Molecular Pathology of Neuro-AIDS (CNS-HIV)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
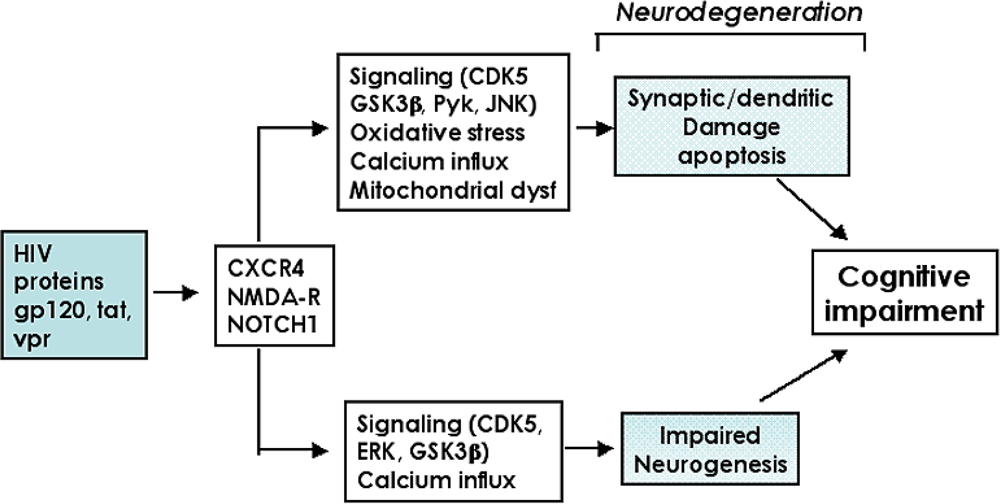
2. Molecular and cellular mechanisms of neurodegeneration in HIV Encephalitis
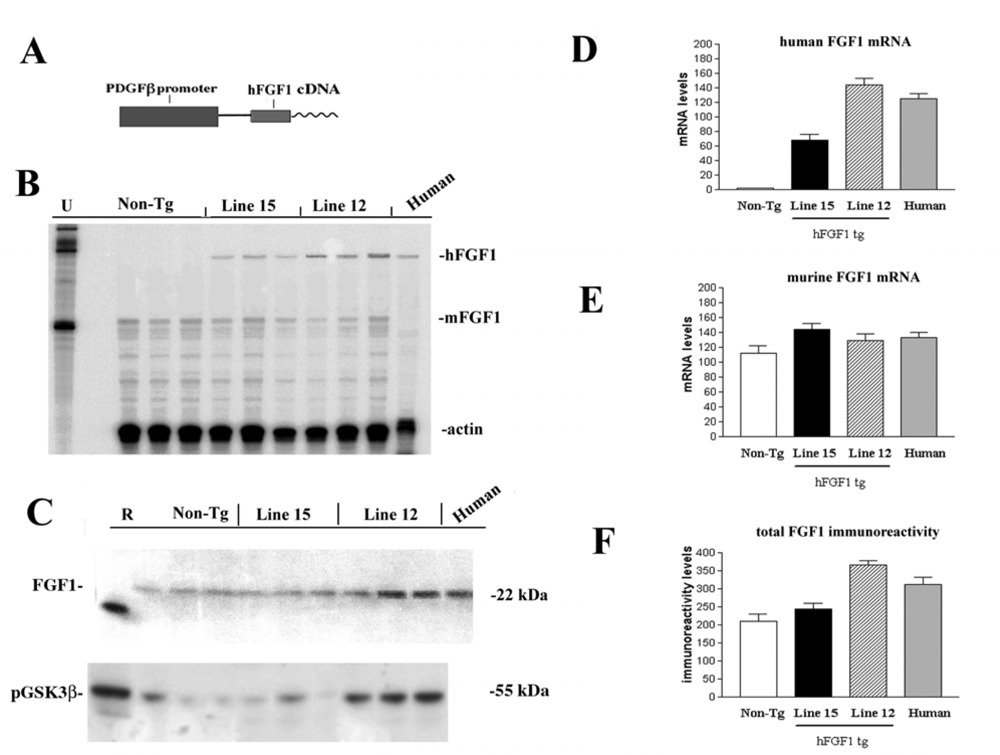
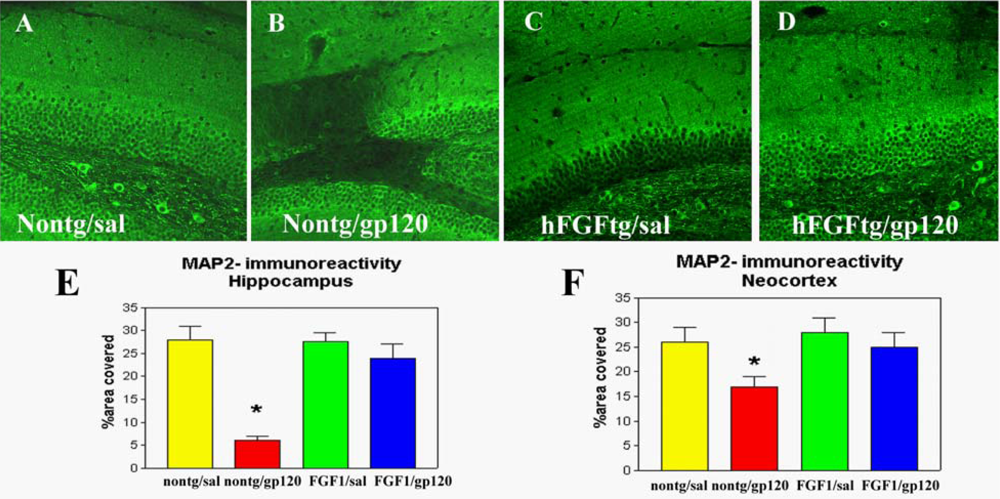
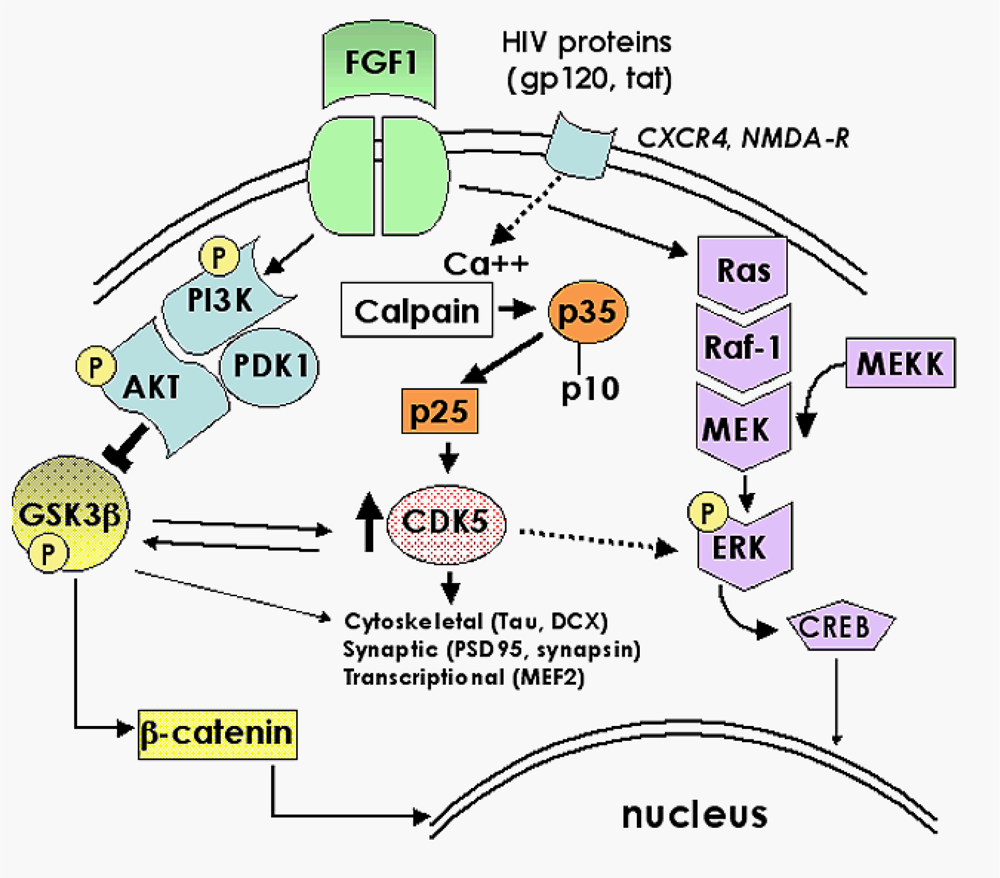
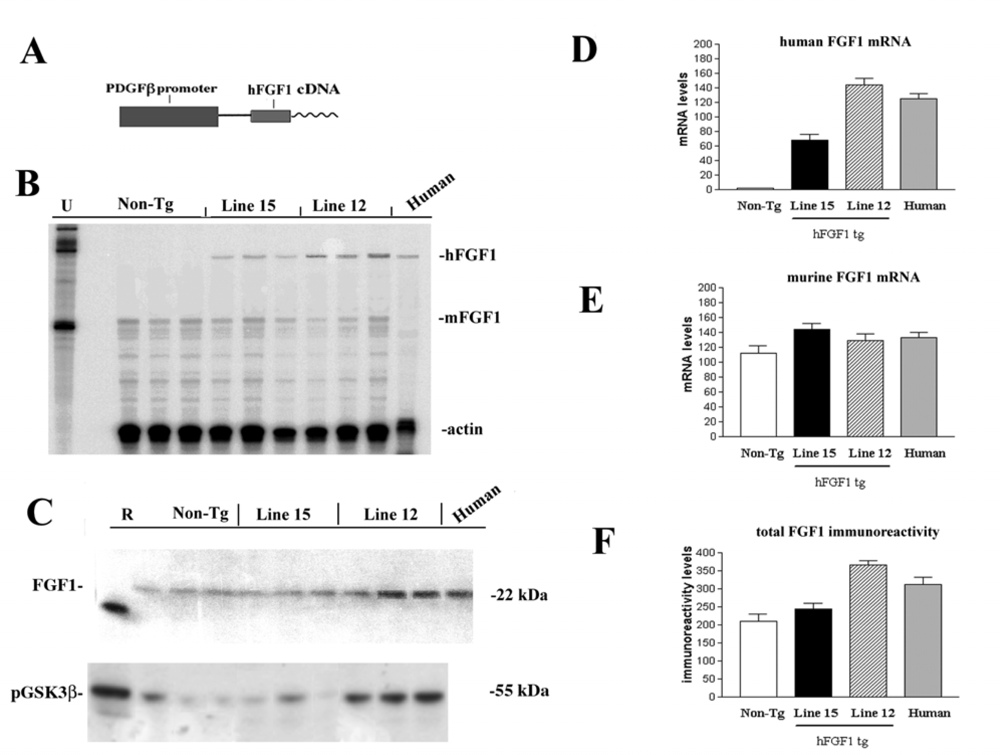
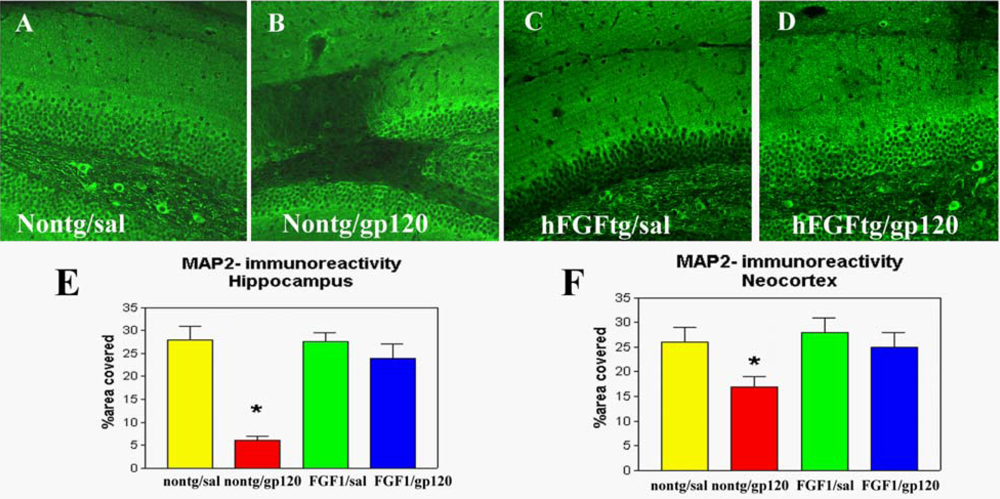
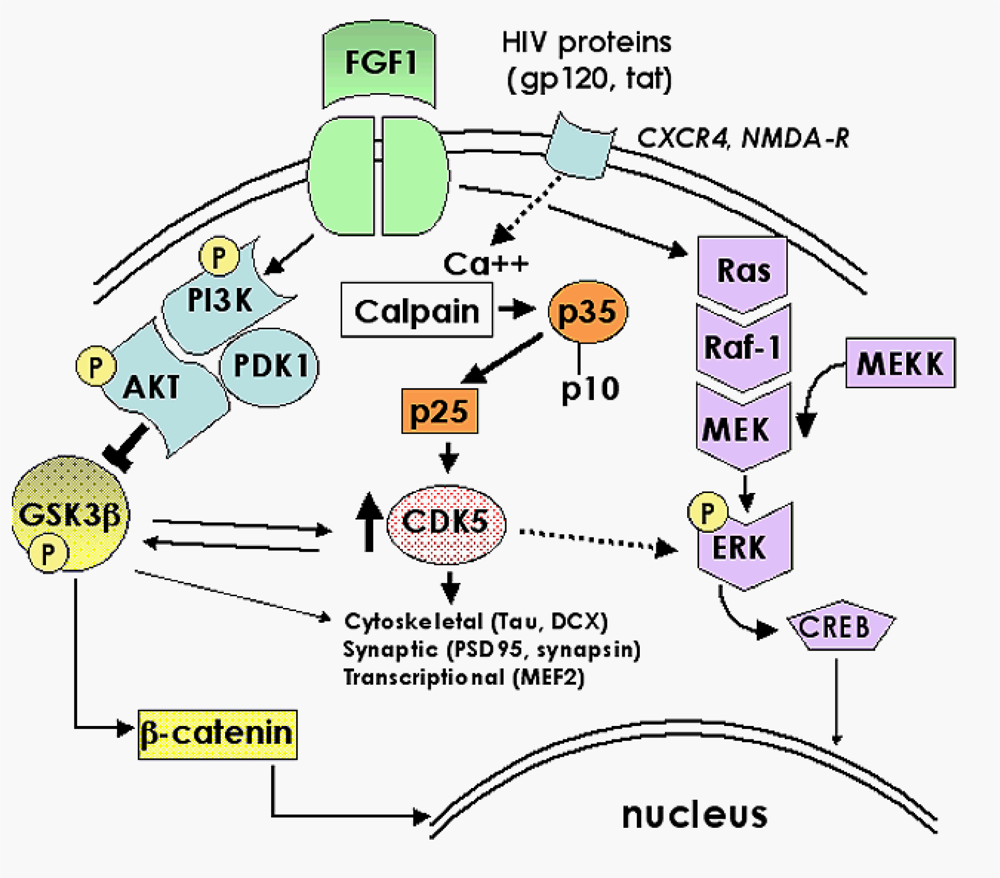
2.1. Role of FGF1-mediated regulation of the GSK3β signaling pathway in the mechanisms of neurodegeneration and neuroprotection in HIVE
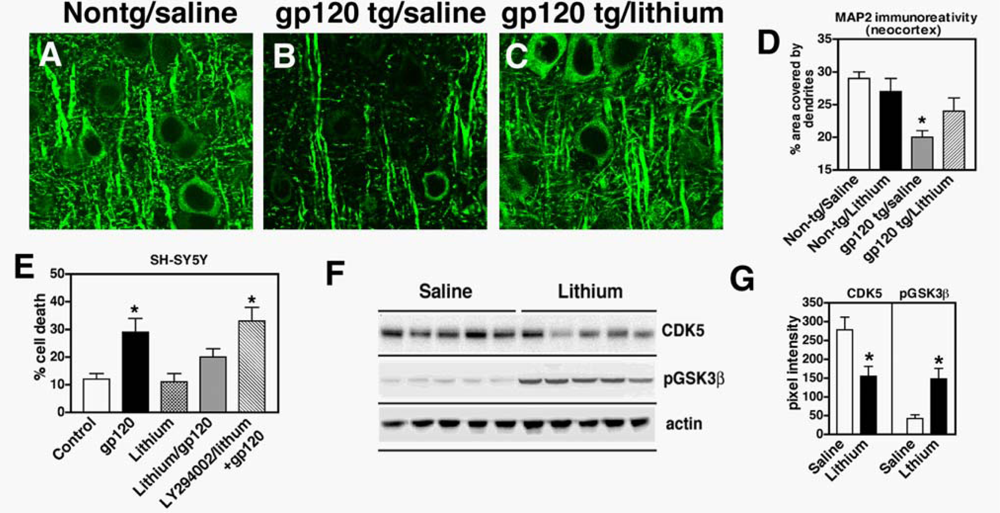
2.2. Neuroprotective effects of GSK3β inhibition in HIV neurotoxicity
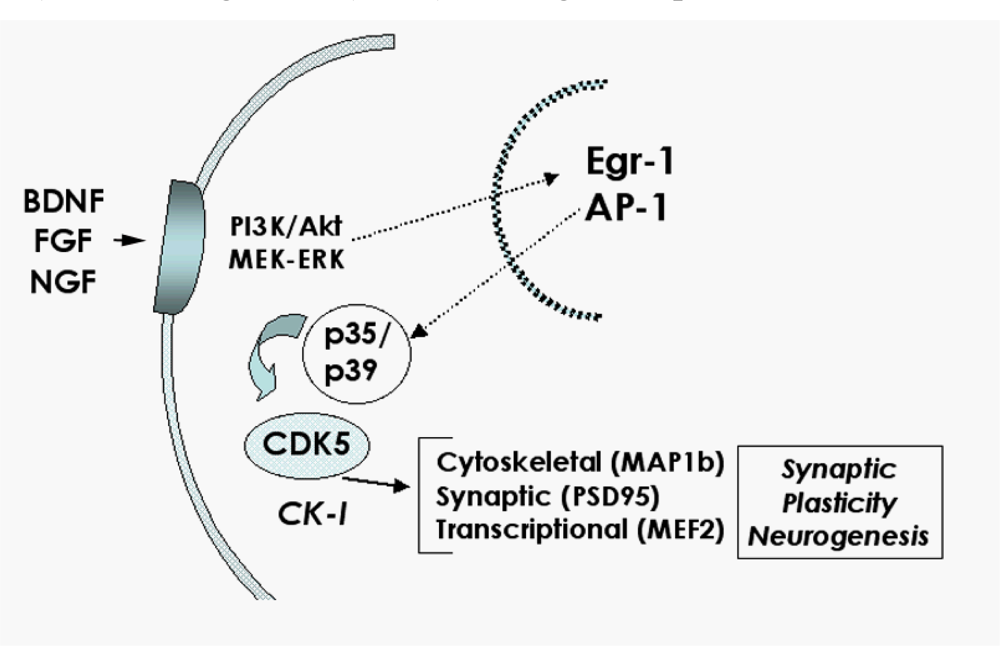
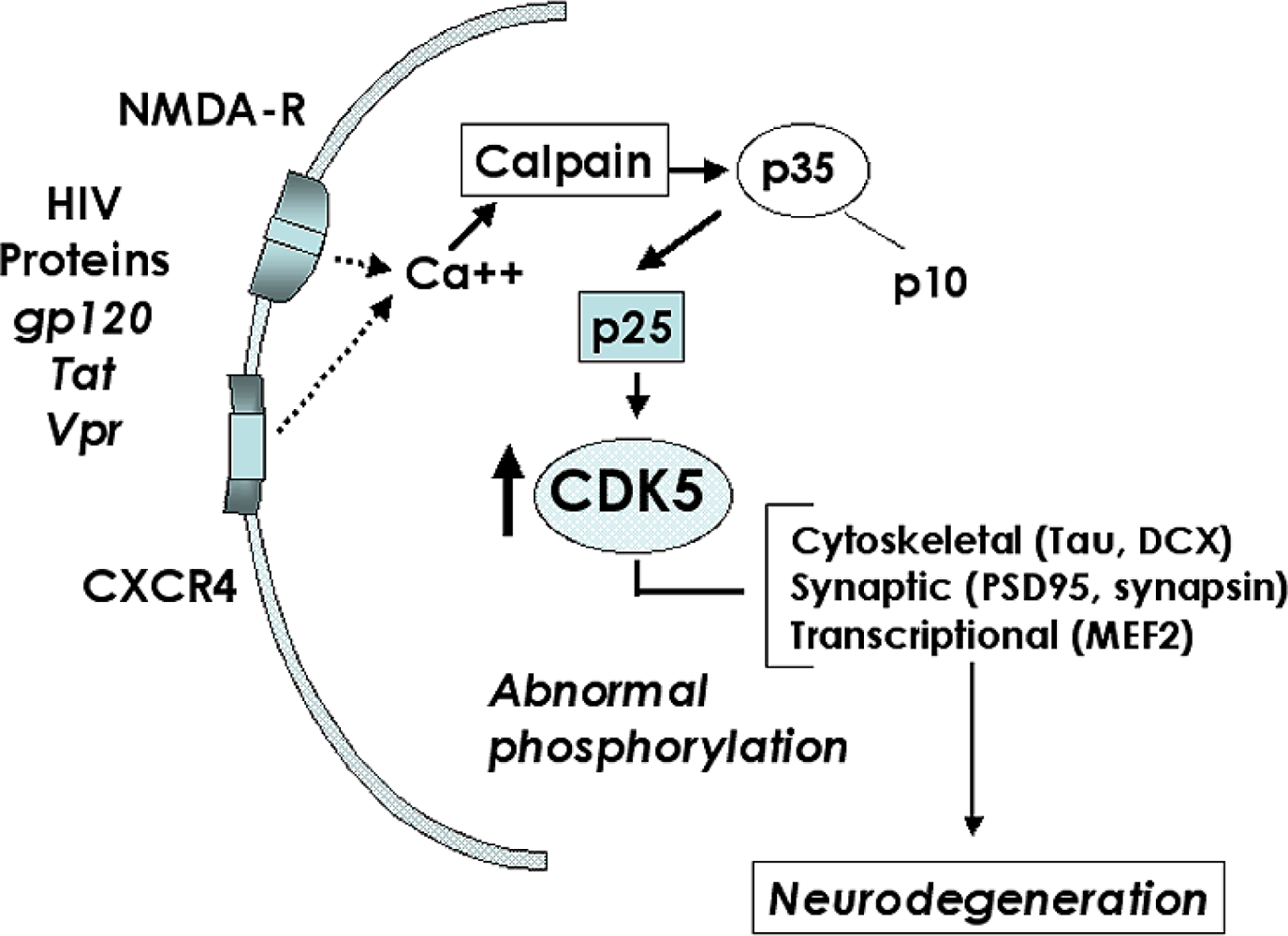
2.3. Role of the CDK5 pathway in the mechanisms of synaptic damage in HIVE
2.4. CDK inhibitors as potential neuroprotective therapies for HACI
3. Conclusions
Acknowledgments
References and Notes
- Gonzalez-Scarano, F; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol 2005, 5, 69–81. [Google Scholar]
- Gendelman, HE; Persidsky, Y; Ghorpade, A; Limoges, J; Stins, M; Fiala, M; Morrisett, R. The neuropathogenesis of the AIDS dementia complex. Aids 1997, 11(Suppl A), S35–S45. [Google Scholar]
- Heaton, R; Grant, I; Butters, N; White, D; Kirson, D; Atkinson, J; McCutchan, J; Taylor, M; Kelly, M; Ellis, R; Wolfson, T; Velin, R; Marcotte, T; Hesselink, J; Jernigan, T; Cahndler, J; Wallace, M; Abramson, I; Group, H. The HNRC 500 - neuropsychology of HIV infection at different disease stages. J. Int. Neuropsych. Soc 1995, 1, 231–251. [Google Scholar]
- Cherner, M; Masliah, E; Ellis, RJ; Marcotte, TD; Moore, DJ; Grant, I; Heaton, RK. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology 2002, 59, 1563–1567. [Google Scholar]
- Wiley, C; Achim, C. HIV encephalitis is the pathologic correlate of dementia in AIDS. Ann. Neurol 1994, 36, 673–676. [Google Scholar]
- Everall, IP; Hansen, LA; Masliah, E. The shifting patterns of HIV encephalitis neuropathology. Neurotox. Res 2005, 8, 51–61. [Google Scholar]
- Bell, JE. An update on the neuropathology of HIV in the HAART era. Histopathology 2004, 45, 549–559. [Google Scholar]
- Gray, F; Chretien, F; Vallat-Decouvelaere, AV; Scaravilli, F. The changing pattern of HIV neuropathology in the HAART era. J. Neuropathol. Exp. Neurol 2003, 62, 429–440. [Google Scholar]
- McArthur, JC; Haughey, N; Gartner, S; Conant, K; Pardo, C; Nath, A; Sacktor, N. Human immunodeficiency virus-associated dementia: an evolving disease. J. Neurovirol 2003, 9, 205–221. [Google Scholar]
- Sacktor, N; McDermott, MP; Marder, K; Schifitto, G; Selnes, OA; McArthur, JC; Stern, Y; Albert, S; Palumbo, D; Kieburtz, K; De Marcaida, JA; Cohen, B; Epstein, L. HIV-associated cognitive impairment before and after the advent of combination therapy. J. Neurovirol 2002, 8, 136–142. [Google Scholar]
