Molecular Neuropathology of Gliomas

Abstract
:1. Introduction
2. Cytogenetic and molecular changes within individual glioma types
2.1. Diffusely infiltrating astrocytic gliomas
2.1.1. Diffuse astrocytoma (WHO grade II)
2.1.2. Anaplastic astrocytoma (WHO grade III)
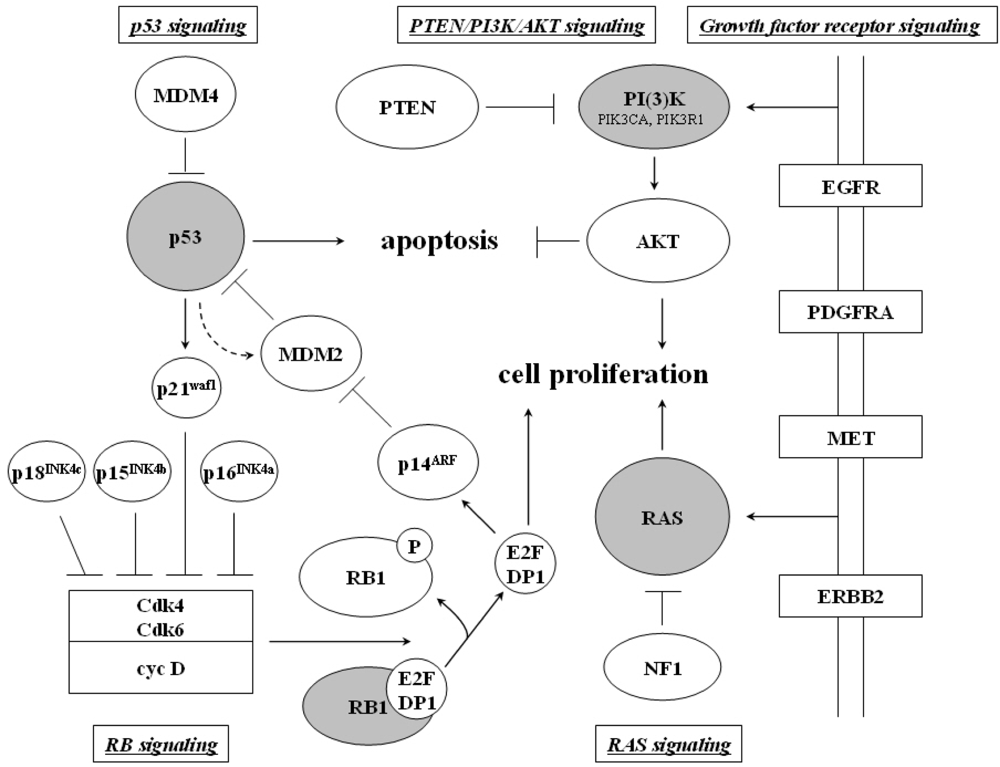
2.1.3. Glioblastoma (WHO grade IV)
2.2. Astrocytic gliomas with more circumscribed growth
2.2.1. Pilocytic astrocytoma (WHO grade I)
2.2.2. Pleomorphic xanthoastrocytoma (WHO grade II)
2.2.3. Subependymal giant cell astrocytoma (WHO grade I)
2.3. Oligodendrogliomas and mixed gliomas
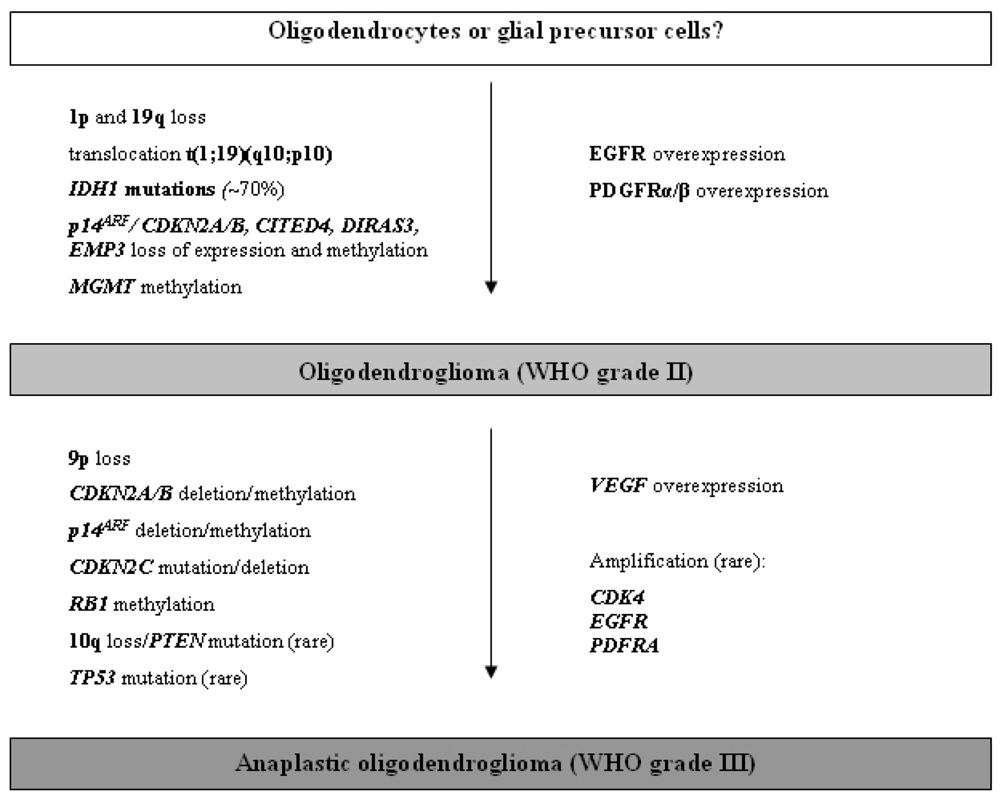
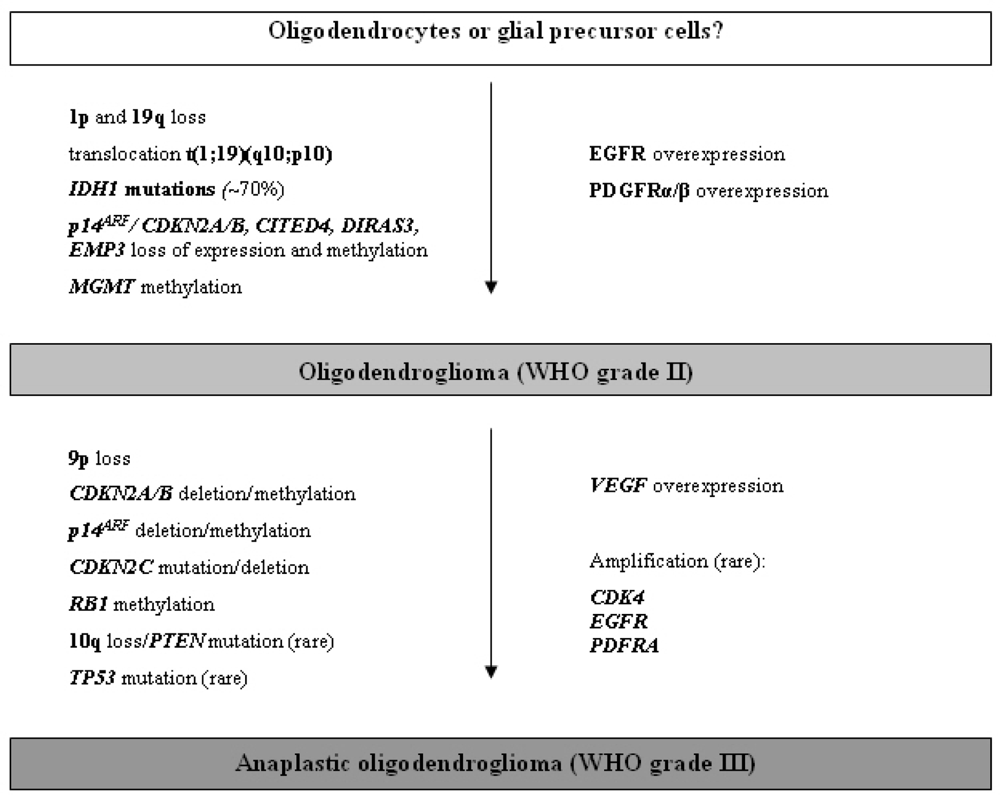
2.3.1. Oligodendroglioma (WHO grade II)
2.3.2. Anaplastic oligodendroglioma (WHO grade III)
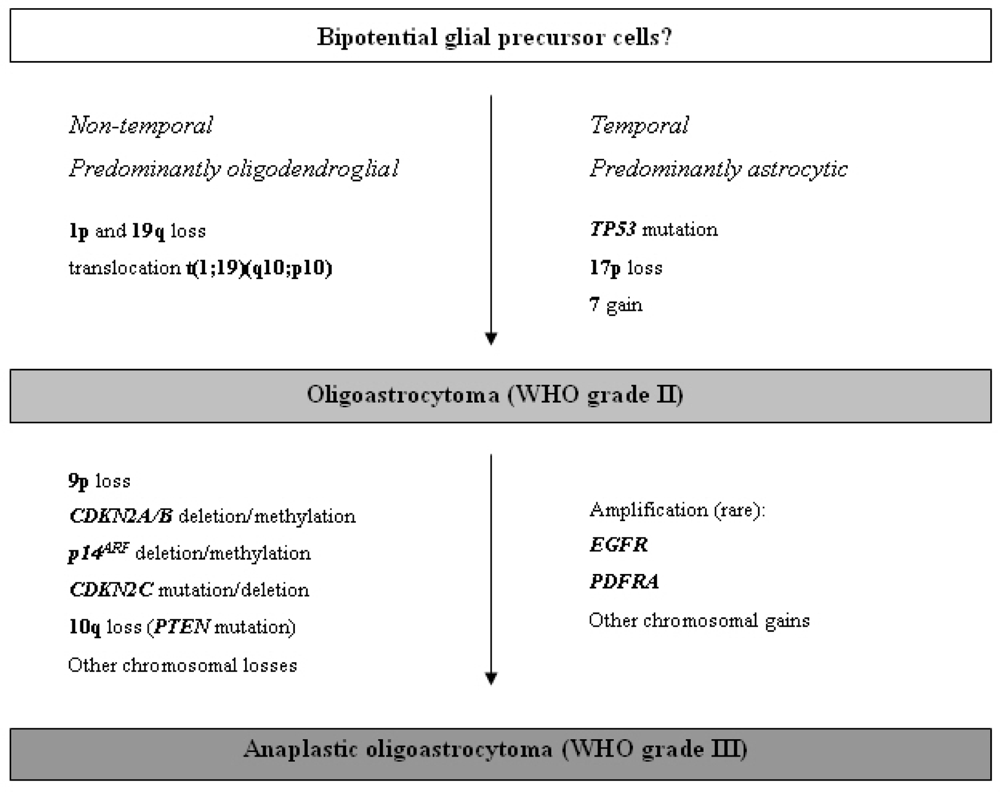
