar]
- Zeeb, M; Max, KE; Weininger, U; Low, C; Sticht, H; Balbach, J. Recognition of T-rich single-stranded DNA by the cold shock protein Bs-CspB in solution. Nucleic Acids Res 2006, 34, 4561–4571. [Google Scholar]
- Aramini, JM; Huang, YJ; Cort, JR; Goldsmith-Fischman, S; Xiao, R; Shih, LY; Ho, CK; Liu, J; Rost, B; Honig, B; Kennedy, MA; Acton, TB; Montelione, GT. Solution NMR structure of the 30S ribosomal protein S28E from Pyrococcus horikoshii. Protein Sci 2003, 12, 2823–2830. [Google Scholar]
- Williamson, RA; Panagiotidou, P; Mott, JD; Howard, MJ. Dynamic characterisation of the netrin-like domain of human type 1 procollagen C-proteinase enhancer and comparison to the N-terminal domain of tissue inhibitor of metalloproteinases (TIMP). Mol. Biosyst 2008, 4, 417–425. [Google Scholar]
- Li, W; Hoffman, DW. Structure and dynamics of translation initiation factor aIF-1A from the archaeon Methanococcus jannaschii determined by NMR spectroscopy. Protein Sci 2001, 10, 2426–2438. [Google Scholar]
- Enggist, E; Thony-Meyer, L; Guntert, P; Pervushin, K. NMR structure of the heme chaperone CcmE reveals a novel functional motif. Structure 2002, 10, 1551–1557. [Google Scholar]
- Schubert, M; Edge, RE; Lario, P; Cook, MA; Strynadka, NC; Mackie, GA; McIntosh, LP. Structural characterization of the RNase E S1 domain and identification of its oligonucleotide-binding and dimerization interfaces. J. Mol. Biol 2004, 341, 37–54. [Google Scholar]
- Gao, G; Semenchenko, V; Arumugam, S; Van Doren, SR. Tissue inhibitor of metalloproteinases-1 undergoes microsecond to millisecond motions at sites of matrix metalloproteinase-induced fit. J. Mol. Biol 2000, 301, 537–552. [Google Scholar]
- Mitton-Fry, RM; Anderson, EM; Theobald, DL; Glustrom, LW; Wuttke, DS. Structural basis for telomeric single-stranded DNA recognition by yeast Cdc13. J. Mol. Biol 2004, 338, 241–255. [Google Scholar]
- Wu, B; Arumugam, S; Gao, G; Lee, GI; Semenchenko, V; Huang, W; Brew, K; Van Doren, SR. NMR structure of tissue inhibitor of metalloproteinases-1 implicates localized induced fit in recognition of matrix metalloproteinases. J. Mol. Biol 2000, 295, 257–268. [Google Scholar]
- Battiste, JL; Pestova, TV; Hellen, CU; Wagner, G. The eIF1A solution structure reveals a large RNA-binding surface important for scanning function. Mol. Cell 2000, 5, 109–119. [Google Scholar]
- Perl, D; Schmid, FX. Electrostatic stabilization of a thermophilic cold shock protein. J. Mol. Biol 2001, 313, 343–357. [Google Scholar]
- Swindells, MB; Orengo, CA; Jones, DT; Hutchinson, EG; Thornton, JM. Contemporary approaches to protein structure classification. Bioessays 1998, 20, 884–891. [Google Scholar]
- Laskowski, RA; Chistyakov, VV; Thornton, JM. PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res 2005, 33, D266–D268. [Google Scholar]
- DeLano, WL. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Hobohm, U; Sander, C. Enlarged representative set of protein structures. Protein Sci 1994, 3, 522–524. [Google Scholar]
- Gasteiger, E; Hoogland, C; Gattiker, A; Duvaud, S; Wilkins, MR; Appel, RD; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2° Structure | # Residues b | # Contacts (raw) | # Contacts (normalized) | Theory B-factor | X-ray B-factor | Hydrophobicity |
|---|---|---|---|---|---|---|
| β-barrel | 3,938 | 18.8 ± 4.6 | 0.62 ± 0.13 | 0.32 ± 0.16 | 0.41 ± 0.17 | 0.50 ± 0.09 |
| strand β1 | 1,085 | 17.8 ± 4.8 | 0.59 ± 0.15 | 0.34 ± 0.16 | 0.44 ± 0.18 | 0.49 ± 0.10 |
| strand β2 | 720 | 19.7 ± 4.6 | 0.64 ± 0.15 | 0.31 ± 0.17 | 0.41 ± 0.16 | 0.51 ± 0.09 |
| strand β3 | 614 | 19.7 ± 4.5 | 0.64 ± 0.14 | 0.30 ± 0.14 | 0.39 ± 0.15 | 0.50 ± 0.08 |
| strand β4 | 804 | 19.0 ± 4.2 | 0.62 ± 0.13 | 0.31 ± 0.15 | 0.40 ± 0.16 | 0.50 ± 0.09 |
| strand β5 | 715 | 18.7 ± 4.4 | 0.61 ± 0.15 | 0.32 ± 0.17 | 0.39 ± 0.17 | 0.49 ± 0.09 |
| βOB | 588 | 14.4 ± 3.9 | 0.48 ± 0.13 | 0.46 ± 0.16 | 0.52 ± 0.21 | 0.44 ± 0.10 |
| accessory 2° | 1,369 | 14.7 ± 5.0 | 0.46 ± 0.15 | 0.54 ± 0.26 | 0.49 ± 0.22 | 0.44 ± 0.10 |
| non-2° | 5,023 | 12.5 ± 5.0 | 0.42 ± 0.17 | 0.57 ± 0.27 | 0.56 ± 0.24 | 0.43 ± 0.10 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guardino, K.M.; Sheftic, S.R.; Slattery, R.E.; Alexandrescu, A.T. Relative Stabilities of Conserved and Non-Conserved Structures in the OB-Fold Superfamily. Int. J. Mol. Sci. 2009, 10, 2412-2430. https://doi.org/10.3390/ijms10052412
Guardino KM, Sheftic SR, Slattery RE, Alexandrescu AT. Relative Stabilities of Conserved and Non-Conserved Structures in the OB-Fold Superfamily. International Journal of Molecular Sciences. 2009; 10(5):2412-2430. https://doi.org/10.3390/ijms10052412
Chicago/Turabian StyleGuardino, Kaitlyn M., Sarah R. Sheftic, Robert E. Slattery, and Andrei T. Alexandrescu. 2009. "Relative Stabilities of Conserved and Non-Conserved Structures in the OB-Fold Superfamily" International Journal of Molecular Sciences 10, no. 5: 2412-2430. https://doi.org/10.3390/ijms10052412