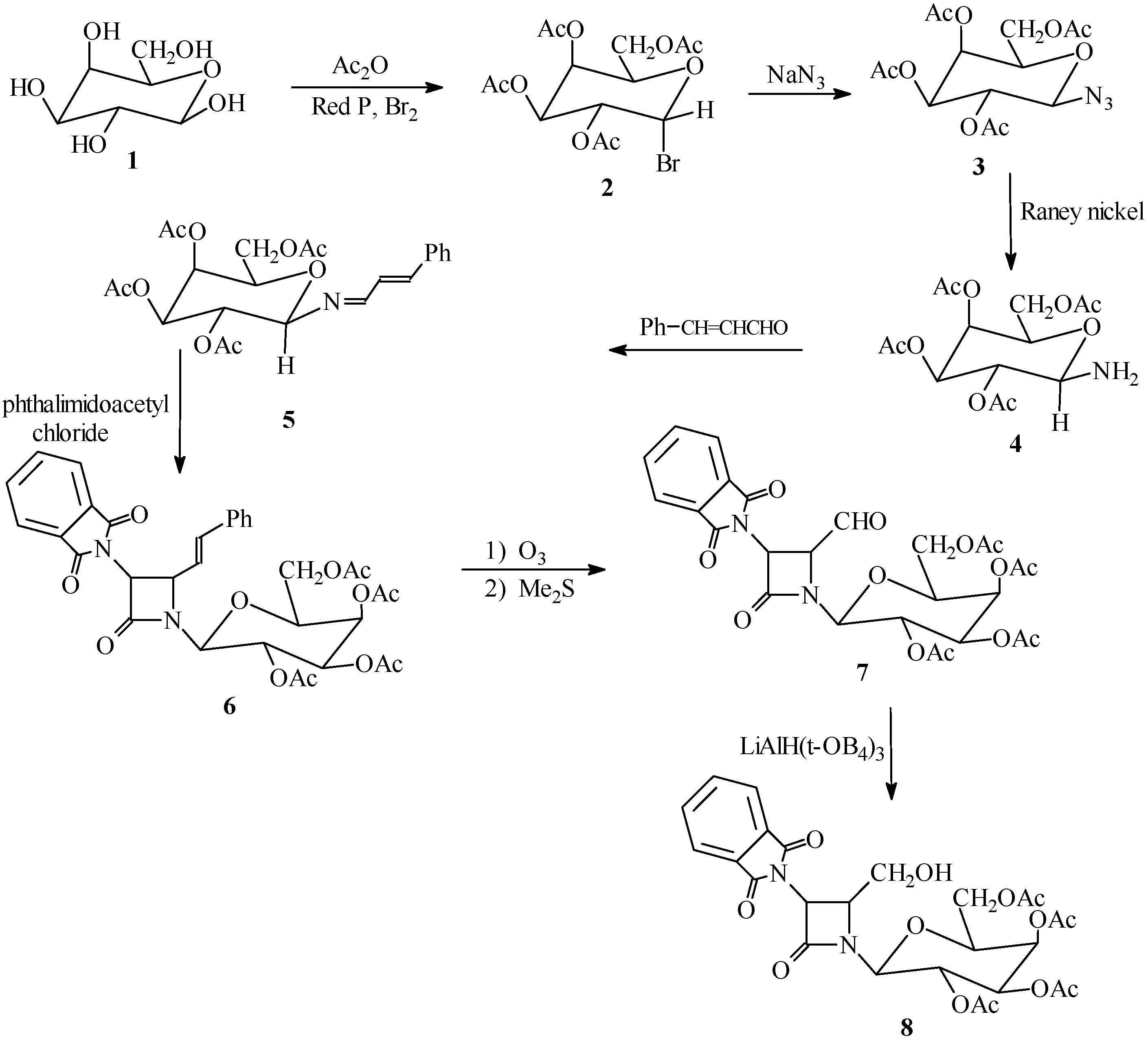
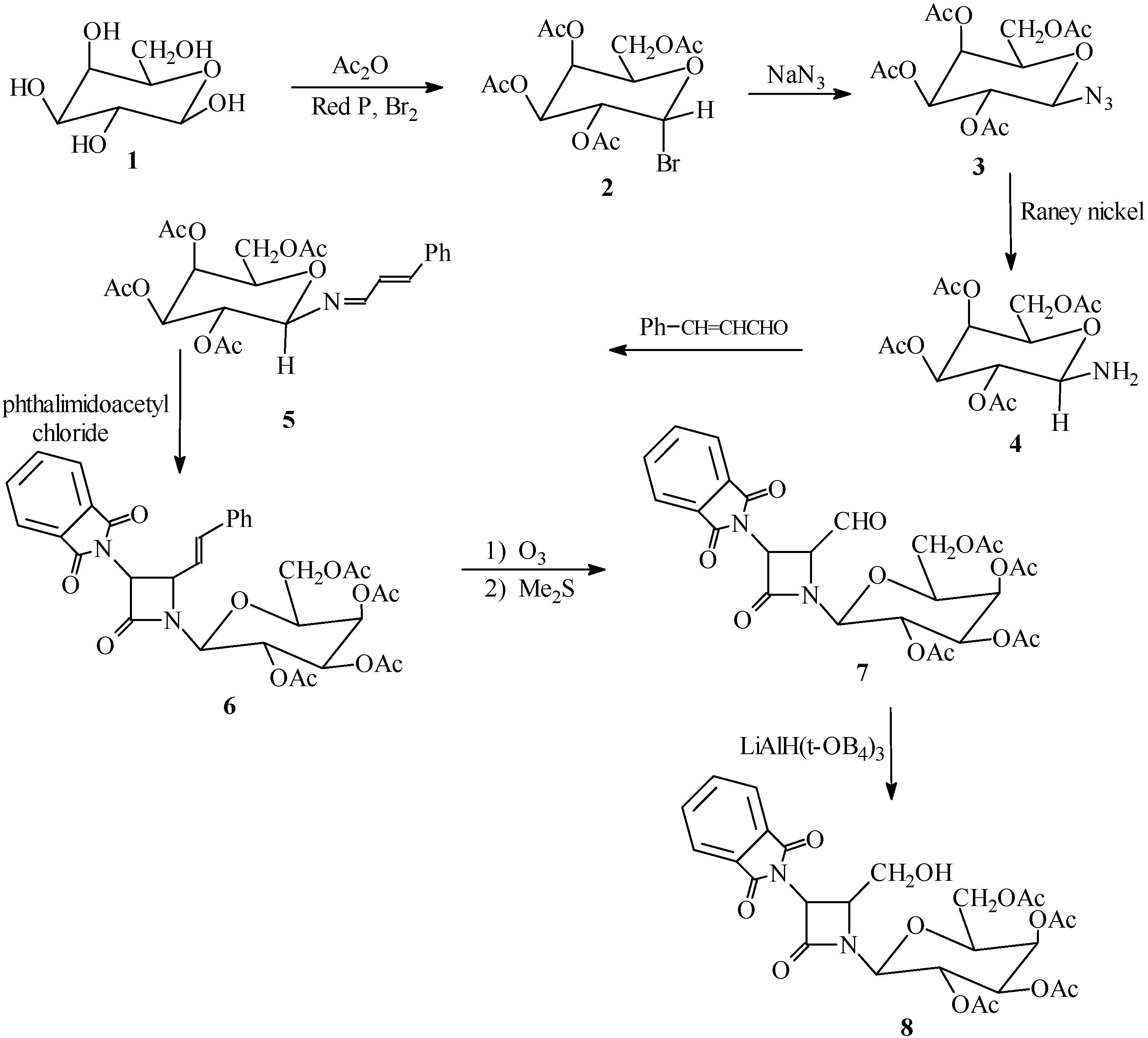
Synthesis of compounds
2,3,4,6-Tetra-O-acetyl-α-D-galactopyranosyl bromide (
2). This compound was prepared according to a literature procedure [
19].
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl azide (3). Sodium azide (0.47 g, 7.30 mmol) was added to a solution of 2 (3.00 g, 7.30 mmol) in 9:1 acetone-water (30 mL) and the mixture was refluxed for 10 hours, then stirred at room temperature for 24 hours. The solvent was evaporated. The residue was taken up in CH2Cl2 (30 mL) and washed with H2O (3 x 30 mL), dried (Na2SO4) and filtered. Evaporation of the solvent under reduced pressure afforded a solid. Recrystallization from ether gave 3 as white crystals (70% yield); m.p. 91-92 °C; IR (cm–1): 2140 (N3), 1740-1750 (OCOCH3); 1H‑NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.95-4.21 (3H, m, H2, CH2OAc), 4.35 (1H, t, H3), 5.05-5.20 (2H, m, H4, H5), 5.95(1H, d, H6 ); Anal.(C14H19N3O9), Calc. C, 45.04; H, 5.13; N, 11.26; O, 38.57%, found: C, 45.00; H, 5.18; N, 11.24; O, 38.55%; MS, m/z (%): 373.11 (M+, 100.00), 374.11 (M+1, 16.20).