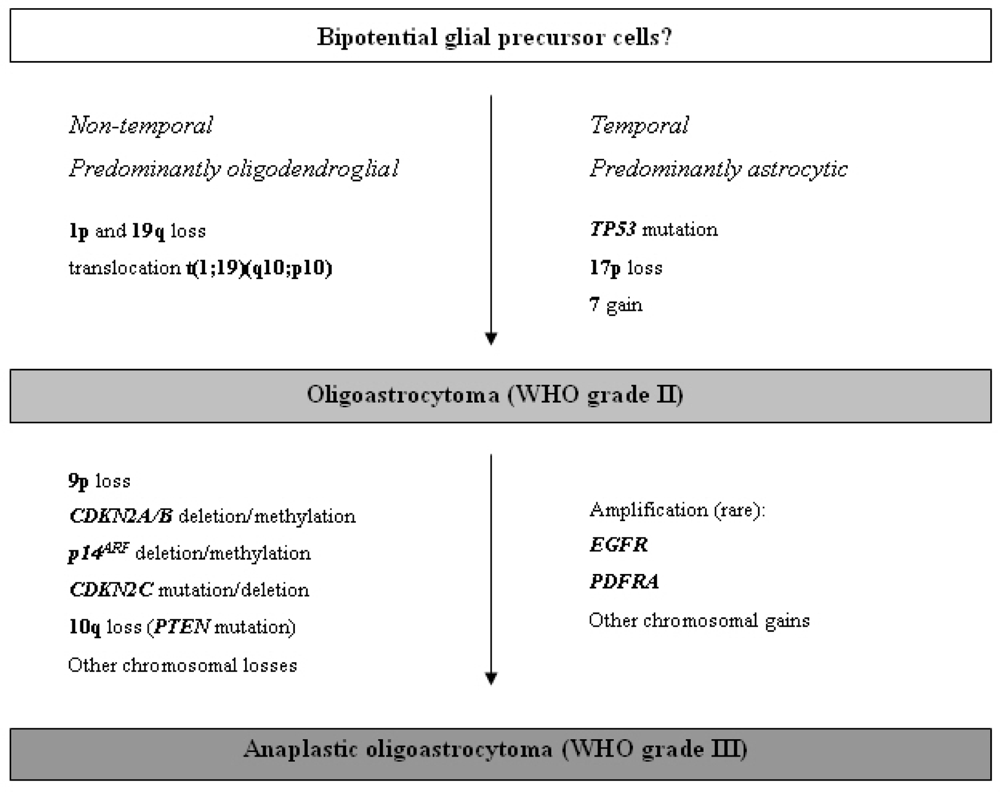
2.3.3. Oligoastrocytomas (WHO grade II or III)
2.4. Gliomas with ependymal differentiation
2.4.1. Ependymoma (WHO grade II)
2.4.2. Anaplastic ependymoma (WHO grade III)
2.4.3. Myxopapillary ependymoma (WHO grade I)
2.4.4. Subependymoma (WHO grade I)
3. Prognostic and predictive relevance of molecular changes
4. Molecular genetics and glioma classification
5. Comprehensive molecular analysis of gliomas by means of high-throughput profiling techniques
6. Conclusions
References
- Ohgaki, H; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. (Berl.) 2005, 109, 93–108. [Google Scholar]
- WHO Classification of Tumours of the Central Nervous System, 3rd Edition; Louis, DN; Ohgaki, H; Wiestler, OD; Cavenee, WK (Eds.) IARC Press: Lyon, France, 2007.