- Maschke, M; Kastrup, O; Esser, S; Ross, B; Hengge, U; Hufnagel, A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J. Neurol. Neurosurg. Psychiatry 2000, 69, 376–380. [Google Scholar]
- Lawrence, DM; Major, EO. HIV-1 and the brain: connections between HIV-1-associated dementia, neuropathology and neuroimmunology. Microbes Infect 2002, 4, 301–308. [Google Scholar]
- Diesing, TS; Swindells, S; Gelbard, H; Gendelman, HE. HIV-1-associated dementia: a basic science and clinical perspective. AIDS Read 2002, 12, 358–368. [Google Scholar]
- Wiley, CA. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol 2003, 13. [Google Scholar]
- Mirra, S; del Rio, C. The fine structure of acquired immunodeficiency syndrome encephalopathy. Arch. Pathol. Lab. Med 1989, 113, 858–865. [Google Scholar]
- Pulliam, L; Herndier, B; Tang, N; McGrath, M. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J. Clin. Invest 1991, 87, 503–512. [Google Scholar]
- Gendelman, H; Lipton, S; Tardieu, M; Bukrinsky, M; Nottet, H. The neuropathogenesis of HIV-1 infection. J. Leukocyte Biol 1994, 56, 389–398. [Google Scholar]
- Langford, D; Masliah, E. Crosstalk between components of the blood brain barrier and cells of the CNS in microglial activation in AIDS. Brain Pathol 2001, 11, 306–312. [Google Scholar]
- Minagar, A; Shapshak, P; Fujimura, R; Ownby, R; Heyes, M; Eisdorfer, C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J. Neurol Sci 2002, 202, 13–23. [Google Scholar]
- Speth, C; Dierich, MP; Sopper, S. HIV-infection of the central nervous system: the tightrope walk of innate immunity. Mol. Immunol 2005, 42, 213–228. [Google Scholar]
- Nath, A. Pathobiology of human immunodeficiency virus dementia. Semin. Neurol 1999, 19, 113–127. [Google Scholar]
- Bellizzi, MJ; Lu, SM; Masliah, E; Gelbard, HA. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest 2005, 115, 3185–3192. [Google Scholar]
- Nath, A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis 2002, 186(Suppl 2), S193–S198. [Google Scholar]
- Kaul, M; Lipton, SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci USA 1999, 96, 8212–8216. [Google Scholar]
- Meucci, O; Fatatis, A; Simen, AA; Bushell, TJ; Gray, PW; Miller, RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14500–14505. [Google Scholar]
- Sanders, VJ; Pittman, CA; White, MG; Wang, G; Wiley, CA; Achim, CL. Chemokines and receptors in HIV encephalitis. Aids 1998, 12, 1021–1026. [Google Scholar]
- Giulian, D; Vaca, K; Noonan, C. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 1990, 250, 1593–1596. [Google Scholar]
- Pulliam, L; Zhou, M; Stubblebine, M; Bitler, CM. Differential modulation of cell death proteins in human brain cells by tumor necrosis factor α and platelet activating factor. J. Neurosci. Res 1998, 54, 530–538. [Google Scholar]