- Cairncross, G; Berkey, B; Shaw, E; Jenkins, R; Scheithauer, B; Brachman, D; Buckner, J; Fink, K; Souhami, L; Laperierre, N; Mehta, M; Curran, W. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J. Clin. Oncol 2006, 24, 2707–2714. [Google Scholar]
- van den Bent, MJ; Carpentier, AF; Brandes, AA; Sanson, M; Taphoorn, MJ; Bernsen, HJ; Frenay, M; Tijssen, CC; Grisold, W; Sipos, L; Haaxma-Reiche, H; Kros, JM; van Kouwenhoven, MC; Vecht, CJ; Allgeier, A; Lacombe, D; Gorlia, T. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: A randomized European Organisation for Research and Treatment of Cancer phase III trial. J. Clin. Oncol 2006, 24, 2715–2722. [Google Scholar]
- Nishizaki, T; Ozaki, S; Harada, K; Ito, H; Arai, H; Beppu, T; Sasaki, K. Investigation of genetic alterations associated with the grade of astrocytic tumor by comparative genomic hybridization. Genes Chromosom. Cancer 1998, 21, 340–346. [Google Scholar]
- Schrock, E; Blume, C; Meffert, MC; du Manoir, S; Bersch, W; Kiessling, M; Lozanowa, T; Thiel, G; Witkowski, R; Ried, T; Cremer, T. Recurrent gain of chromosome arm 7q in low-grade astrocytic tumors studied by comparative genomic hybridization. Genes Chromosom. Cancer 1996, 15, 199–205. [Google Scholar]
- Reifenberger, G; Collins, VP. Pathology and molecular genetics of astrocytic gliomas. J. Mol. Med 2004, 82, 656–670. [Google Scholar]
- Ichimura, K; Bolin, MB; Goike, HM; Schmidt, EE; Moshref, A; Collins, VP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res 2000, 60, 417–424. [Google Scholar]
- Balss, J; Meyer, J; Mueller, W; Korshunov, A; Hartmann, C; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008, 116, 597–602. [Google Scholar]
- Parsons, DW; Jones, S; Zhang, X; Lin, JC; Leary, RJ; Angenendt, P; Mankoo, P; Carter, H; Siu, IM; Gallia, GL; Olivi, A; McLendon, R; Rasheed, BA; Keir, S; Nikolskaya, T; Nikolsky, Y; Busam, DA; Tekleab, H; Diaz, LA, Jr; Hartigan, J; Smith, DR; Strausberg, RL; Marie, SK; Shinjo, SM; Yan, H; Riggins, GJ; Bigner, DD; Karchin, R; Papadopoulos, N; Parmigiani, G; Vogelstein, B; Velculescu, VE; Kinzler, KW. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar]
- Watanabe, K; Peraud, A; Gratas, C; Wakai, S; Kleihues, P; Ohgaki, H. p53 and PTEN gene mutations in gemistocytic astrocytomas. Acta Neuropathol. (Berl.) 1998, 95, 559–564. [Google Scholar]
- Watanabe, T; Katayama, Y; Yoshino, A; Yachi, K; Ohta, T; Ogino, A; Komine, C; Fukushima, T. Aberrant hypermethylation of p14ARF and O6-methylguanine-DNA methyltransferase genes in astrocytoma progression. Brain Pathol 2007, 17, 5–10. [Google Scholar]
- Waha, A; Guntner, S; Huang, TH; Yan, PS; Arslan, B; Pietsch, T; Wiestler, OD. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia 2005, 7, 193–199. [Google Scholar]
- Kunitz, A; Wolter, M; van den Boom, J; Felsberg, J; Tews, B; Hahn, M; Benner, A; Sabel, M; Lichter, P; Reifenberger, G; von Deimling, A; Hartmann, C. DNA hypermethylation and Aberrant Expression of the EMP3 Gene at 19q13.3 in Human Gliomas. Brain Pathol 2007, 17, 363–370. [Google Scholar]
- Hermanson, M; Funa, K; Hartman, M; Claesson-Welsh, L; Heldin, CH; Westermark, B; Nister, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 1992, 52, 3213–3219. [Google Scholar]
- Fleming, TP; Saxena, A; Clark, WC; Robertson, JT; Oldfield, EH; Aaronson, SA; Ali, IU. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 1992, 52, 4550–4553. [Google Scholar]
- Reifenberger, G; Reifenberger, J; Ichimura, K; Meltzer, PS; Collins, VP. Amplification of multiple genes from chromosomal region 12q13–14 in human malignant gliomas: preliminary mapping of the amplicons shows preferential involvement of CDK4, SAS, and MDM2. Cancer Res 1994, 54, 4299–4303. [Google Scholar]
- Ichimura, K; Schmidt, EE; Goike, HM; Collins, VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996, 13, 1065–1072. [Google Scholar]
- Schmidt, EE; Ichimura, K; Reifenberger, G; Collins, VP. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res 1994, 54, 6321–6324. [Google Scholar]
- Smith, JS; Tachibana, I; Passe, SM; Huntley, BK; Borell, TJ; Iturria, N; O’Fallon, JR; Schaefer, PL; Scheithauer, BW; James, CD; Buckner, JC; Jenkins, RB. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J. Natl. Cancer Inst 2001, 93, 1246–1256. [Google Scholar]
- Ohgaki, H; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol 2007, 170, 1445–1453. [Google Scholar]
- Nakamura, M; Watanabe, T; Klangby, U; Asker, C; Wiman, K; Yonekawa, Y; Kleihues, P; Ohgaki, H. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol 2001, 11, 159–168. [Google Scholar]
- Reifenberger, G; Ichimura, K; Reifenberger, J; Elkahloun, AG; Meltzer, PS; Collins, VP. Refined mapping of 12q13–q15 amplicons in human malignant gliomas suggests CDK4/SAS and MDM2 as independent amplification targets. Cancer Res 1996, 56, 5141–5145. [Google Scholar]
- Riemenschneider, MJ; Buschges, R; Wolter, M; Reifenberger, J; Bostrom, J; Kraus, JA; Schlegel, U; Reifenberger, G. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res 1999, 59, 6091–6096. [Google Scholar]
- Riemenschneider, MJ; Knobbe, CB; Reifenberger, G. Refined mapping of 1q32 amplicons in malignant gliomas confirms MDM4 as the main amplification target. Int. J. Cancer 2003, 104, 752–757. [Google Scholar]
- Burger, PC; Pearl, DK; Aldape, K; Yates, AJ; Scheithauer, BW; Passe, SM; Jenkins, RB; James, CD. Small cell architecture--a histological equivalent of EGFR amplification in glioblastoma multiforme? J. Neuropathol. Exp. Neurol 2001, 60, 1099–1104. [Google Scholar]
- Nakamura, M; Yang, F; Fujisawa, H; Yonekawa, Y; Kleihues, P; Ohgaki, H. Loss of heterozygosity on chromosome 19 in secondary glioblastomas. J. Neuropathol. Exp. Neurol 2000, 59, 539–543. [Google Scholar]
- Tepel, M; Roerig, P; Wolter, M; Gutmann, DH; Perry, A; Reifenberger, G; Riemenschneider, MJ. Frequent promoter hypermethylation and transcriptional downregulation of the NDRG2 gene at 14q11.2 in primary glioblastoma. Int. J. Cancer 2008, 123, 2080–2086. [Google Scholar]
- Mueller, W; Nutt, CL; Ehrich, M; Riemenschneider, MJ; von Deimling, A; van den Boom, D; Louis, DN. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene 2007, 26, 583–593. [Google Scholar]
- Riemenschneider, MJ; Betensky, RA; Pasedag, SM; Louis, DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res 2006, 66, 5618–5623. [Google Scholar]
- Riemenschneider, MJ; Mueller, W; Betensky, RA; Mohapatra, G; Louis, DN. In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am. J. Pathol 2005, 167, 1379–1387. [Google Scholar]
- Gerson, SL. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar]
- Esteller, M; Garcia-Foncillas, J; Andion, E; Goodman, SN; Hidalgo, OF; Vanaclocha, V; Baylin, SB; Herman, JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med 2000, 343, 1350–1354. [Google Scholar]
- Hegi, ME; Diserens, AC; Gorlia, T; Hamou, MF; de Tribolet, N; Weller, M; Kros, JM; Hainfellner, JA; Mason, W; Mariani, L; Bromberg, JE; Hau, P; Mirimanoff, RO; Cairncross, JG; Janzer, RC; Stupp, R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 2005, 352, 997–1003. [Google Scholar]
- Herrlinger, U; Rieger, J; Koch, D; Loeser, S; Blaschke, B; Kortmann, RD; Steinbach, JP; Hundsberger, T; Wick, W; Meyermann, R; Tan, TC; Sommer, C; Bamberg, M; Reifenberger, G; Weller, M. Phase II trial of lomustine plus temozolomide chemotherapy in addition to radiotherapy in newly diagnosed glioblastoma: UKT-03. J. Clin. Oncol 2006, 24, 4412–4417. [Google Scholar]
- Meyer-Puttlitz, B; Hayashi, Y; Waha, A; Rollbrocker, B; Bostrom, J; Wiestler, OD; Louis, DN; Reifenberger, G; von Deimling, A. Molecular genetic analysis of giant cell glioblastomas. Am. J. Pathol 1997, 151, 853–857. [Google Scholar]
- Peraud, A; Watanabe, K; Schwechheimer, K; Yonekawa, Y; Kleihues, P; Ohgaki, H. Genetic profile of the giant cell glioblastoma. Lab. Invest 1999, 79, 123–129. [Google Scholar]
- Reis, RM; Konu-Lebleblicioglu, D; Lopes, JM; Kleihues, P; Ohgaki, H. Genetic profile of gliosarcomas. Am. J. Pathol 2000, 156, 425–432. [Google Scholar]
- Actor, B; Cobbers, JM; Buschges, R; Wolter, M; Knobbe, CB; Lichter, P; Reifenberger, G; Weber, RG. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosom. Cancer 2002, 34, 416–427. [Google Scholar]
- Rickert, CH; Paulus, W. No chromosomal imbalances detected by comparative genomic hybridisation in subependymal giant cell astrocytomas. Acta Neuropathol. (Berl.) 2002, 104, 206–208. [Google Scholar]
- Giangaspero, F; Kaulich, K; Cenacchi, G; Cerasoli, S; Lerch, KD; Breu, H; Reuter, T; Reifenberger, G. Lipoastrocytoma: a rare low-grade astrocytoma variant of pediatric age. Acta Neuropathol 2002, 103, 152–156. [Google Scholar]
- Kepes, JJ; Fulling, KH; Garcia, JH. The clinical significance of “adenoid” formations of neoplastic astrocytes, imitating metastatic carcinoma, in gliosarcomas. A review of five cases. Clin. Neuropathol 1982, 1, 139–150. [Google Scholar]
- Mathews, T; Moossy, J. Gliomas containing bone and cartilage. J. Neuropathol. Exp. Neurol 1974, 33, 456–471. [Google Scholar]
- Mork, SJ; Rubinstein, LJ; Kepes, JJ; Perentes, E; Uphoff, DF. Patterns of epithelial metaplasia in malignant gliomas. II. Squamous differentiation of epithelial-like formations in gliosarcomas and glioblastomas. J. Neuropathol. Exp. Neurol 1988, 47, 101–118. [Google Scholar]
- Sanoudou, D; Tingby, O; Ferguson-Smith, MA; Collins, VP; Coleman, N. Analysis of pilocytic astrocytoma by comparative genomic hybridization. Br. J. Cancer 2000, 82, 1218–1222. [Google Scholar]
- Pfister, S; Janzarik, WG; Remke, M; Ernst, A; Werft, W; Becker, N; Toedt, G; Wittmann, A; Kratz, C; Olbrich, H; Ahmadi, R; Thieme, B; Joos, S; Radlwimmer, B; Kulozik, A; Pietsch, T; Herold-Mende, C; Gnekow, A; Reifenberger, G; Korshunov, A; Scheurlen, W; Omran, H; Lichter, P. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J. Clin. Invest 2008, 118, 1739–1749. [Google Scholar]
- Kluwe, L; Hagel, C; Tatagiba, M; Thomas, S; Stavrou, D; Ostertag, H; von Deimling, A; Mautner, VF. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J. Neuropathol. Exp. Neurol 2001, 60, 917–920. [Google Scholar]
- Wimmer, K; Eckart, M; Meyer-Puttlitz, B; Fonatsch, C; Pietsch, T. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. J. Neuropathol. Exp. Neurol 2002, 61, 896–902. [Google Scholar]