- Pulliam, L; Clarke, JA; McGuire, D; McGrath, MS. Investigation of HIV-infected macrophage neurotoxin production from patients with AIDS dementia. Adv. Neuroimmunol 1994, 4, 195–198. [Google Scholar]
- Wang, Z; Trillo-Pazos, G; Kim, SY; Canki, M; Morgello, S; Sharer, LR; Gelbard, HA; Su, ZZ; Kang, DC; Brooks, AI; Fisher, PB; Volsky, DJ. Effects of human immunodeficiency virus type 1 on astrocyte gene expression and function: potential role in neuropathogenesis. J Neurovirol 2004, 10(Suppl 1), 25–32. [Google Scholar]
- Brandimarti, R; Khan, MZ; Fatatis, A; Meucci, O. Regulation of cell cycle proteins by chemokine receptors: A novel pathway in human immunodeficiency virus neuropathogenesis? J Neurovirol 2004, 10(Suppl 1), 108–112. [Google Scholar]
- Martin-Garcia, J; Kolson, DL; Gonzalez-Scarano, F. Chemokine receptors in the brain: their role in HIV infection and pathogenesis. Aids 2002, 16, 1709–1730. [Google Scholar]
- Brew, B; Rosenblum, M; Cronin, K; Price, R. AIDS dementia comples and HIV-1 brain infection: clinical-virological correlations. Ann. Neurol 1995, 38, 563–570. [Google Scholar]
- Glass, J; Fedor, H; Wesselingh, S; McArthur, J. Immunocytochemical quantification of human immunodeficiency virus in the brain: correlations with dementia. Ann. Neurol 1995, 38, 755–762. [Google Scholar]
- McArthur, JC; McClernon, DR; Cronin, MF; Nance-Sproson, TE; Saah, AJ; St Clair, M; Lanier, ER. Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Ann. Neurol 1997, 42, 689–698. [Google Scholar]
- Masliah, E; Heaton, RK; Marcotte, TD; Ellis, RJ; Wiley, CA; Mallory, M; Achim, CL; McCutchan, JA; Nelson, JA; Atkinson, JH; Grant, I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann. Neurol 1997, 42, 963–972. [Google Scholar]
- Masliah, E; Ge, N; Achim, C; Wiley, C. Differential vulnerability of calbindin-immunoreactive neurons in HIV encephalitis. J. Neuropathol. Exp. Neurol 1995, 54, 350–357. [Google Scholar]
- Langford, TD; Letendre, SL; Marcotte, TD; Ellis, RJ; McCutchan, JA; Grant, I; Mallory, ME; Hansen, LA; Archibald, S; Jernigan, T; Masliah, E. Severe, demyelinating leukoencephalopathy in AIDS patients on antiretroviral therapy. AIDS 2002, 16, 1019–1029. [Google Scholar]
- Moore, DJ; Masliah, E; Rippeth, JD; Gonzalez, R; Carey, CL; Cherner, M; Ellis, RJ; Achim, CL; Marcotte, TD; Heaton, RK; Grant, I. Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. Aids 2006, 20, 879–887. [Google Scholar]
- Tran, PB; Miller, RJ. HIV-1, chemokines and neurogenesis. Neurotox. Res 2005, 8, 149–158. [Google Scholar]
- Krathwohl, MD; Kaiser, JL. HIV-1 promotes quiescence in human neural progenitor cells. J. Infect. Dis 2004, 190, 216–226. [Google Scholar]
- Lawrence, DM; Durham, LC; Schwartz, L; Seth, P; Marie, D; Major, EO. Human immunodeficiency virus type 1 infection of human brain-derived progenitor cells. J. Virol 2004, 78, 7319–7328. [Google Scholar]