- Lang, FF; Miller, DC; Pisharody, S; Koslow, M; Newcomb, EW. High frequency of p53 protein accumulation without p53 gene mutation in human juvenile pilocytic, low grade and anaplastic astrocytomas. Oncogene 1994, 9, 949–954. [Google Scholar]
- Ohgaki, H; Eibl, RH; Schwab, M; Reichel, MB; Mariani, L; Gehring, M; Petersen, I; Holl, T; Wiestler, OD; Kleihues, P. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol. Carcinog 1993, 8, 74–80. [Google Scholar]
- Jones, DT; Kocialkowski, S; Liu, L; Pearson, DM; Backlund, LM; Ichimura, K; Collins, VP. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008, 68, 8673–8677. [Google Scholar]
- Bar, EE; Lin, A; Tihan, T; Burger, PC; Eberhart, CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol 2008, 67, 878–887. [Google Scholar]
- Deshmukh, H; Yeh, TH; Yu, J; Sharma, MK; Perry, A; Leonard, JR; Watson, MA; Gutmann, DH; Nagarajan, R. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene 2008, 27, 4745–4751. [Google Scholar]
- Tihan, T; Fisher, PG; Kepner, JL; Godfraind, C; McComb, RD; Goldthwaite, PT; Burger, PC. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J. Neuropathol. Exp. Neurol 1999, 58, 1061–1068. [Google Scholar]
- Rodriguez, FJ; Giannini, C; Asmann, YW; Sharma, MK; Perry, A; Tibbetts, KM; Jenkins, RB; Scheithauer, BW; Anant, S; Jenkins, S; Eberhart, CG; Sarkaria, JN; Gutmann, DH. Gene expression profiling of NF-1-associated and sporadic pilocytic astrocytoma identifies aldehyde dehydrogenase 1 family member L1 (ALDH1L1) as an underexpressed candidate biomarker in aggressive subtypes. J. Neuropathol. Exp. Neurol 2008, 67, 1194–1204. [Google Scholar]
- Weber, RG; Hoischen, A; Ehrler, M; Zipper, P; Kaulich, K; Blaschke, B; Becker, AJ; Weber-Mangal, S; Jauch, A; Radlwimmer, B; Schramm, J; Wiestler, OD; Lichter, P; Reifenberger, G. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 2007, 26, 1088–1097. [Google Scholar]
- Giannini, C; Hebrink, D; Scheithauer, BW; Dei Tos, AP; James, CD. Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics 2001, 3, 159–162. [Google Scholar]
- Kaulich, K; Blaschke, B; Numann, A; von Deimling, A; Wiestler, OD; Weber, RG; Reifenberger, G. Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas. J. Neuropathol. Exp. Neurol 2002, 61, 1092–1099. [Google Scholar]
- Chan, JA; Zhang, H; Roberts, PS; Jozwiak, S; Wieslawa, G; Lewin-Kowalik, J; Kotulska, K; Kwiatkowski, DJ. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J. Neuropathol. Exp. Neurol 2004, 63, 1236–1242. [Google Scholar]
- Riemenschneider, MJ; Koy, TH; Reifenberger, G. Expression of oligodendrocyte lineage genes in oligodendroglial and astrocytic gliomas. Acta Neuropathol 2004, 107, 277–282. [Google Scholar]
- Bouvier, C; Bartoli, C; Aguirre-Cruz, L; Virard, I; Colin, C; Fernandez, C; Gouvernet, J; Figarella-Branger, D. Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J. Neurosurg 2003, 99, 344–350. [Google Scholar]
- Ohnishi, A; Sawa, H; Tsuda, M; Sawamura, Y; Itoh, T; Iwasaki, Y; Nagashima, K. Expression of the oligodendroglial lineage-associated markers Olig1 and Olig2 in different types of human gliomas. J. Neuropathol. Exp. Neurol 2003, 62, 1052–1059. [Google Scholar]
- Reifenberger, J; Reifenberger, G; Liu, L; James, CD; Wechsler, W; Collins, VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am. J. Pathol 1994, 145, 1175–1190. [Google Scholar]
- Griffin, CA; Burger, P; Morsberger, L; Yonescu, R; Swierczynski, S; Weingart, JD; Murphy, KM. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J. Neuropathol. Exp. Neurol 2006, 65, 988–994. [Google Scholar]
- Jenkins, RB; Blair, H; Ballman, KV; Giannini, C; Arusell, RM; Law, M; Flynn, H; Passe, S; Felten, S; Brown, PD; Shaw, EG; Buckner, JC. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006, 66, 9852–9861. [Google Scholar]
- Boulay, JL; Miserez, AR; Zweifel, C; Sivasankaran, B; Kana, V; Ghaffari, A; Luyken, C; Sabel, M; Zerrouqi, A; Wasner, M; Van Meir, E; Tolnay, M; Reifenberger, G; Merlo, A. Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumors. PLoS ONE 2007, 2, e576. [Google Scholar]
- Barbashina, V; Salazar, P; Holland, EC; Rosenblum, MK; Ladanyi, M. Allelic losses at 1p36 and 19q13 in gliomas: Correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin. Cancer Res 2005, 11, 1119–1128. [Google Scholar]
- Dong, S; Pang, JC; Hu, J; Zhou, LF; Ng, HK. Transcriptional inactivation of TP73 expression in oligodendroglial tumors. Int. J. Cancer 2002, 98, 370–375. [Google Scholar]
- McDonald, JM; Dunlap, S; Cogdell, D; Dunmire, V; Wei, Q; Starzinski-Powitz, A; Sawaya, R; Bruner, J; Fuller, GN; Aldape, K; Zhang, W. The SHREW1 gene, frequently deleted in oligodendrogliomas, functions to inhibit cell adhesion and migration. Cancer Biol. Ther 2006, 5, 300–304. [Google Scholar]
- McDonald, JM; Dunmire, V; Taylor, E; Sawaya, R; Bruner, J; Fuller, GN; Aldape, K; Zhang, W. Attenuated expression of DFFB is a hallmark of oligodendrogliomas with 1p-allelic loss. Mol. Cancer 2005, 4, 35. [Google Scholar]
- Reifenberger, G; Louis, DN. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J. Neuropathol. Exp. Neurol 2003, 62, 111–126. [Google Scholar]
- Riemenschneider, MJ; Reifenberger, J; Reifenberger, G. Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int. J. Cancer 2008, 122, 2503–2510. [Google Scholar]
- Tews, B; Roerig, P; Hartmann, C; Hahn, M; Felsberg, J; Blaschke, B; Sabel, M; Kunitz, A; Toedt, G; Neben, K; Benner, A; Deimling, A; Reifenberger, G; Lichter, P. Hypermethylation and transcriptional downregulation of the CITED4 gene at 1p34.2 in oligodendroglial tumours with allelic losses on 1p and 19q. Oncogene 2007, 26, 5010–5016. [Google Scholar]
- Wolf, RM; Draghi, N; Liang, X; Dai, C; Uhrbom, L; Eklof, C; Westermark, B; Holland, EC; Resh, MD. p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev 2003, 17, 476–487. [Google Scholar]
- Alaminos, M; Davalos, V; Ropero, S; Setien, F; Paz, MF; Herranz, M; Fraga, MF; Mora, J; Cheung, NK; Gerald, WL; Esteller, M. EMP3, a myelin-related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res 2005, 65, 2565–2571. [Google Scholar]
- Hong, C; Bollen, AW; Costello, JF. The contribution of genetic and epigenetic mechanisms to gene silencing in oligodendrogliomas. Cancer Res 2003, 63, 7600–7605. [Google Scholar]
- Trouillard, O; Aguirre-Cruz, L; Hoang-Xuan, K; Marie, Y; Delattre, JY; Sanson, M. Parental 19q loss and PEG3 expression in oligodendrogliomas. Cancer Genet. Cytogenet 2004, 151, 182–183. [Google Scholar]
- Jeuken, JW; von Deimling, A; Wesseling, P. Molecular pathogenesis of oligodendroglial tumors. J. Neurooncol 2004, 70, 161–181. [Google Scholar]
- Trost, D; Ehrler, M; Fimmers, R; Felsberg, J; Sabel, MC; Kirsch, L; Schramm, J; Wiestler, OD; Reifenberger, G; Weber, RG. Identification of genomic aberrations associated with shorter overall survival in patients with oligodendroglial tumors. Int. J. Cancer 2007, 120, 2368–2376. [Google Scholar]
- Watanabe, T; Nakamura, M; Yonekawa, Y; Kleihues, P; Ohgaki, H. Promoter hypermethylation and homozygous deletion of the p14ARF and p16INK4a genes in oligodendrogliomas. Acta Neuropathol 2001, 101, 185–189. [Google Scholar]
- Wolter, M; Reifenberger, J; Blaschke, B; Ichimura, K; Schmidt, EE; Collins, VP; Reifenberger, G. Oligodendroglial tumors frequently demonstrate hypermethylation of the CDKN2A (MTS1, p16INK4a), p14ARF, and CDKN2B (MTS2, p15INK4b) tumor suppressor genes. J. Neuropathol. Exp. Neurol 2001, 60, 1170–1180. [Google Scholar]
- Alonso, ME; Bello, MJ; Gonzalez-Gomez, P; Arjona, D; Lomas, J; de Campos, JM; Isla, A; Sarasa, JL; Rey, JA. Aberrant promoter methylation of multiple genes in oligodendrogliomas and ependymomas. Cancer Genet. Cytogenet 2003, 144, 134–142. [Google Scholar]
- Dong, SM; Pang, JC; Poon, WS; Hu, J; To, KF; Chang, AR; Ng, HK. Concurrent hypermethylation of multiple genes is associated with grade of oligodendroglial tumors. J. Neuropathol. Exp. Neurol 2001, 60, 808–816. [Google Scholar]
- McLendon, RE; Herndon, JE, 2nd; West, B; Reardon, D; Wiltshire, R; Rasheed, BK; Quinn, J; Friedman, HS; Friedman, AH; Bigner, DD. Survival analysis of presumptive prognostic markers among oligodendrogliomas. Cancer 2005, 104, 1693–1699. [Google Scholar]
- Mollemann, M; Wolter, M; Felsberg, J; Collins, VP; Reifenberger, G. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int. J. Cancer 2005, 113, 379–385. [Google Scholar]
- Di Rocco, F; Carroll, RS; Zhang, J; Black, PM. Platelet-derived growth factor and its receptor expression in human oligodendrogliomas. Neurosurgery 1998, 42, 341–346. [Google Scholar]
- Fallon, KB; Palmer, CA; Roth, KA; Nabors, LB; Wang, W; Carpenter, M; Banerjee, R; Forsyth, P; Rich, K; Perry, A. Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J. Neuropathol. Exp. Neurol 2004, 63, 314–322. [Google Scholar]
- Hoang-Xuan, K; He, J; Huguet, S; Mokhtari, K; Marie, Y; Kujas, M; Leuraud, P; Capelle, L; Delattre, JY; Poirier, J; Broet, P; Sanson, M. Molecular heterogeneity of oligodendrogliomas suggests alternative pathways in tumor progression. Neurology 2001, 57, 1278–1281. [Google Scholar]
- Sasaki, H; Zlatescu, MC; Betensky, RA; Ino, Y; Cairncross, JG; Louis, DN. PTEN is a target of chromosome 10q loss in anaplastic oligodendrogliomas and PTEN alterations are associated with poor prognosis. Am. J. Pathol 2001, 159, 359–367. [Google Scholar]
- Broderick, DK; Di, C; Parrett, TJ; Samuels, YR; Cummins, JM; McLendon, RE; Fults, DW; Velculescu, VE; Bigner, DD; Yan, H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res 2004, 64, 5048–5050. [Google Scholar]
- Cairncross, JG; Ueki, K; Zlatescu, MC; Lisle, DK; Finkelstein, DM; Hammond, RR; Silver, JS; Stark, PC; Macdonald, DR; Ino, Y; Ramsay, DA; Louis, DN. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J. Natl. Cancer Inst 1998, 90, 1473–1479. [Google Scholar]
- Smith, JS; Perry, A; Borell, TJ; Lee, HK; O’Fallon, J; Hosek, SM; Kimmel, D; Yates, A; Burger, PC; Scheithauer, BW; Jenkins, RB. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J. Clin. Oncol 2000, 18, 636–645. [Google Scholar]
- Felsberg, J; Erkwoh, A; Sabel, MC; Kirsch, L; Fimmers, R; Blaschke, B; Schlegel, U; Schramm, J; Wiestler, OD; Reifenberger, G. Oligodendroglial tumors: Refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol 2004, 14, 121–130. [Google Scholar]