- van Marle, G; Antony, JM; Silva, C; Sullivan, A; Power, C. Aberrant cortical neurogenesis in a pediatric neuroAIDS model: neurotrophic effects of growth hormone. Aids 2005, 19, 1781–1791. [Google Scholar]
- Gage, FH; Kempermann, G; Palmer, TD; Peterson, DA; Ray, J. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol 1998, 36, 249–266. [Google Scholar]
- Aimone, JB; Wiles, J; Gage, FH. Potential role for adult neurogenesis in the encoding of time in new memories. Nat. Neurosci 2006, 9, 723–727. [Google Scholar]
- Olson, AK; Eadie, BD; Ernst, C; Christie, BR. Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus 2006, 16, 250–260. [Google Scholar]
- Bruel-Jungerman, E; Laroche, S; Rampon, C. New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur. J. Neurosci 2005, 21, 513–521. [Google Scholar]
- Brown, J; Cooper-Kuhn, CM; Kempermann, G; Van Praag, H; Winkler, J; Gage, FH; Kuhn, HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur. J. Neurosci 2003, 17, 2042–2046. [Google Scholar]
- Lie, DC; Colamarino, SA; Song, HJ; Desire, L; Mira, H; Consiglio, A; Lein, ES; Jessberger, S; Lansford, H; Dearie, AR; Gage, FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar]
- Jessberger, S; Aigner, S; Clemenson, GD, Jr; Toni, N; Lie, DC; Karalay, O; Overall, R; Kempermann, G; Gage, FH. Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol 2008, 6, e272. [Google Scholar]
- Kaul, M; Garden, GA; Lipton, SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001, 410, 988–994. [Google Scholar]
- Haughey, NJ; Nath, A; Mattson, MP; Slevin, JT; Geiger, JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem 2001, 78, 457–467. [Google Scholar]
- Maragos, WF; Young, KL; Turchan, JT; Guseva, M; Pauly, JR; Nath, A; Cass, WA. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J. Neurochem 2002, 83, 955–963. [Google Scholar]
- Turchan, J; Pocernich, CB; Gairola, C; Chauhan, A; Schifitto, G; Butterfield, DA; Buch, S; Narayan, O; Sinai, A; Geiger, J; Berger, JR; Elford, H; Nath, A. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 2003, 60, 307–314. [Google Scholar]
- Haughey, NJ; Mattson, MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr 2002, 31(Suppl 2), S55–61. [Google Scholar]
- Mattson, MP. Oxidative stress, perturbed calcium homeostasis, and immune dysfunction in Alzheimer’s disease. J. Neurovirol 2002, 8, 539–550. [Google Scholar]
- Darbinian, N; Darbinyan, A; Czernik, M; Peruzzi, F; Khalili, K; Reiss, K; Gordon, J; Amini, S. HIV-1 Tat inhibits NGF-induced Egr-1 transcriptional activity and consequent p35 expression in neural cells. J. Cell. Physiol 2008, 216, 128–134. [Google Scholar]
- Wang, Y; White, MG; Akay, C; Chodroff, RA; Robinson, J; Lindl, KA; Dichter, MA; Qian, Y; Mao, Z; Kolson, DL; Jordan-Sciutto, KL. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J. Neurochem 2007, 103, 439–455. [Google Scholar]