- Kanner, AA; Staugaitis, SM; Castilla, EA; Chernova, O; Prayson, RA; Vogelbaum, MA; Stevens, G; Peereboom, D; Suh, J; Lee, SY; Tubbs, RR; Barnett, GH. The impact of genotype on outcome in oligodendroglioma: validation of the loss of chromosome arm 1p as an important factor in clinical decision making. J. Neurosurg 2006, 104, 542–550. [Google Scholar]
- Kujas, M; Lejeune, J; Benouaich-Amiel, A; Criniere, E; Laigle-Donadey, F; Marie, Y; Mokhtari, K; Polivka, M; Bernier, M; Chretien, F; Couvelard, A; Capelle, L; Duffau, H; Cornu, P; Broet, P; Thillet, J; Carpentier, AF; Sanson, M; Hoang-Xuan, K; Delattre, JY. Chromosome 1p loss: a favorable prognostic factor in low-grade gliomas. Ann. Neurol 2005, 58, 322–326. [Google Scholar]
- Hoang-Xuan, K; Capelle, L; Kujas, M; Taillibert, S; Duffau, H; Lejeune, J; Polivka, M; Criniere, E; Marie, Y; Mokhtari, K; Carpentier, AF; Laigle, F; Simon, JM; Cornu, P; Broet, P; Sanson, M; Delattre, JY. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions. J. Clin. Oncol 2004, 22, 3133–3138. [Google Scholar]
- Levin, N; Lavon, I; Zelikovitsh, B; Fuchs, D; Bokstein, F; Fellig, Y; Siegal, T. Progressive low-grade oligodendrogliomas: response to temozolomide and correlation between genetic profile and O6-methylguanine DNA methyltransferase protein expression. Cancer 2006, 106, 1759–1765. [Google Scholar]
- Weller, M; Berger, H; Hartmann, C; Schramm, J; Westphal, M; Simon, M; Goldbrunner, R; Krex, D; Steinbach, JP; Ostertag, CB; Loeffler, M; Pietsch, T; von Deimling, A. Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin. Cancer Res 2007, 13, 6933–6937. [Google Scholar]
- Kraus, JA; Koopmann, J; Kaskel, P; Maintz, D; Brandner, S; Schramm, J; Louis, DN; Wiestler, OD; von Deimling, A. Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J. Neuropathol. Exp. Neurol 1995, 54, 91–95. [Google Scholar]
- Qu, M; Olofsson, T; Sigurdardottir, S; You, C; Kalimo, H; Nister, M; Smits, A; Ren, ZP. Genetically distinct astrocytic and oligodendroglial components in oligoastrocytomas. Acta Neuropathol 2007, 113, 129–136. [Google Scholar]
- Miller, CR; Dunham, CP; Scheithauer, BW; Perry, A. Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J. Clin. Oncol 2006, 24, 5419–5426. [Google Scholar]
- Carter, M; Nicholson, J; Ross, F; Crolla, J; Allibone, R; Balaji, V; Perry, R; Walker, D; Gilbertson, R; Ellison, DW. Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br. J. Cancer 2002, 86, 929–939. [Google Scholar]
- Dyer, S; Prebble, E; Davison, V; Davies, P; Ramani, P; Ellison, D; Grundy, R. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am. J. Pathol 2002, 161, 2133–2141. [Google Scholar]
- Jeuken, JW; Sprenger, SH; Gilhuis, J; Teepen, HL; Grotenhuis, AJ; Wesseling, P. Correlation between localization, age, and chromosomal imbalances in ependymal tumours as detected by CGH. J. Pathol 2002, 197, 238–244. [Google Scholar]
- Mendrzyk, F; Korshunov, A; Benner, A; Toedt, G; Pfister, S; Radlwimmer, B; Lichter, P. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin. Cancer Res 2006, 12, 2070–2079. [Google Scholar]
- Taylor, MD; Poppleton, H; Fuller, C; Su, X; Liu, Y; Jensen, P; Magdaleno, S; Dalton, J; Calabrese, C; Board, J; Macdonald, T; Rutka, J; Guha, A; Gajjar, A; Curran, T; Gilbertson, RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005, 8, 323–335. [Google Scholar]
- Ebert, C; von Haken, M; Meyer-Puttlitz, B; Wiestler, OD; Reifenberger, G; Pietsch, T; von Deimling, A. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am. J. Pathol 1999, 155, 627–632. [Google Scholar]
- Kraus, JA; de Millas, W; Sorensen, N; Herbold, C; Schichor, C; Tonn, JC; Wiestler, OD; von Deimling, A; Pietsch, T. Indications for a tumor suppressor gene at 22q11 involved in the pathogenesis of ependymal tumors and distinct from hSNF5/INI1. Acta Neuropathol 2001, 102, 69–74. [Google Scholar]
- Ohgaki, H; Eibl, RH; Wiestler, OD; Yasargil, MG; Newcomb, EW; Kleihues, P. p53 mutations in nonastrocytic human brain tumors. Cancer Res 1991, 51, 6202–6205. [Google Scholar]
- Hamilton, DW; Lusher, ME; Lindsey, JC; Ellison, DW; Clifford, SC. Epigenetic inactivation of the RASSF1A tumour suppressor gene in ependymoma. Cancer Lett 2005, 227, 75–81. [Google Scholar]
- Rousseau, E; Ruchoux, MM; Scaravilli, F; Chapon, F; Vinchon, M; De Smet, C; Godfraind, C; Vikkula, M. CDKN2A, CDKN2B and p14ARF are frequently and differentially methylated in ependymal tumours. Neuropathol. Appl. Neurobiol 2003, 29, 574–583. [Google Scholar]
- Gilbertson, RJ; Bentley, L; Hernan, R; Junttila, TT; Frank, AJ; Haapasalo, H; Connelly, M; Wetmore, C; Curran, T; Elenius, K; Ellison, DW. ERBB receptor signaling promotes ependymoma cell proliferation and represents a potential novel therapeutic target for this disease. Clin. Cancer Res 2002, 8, 3054–3064. [Google Scholar]
- Hirose, Y; Aldape, K; Bollen, A; James, CD; Brat, D; Lamborn, K; Berger, M; Feuerstein, BG. Chromosomal abnormalities subdivide ependymal tumors into clinically relevant groups. Am. J. Pathol 2001, 158, 1137–1143. [Google Scholar]
- Korshunov, A; Neben, K; Wrobel, G; Tews, B; Benner, A; Hahn, M; Golanov, A; Lichter, P. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am. J. Pathol 2003, 163, 1721–1727. [Google Scholar]