- Sui, Z; Sniderhan, LF; Fan, S; Kazmierczak, K; Reisinger, E; Kovacs, AD; Potash, MJ; Dewhurst, S; Gelbard, HA; Maggirwar, SB. Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3β to antagonize nuclear factor-kappaB survival pathway in neurons. Eur. J. Neurosci 2006, 23, 2623–2634. [Google Scholar]
- Maggirwar, SB; Tong, N; Ramirez, S; Gelbard, HA; Dewhurst, S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3β contributes to Tat-mediated neurotoxicity. J. Neurochem 1999, 73, 578–586. [Google Scholar]
- Lannuzel, A; Barnier, JV; Hery, C; Huynh, VT; Guibert, B; Gray, F; Vincent, JD; Tardieu, M. Human immunodeficiency virus type 1 and its coat protein gp120 induce apoptosis and activate JNK and ERK mitogen-activated protein kinases in human neurons. Ann. Neurol 1997, 42, 847–856. [Google Scholar]
- Rusnati, M; Urbinati, C; Musulin, B; Ribatti, D; Albini, A; Noonan, D; Marchisone, C; Waltenberger, J; Presta, M. Activation of endothelial cell mitogen activated protein kinase ERK(l/2) by extracellular HIV-1 Tat protein. Endothelium 2001, 8, 65–74. [Google Scholar]
- Hashimoto, M; Sagara, Y; Everall, IP; Mallory, M; Everson, A; Langford, D; Masliah, E. Fibroblast growth factor 1 regulates signaling via the GSK3β pathway: implications for neuroprotection. J. Biol. Chem 2002, 277, 32985–32991. [Google Scholar]
- Langford, D; Hurford, R; Hashimoto, M; Digicaylioglu, M; Masliah, E. Signalling crosstalk in FGF2-mediated protection of endothelial cells from HIV-gp120. BMC Neurosci 2005, 6(1), 8. [Google Scholar]
- Tong, N; Sanchez, JF; Maggirwar, SB; Ramirez, SH; Guo, H; Dewhurst, S; Gelbard, HA. Activation of glycogen synthase kinase 3 β (GSK-3β) by platelet activating factor mediates migration and cell death in cerebellar granule neurons. Eur. J. Neurosci 2001, 13, 1913–1922. [Google Scholar]
- Alirezaei, M; Watry, DD; Flynn, CF; Kiosses, WB; Masliah, E; Williams, BR; Kaul, M; Lipton, SA; Fox, HS. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J. Neurosci 2007, 27, 11047–11055. [Google Scholar]
- Del Corno, M; Liu, QH; Schols, D; de Clercq, E; Gessani, S; Freedman, BD; Collman, RG. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood 2001, 98, 2909–2916. [Google Scholar]
- Yi, Y; Lee, C; Liu, QH; Freedman, BD; Collman, RG. Chemokine receptor utilization and macrophage signaling by human immunodeficiency virus type 1 gp120: Implications for neuropathogenesis. J. Neurovirol. 2004, 10(Suppl 1), 91–96. [Google Scholar]
- Everall, IP; Trillo-Pazos, G; Bell, C; Mallory, M; Sanders, V; Masliah, E. Amelioration of neurotoxic effects of HIV envelope protein gp120 by fibroblast growth factor: a strategy for neuroprotection. J. Neuropathol. Exp. Neurol 2001, 60, 293–301. [Google Scholar]
- Sanders, V; Everall, IP; Johnson, RW; Masliah, E; Group, t. H. Fibroblast growth factor modulates HIV co-receptor expression by neural cells. J. Neurosci. Res 2000, 59, 671–679. [Google Scholar]
- Reuss, B; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell. Tissue Res 2003, 313, 139–157. [Google Scholar]
- Presta, M; Dell’Era, P; Mitola, S; Moroni, E; Ronca, R; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005, 16, 159–178. [Google Scholar] [Green Version]
- Langford, D; Masliah, E. Role of trophic factors on neuroimmunity in neurodegenerative infectious diseases. J. Neurovirol 2002, 8, 625–638. [Google Scholar]
- Brady, MJ; Bourbonais, FJ; Saltiel, AR. The activation of glycogen synthase by insulin switches from kinase inhibition to phosphatase activation during adipogenesis in 3T3-L1 cells. J. Biol. Chem 1998, 273, 14063–14066. [Google Scholar]
- Saito, Y; Vandenheede, JR; Cohen, P. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochem J 1994, 303, 27–31. [Google Scholar]
- Everall, IP; Bell, C; Mallory, M; Langford, D; Adame, A; Rockestein, E; Masliah, E. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol. Cell. Neurosci 2002, 21, 493–501. [Google Scholar]
- Rockenstein, E; Torrance, M; Adame, A; Mante, M; Bar-on, P; Rose, JB; Crews, L; Masliah, E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci 2007, 27, 1981–1991. [Google Scholar]
- Dewhurst, S; Maggirwar, SB; Schifitto, G; Gendelman, HE; Gelbard, HA. Glycogen synthase kinase 3 β (GSK-3β) as a therapeutic target in neuroAIDS. J. Neuroimmune Pharmacol 2007, 2, 93–96. [Google Scholar]
- Ances, BM; Letendre, SL; Alexander, T; Ellis, RJ. Role of psychiatric medications as adjunct therapy in the treatment of HIV associated neurocognitive disorders. Int. Rev. Psychiatry 2008, 20, 89–93. [Google Scholar]
- Schifitto, G; Peterson, DR; Zhong, J; Ni, H; Cruttenden, K; Gaugh, M; Gendelman, HE; Boska, M; Gelbard, H. Valproic acid adjunctive therapy for HIV-associated cognitive impairment: a first report. Neurology 2006, 66, 919–921. [Google Scholar]
- Dou, H; Ellison, B; Bradley, J; Kasiyanov, A; Poluektova, LY; Xiong, H; Maggirwar, S; Dewhurst, S; Gelbard, HA; Gendelman, HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J. Neurosci 2005, 25, 8375–8385. [Google Scholar]
- Dou, H; Birusingh, K; Faraci, J; Gorantla, S; Poluektova, LY; Maggirwar, SB; Dewhurst, S; Gelbard, HA; Gendelman, HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J. Neurosci 2003, 23, 9162–9170. [Google Scholar]
- Letendre, SL; Woods, SP; Ellis, RJ; Atkinson, JH; Masliah, E; van den Brande, G; Durelle, J; Grant, I; Everall, I. Lithium improves HIV-associated neurocognitive impairment. Aids 2006, 20, 1885–1888. [Google Scholar]
- Jorda, EG; Verdaguer, E; Canudas, AM; Jimenez, A; Garcia de Arriba, S; Allgaier, C; Pallas, M; Camins, A. Implication of cyclin-dependent kinase 5 in the neuroprotective properties of lithium. Neuroscience 2005, 134, 1001–1011. [Google Scholar]