- Scheil, S; Bruderlein, S; Eicker, M; Herms, J; Herold-Mende, C; Steiner, HH; Barth, TF; Moller, P. Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 2001, 11, 133–143. [Google Scholar]
- Mahler-Araujo, MB; Sanoudou, D; Tingby, O; Liu, L; Coleman, N; Ichimura, K; Collins, VP. Structural genomic abnormalities of chromosomes 9 and 18 in myxopapillary ependymomas. J. Neuropathol. Exp. Neurol 2003, 62, 927–935. [Google Scholar]
- Dal Cin, P; Van den Berghe, H; Buonamici, L; Losi, L; Roncaroli, F; Calbucci, F. Cytogenetic investigation in subependymoma. Cancer Genet. Cytogenet 1999, 108, 84. [Google Scholar]
- Sasaki, H; Zlatescu, MC; Betensky, RA; Johnk, LB; Cutone, AN; Cairncross, JG; Louis, DN. Histopathological-molecular genetic correlations in referral pathologist-diagnosed low-grade “oligodendroglioma”. J. Neuropathol. Exp. Neurol 2002, 61, 58–63. [Google Scholar]
- Roerig, P; Nessling, M; Radlwimmer, B; Joos, S; Wrobel, G; Schwaenen, C; Reifenberger, G; Lichter, P. Molecular classification of human gliomas using matrix-based comparative genomic hybridization. Int. J. Cancer 2005, 117, 95–103. [Google Scholar]
- Nutt, CL; Mani, DR; Betensky, RA; Tamayo, P; Cairncross, JG; Ladd, C; Pohl, U; Hartmann, C; McLaughlin, ME; Batchelor, TT; Black, PM; von Deimling, A; Pomeroy, SL; Golub, TR; Louis, DN. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 2003, 63, 1602–1607. [Google Scholar]
- Freije, WA; Castro-Vargas, FE; Fang, Z; Horvath, S; Cloughesy, T; Liau, LM; Mischel, PS; Nelson, SF. Gene expression profiling of gliomas strongly predicts survival. Cancer Res 2004, 64, 6503–6510. [Google Scholar]
- Phillips, HS; Kharbanda, S; Chen, R; Forrest, WF; Soriano, RH; Wu, TD; Misra, A; Nigro, JM; Colman, H; Soroceanu, L; Williams, PM; Modrusan, Z; Feuerstein, BG; Aldape, K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar]
- Lee, Y; Scheck, AC; Cloughesy, TF; Lai, A; Dong, J; Farooqi, HK; Liau, LM; Horvath, S; Mischel, PS; Nelson, SF. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genomics 2008, 1, 52. [Google Scholar]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Li, Y; Bollag, G; Clark, R; Stevens, J; Conroy, L; Fults, D; Ward, K; Friedman, E; Samowitz, W; Robertson, M; et al. Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell 1992, 69, 275–281. [Google Scholar]
- Mizoguchi, M; Nutt, CL; Mohapatra, G; Louis, DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 2004, 14, 372–377. [Google Scholar]
- Cahill, DP; Levine, KK; Betensky, RA; Codd, PJ; Romany, CA; Reavie, LB; Batchelor, TT; Futreal, PA; Stratton, MR; Curry, WT; Iafrate, AJ; Louis, DN. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res 2007, 13, 2038–2045. [Google Scholar]
- Hunter, C; Smith, R; Cahill, DP; Stephens, P; Stevens, C; Teague, J; Greenman, C; Edkins, S; Bignell, G; Davies, H; O’Meara, S; Parker, A; Avis, T; Barthorpe, S; Brackenbury, L; Buck, G; Butler, A; Clements, J; Cole, J; Dicks, E; Forbes, S; Gorton, M; Gray, K; Halliday, K; Harrison, R; Hills, K; Hinton, J; Jenkinson, A; Jones, D; Kosmidou, V; Laman, R; Lugg, R; Menzies, A; Perry, J; Petty, R; Raine, K; Richardson, D; Shepherd, R; Small, A; Solomon, H; Tofts, C; Varian, J; West, S; Widaa, S; Yates, A; Easton, DF; Riggins, G; Roy, JE; Levine, KK; Mueller, W; Batchelor, TT; Louis, DN; Stratton, MR; Futreal, PA; Wooster, R. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006, 66, 3987–3991. [Google Scholar]
- Murat, A; Migliavacca, E; Gorlia, T; Lambiv, WL; Shay, T; Hamou, MF; de Tribolet, N; Regli, L; Wick, W; Kouwenhoven, MC; Hainfellner, JA; Heppner, FL; Dietrich, PY; Zimmer, Y; Cairncross, JG; Janzer, RC; Domany, E; Delorenzi, M; Stupp, R; Hegi, ME. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol 2008, 26, 3015–3024. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor type | WHO grade |
|---|---|
| Diffusely infiltrating astrocytic gliomas | |
| Diffuse astrocytoma | II |
| Anaplastic astrocytoma | III |
| Glioblastoma | IV |
| Giant cell glioblastoma | IV |
| Gliosarcoma
| IV
|
| Astrocytic gliomas with more circumscribed growth | |
| Pilocytic astrocytoma | I |
| Pilomyxoid astrocytoma | II |
| Pleomorphic xanthoastrocytoma | II |
| Subependymal giant cell astrocytoma | I
|
| Oligodendrogliomas and mixed gliomas | |
| Oligodendroglioma | II |
| Anaplastic oligodendroglioma | III |
| Oligoastrocytoma | II |
| Anaplastic oligoastrocytoma | III
|
| Gliomas with ependymal differentiation | |
| Subependymoma | I |
| Myxopapillary ependymoma | I |
| Ependymoma | II |
| Anaplastic ependymoma | III |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Riemenschneider, M.J.; Reifenberger, G. Molecular Neuropathology of Gliomas. Int. J. Mol. Sci. 2009, 10, 184-212. https://doi.org/10.3390/ijms10010184
Riemenschneider MJ, Reifenberger G. Molecular Neuropathology of Gliomas. International Journal of Molecular Sciences. 2009; 10(1):184-212. https://doi.org/10.3390/ijms10010184
Chicago/Turabian StyleRiemenschneider, Markus J., and Guido Reifenberger. 2009. "Molecular Neuropathology of Gliomas" International Journal of Molecular Sciences 10, no. 1: 184-212. https://doi.org/10.3390/ijms10010184