- Masliah, E; Roberts, ES; Langford, D; Everall, I; Crews, L; Adame, A; Rockenstein, E; Fox, HS. Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J. Neuroimmunol 2004, 157, 163–175. [Google Scholar]
- Fischer, A; Sananbenesi, F; Spiess, J; Radulovic, J. Cdk5 in the adult non-demented brain. Curr. Drug Targets CNS Neurol. Disord 2003, 2, 375–381. [Google Scholar]
- Johansson, JU; Lilja, L; Chen, XL; Higashida, H; Meister, B; Noda, M; Zhong, ZG; Yokoyama, S; Berggren, PO; Bark, C. Cyclin-dependent kinase 5 activators p35 and p39 facilitate formation of functional synapses. Brain Res. Mol. Brain Res 2005, 138, 215–227. [Google Scholar]
- Lagace, DC; Benavides, DR; Kansy, JW; Mapelli, M; Greengard, P; Bibb, JA; Eisch, AJ. Cdk5 is essential for adult hippocampal neurogenesis. Proc. Natl. Acad. Sci USA 2008, 105, 18567–18571. [Google Scholar]
- Xie, Z; Sanada, K; Samuels, BA; Shih, H; Tsai, LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 2003, 114, 469–482. [Google Scholar]
- Samuels, BA; Hsueh, YP; Shu, T; Liang, H; Tseng, HC; Hong, CJ; Su, SC; Volker, J; Neve, RL; Yue, DT; Tsai, LH. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron 2007, 56, 823–837. [Google Scholar]
- Fischer, A; Sananbenesi, F; Pang, PT; Lu, B; Tsai, LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 2005, 48, 825–838. [Google Scholar]
- Tang, X; Wang, X; Gong, X; Tong, M; Park, D; Xia, Z; Mao, Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J. Neurosci 2005, 25, 4823–4834. [Google Scholar]
- Dhavan, R; Tsai, LH. A decade of CDK5. Nat. Rev. Mol. Cell Biol 2001, 2, 749–759. [Google Scholar]
- Shelton, SB; Johnson, GV. Cyclin-dependent kinase-5 in neurodegeneration. J. Neurochem 2004, 88, 1313–1326. [Google Scholar]
- Smith, DS; Greer, PL; Tsai, LH. Cdk5 on the brain. Cell Growth Differ 2001, 12, 277–283. [Google Scholar]
- Sherr, CJ; Roberts, JM. CDK inhibitors: positive and negative regulators of Gl-phase progression. Genes Dev 1999, 13, 1501–12. [Google Scholar]
- Hisanaga, S; Saito, T. The regulation of cyclin-dependent kinase 5 activity through the metabolism of p35 or p39 Cdk5 activator. Neurosignals 2003, 12, 221–229. [Google Scholar]
- Ko, J; Humbert, S; Bronson, RT; Takahashi, S; Kulkarni, AB; Li, E; Tsai, LH. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci 2001, 21, 6758–6771. [Google Scholar]
- Cicero, S; Herrup, K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J. Neurosci 2005, 25, 9658–9668. [Google Scholar]
- Zhu, YS; Saito, T; Asada, A; Maekawa, S; Hisanaga, S. Activation of latent cyclin-dependent kinase 5 (Cdk5)-p35 complexes by membrane dissociation. J. Neurochem 2005, 94, 1535–1545. [Google Scholar]
- Sharma, P; Sharma, M; Amin, ND; Albers, RW; Pant, HC. Regulation of cyclin-dependent kinase 5 catalytic activity by phosphorylation. Proc. Natl. Acad. Sci USA 1999, 96, 11156–11160. [Google Scholar]
- Rosales, J; Han, B; Lee, KY. Cdk7 functions as a cdk5 activating kinase in brain. Cell Physiol Biochem 2003, 13, 285–296. [Google Scholar]
- Tokuoka, H; Saito, T; Yorifuji, H; Wei, F; Kishimoto, T; Hisanaga, S. Brain-derived neurotrophic factor-induced phosphorylation of neurofilament-H subunit in primary cultures of embryo rat cortical neurons. J Cell Sci 2000, 113, 1059–1068. [Google Scholar]
- Paglini, G; Pigino, G; Kunda, P; Morfini, G; Maccioni, R; Quiroga, S; Ferreira, A; Caceres, A. Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J. Neurosci 1998, 18, 9858–9869. [Google Scholar]
- Copani, A; Uberti, D; Sortino, MA; Bruno, V; Nicoletti, F; Memo, M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 2001, 24, 25–31. [Google Scholar]
- Patrick, GN; Zukerberg, L; Nikolic, M; de la Monte, S; Dikkes, P; Tsai, LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar]
- Neve, RL; McPhie, DL. The cell cycle as a therapeutic target for Alzheimer’s disease. Pharmacol. Ther 2006, 111, 99–113. [Google Scholar]
- Lee, MS; Kwon, YT; Li, M; Peng, J; Friedlander, RM; Tsai, LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar]
- Nath, A; Haughey, NJ; Jones, M; Anderson, C; Bell, JE; Geiger, JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann. Neurol 2000, 47, 186–194. [Google Scholar]
- Alisky, JM. Could cholinesterase inhibitors and memantine alleviate HIV dementia? J. Acquir. Immune Defic. Syndr 2005, 38, 113–114. [Google Scholar]
- Manji, H; Miller, R. The neurology of HIV infection. J Neurol Neurosurg Psychiatry 2004, 75(Suppl 1), i29–35. [Google Scholar]
- Schang, LM. Cyclin-dependent kinases as cellular targets for antiviral drugs. J. Antimicrob. Chemother 2002, 50, 779–792. [Google Scholar]
- Schwartz, GK; Shah, MA. Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol 2005, 23, 9408–9421. [Google Scholar]
- Vita, M; Abdel-Rehim, M; Olofsson, S; Hassan, Z; Meurling, L; Siden, A; Siden, M; Pettersson, T; Hassan, M. Tissue distribution, pharmacokinetics and identification of roscovitine metabolites in rat. Eur. J. Pharm. Sci 2005, 25, 91–103. [Google Scholar]
- Di Giovanni, S; Movsesyan, V; Ahmed, F; Cernak, I; Schinelli, S; Stoica, B; Faden, AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8338. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Crews, L.; Patrick, C.; Achim, C.L.; Everall, I.P.; Masliah, E. Molecular Pathology of Neuro-AIDS (CNS-HIV). Int. J. Mol. Sci. 2009, 10, 1045-1063. https://doi.org/10.3390/ijms10031045
Crews L, Patrick C, Achim CL, Everall IP, Masliah E. Molecular Pathology of Neuro-AIDS (CNS-HIV). International Journal of Molecular Sciences. 2009; 10(3):1045-1063. https://doi.org/10.3390/ijms10031045
Chicago/Turabian StyleCrews, Leslie, Christina Patrick, Cristian L. Achim, Ian P. Everall, and Eliezer Masliah. 2009. "Molecular Pathology of Neuro-AIDS (CNS-HIV)" International Journal of Molecular Sciences 10, no. 3: 1045-1063. https://doi.org/10.3390/ijms10031045