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl amine (4). To a solution of 3 (6.00 g, 16.07 mmol) in ethyl acetate (50 mL) was added Raney Ni (6.00 g) and the reaction mixture was refluxed for 2.5 hours. The resulting solid was filtered and washed with ether. Recrystallization from ether gave 4 as yellow crystals in 90% yield; m.p. 120-122 °C. IR (cm–1): 3400, 3330 (NH2), 1730-1740 (OCOCH3); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.95-4.21 (3H, m, H2, CH2OAc), 4.35 (1H, t, H3), 4.55 (2H, d, NH2), 5.05-5.20 (2H, m, H4, H5), 5.95 (1H, d, H6); Anal., Calc. for C14H21NO9: C, 48.41; H, 6.09; N, 4.03; O, 41.46%, found: C, 48.38; H, 6.05; N, 4.01; O, 41.42%; MS, m/z (%): 347.12 (M+, 100.00), 348.12 (M+1, 15.90).
β-D-galactopyranosylamino-(N-cinnamylidene)-2,3,4,6-tetra-O-acetate (5). A mixture of compound 4 (3.47 g, 10.00 mmol), cinnamaldehyde (1.32 g, 10.00 mmol) and Na2SO4 (15.00 g, 105.61 mmol) in dry CH2Cl2 (40 mL) was refluxed for 5 hours. The resulting mixture was filtered and washed with H2O (3 x 30 mL). The organic layer was separated and dried (Na2SO4). Evaporation of the solvent under reduced pressure gave 5 as a solid which was recrystalized from ether-hexane (1:1) (3.69g, 80.00%); m.p.110-112 °C; IR (cm-1): 1740 (OCOCH3), 1685 (C=C), 1680 (C=N); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 4.20-4.25 (3H, m, H2 and CH2OAc), 4.35 (1H, t, H3), 5.05-5.2 (2H, m, H4, H5), 5.60 (1H, dd, J=8.00 and 16.00 Hz, CH=CHPh), 5.95 (1H, d, H6), 6.60 (1H, d, CH=CHPh), 7.10-7.30 (5H, m, Ph), 7.50 (1H, d, N=CH); Anal. Calc. for C23H27NO9: C, 59.86;H, 5.90; N, 3.04; O, 31.20%, found: C, 59.80; H, 6.10; N, 3.00; O, 31.10%; MS, m/z: 461.11 (M+).
1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-phthalimido-4-styryl]azetidin-2-one (6). A solution of phthalimidoacetyl chloride (7.00 g, 35.00 mmol) in dry CH2Cl2 (50 mL) was added dropwise to a solution of Schiff base 5 (4.61g, 10.00 mmol) and triethylamine (1.01g, 10 mmol) in dry CH2Cl2 (100 mL) at -10 °C. After the addition was complete, the solution was stirred at room temperature for 24 hours and then washed with saturated NaHCO3 solution (2 x 70 mL), water (3 x 50 mL) and brine (70 mL). The organic layer was separated, dried (Na2SO4), filtered and evaporated. Recrystallization from hexane-ether (1:1) gave 6 in 75% yield; m.p. 95-97 °C; IR (cm-1): 1780 (β‑lactam CO), 1760, 1735 (phthalimido CO), 1740 (OCOCH3); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.95-4.20 (3H, m, H2,CH2OAc), 4.35 (1H, t, H3), 4.50 (1H, dd, J=5 and J=8Hz, CH‑CH=CH), 4.90 (1H, d, J=5, CH-N(CO)2), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6), 6.20 (1H, dd, J=8 and J= 16, CH-CH=CHPh), 6.40 (1H, d, CH=CHPh), 7.10-7.25 (5H, m, Ph), 7.60-7.95 (4H, m, C6H4(CO)2N); Anal. Calc. for C33H33N2O12: C, 61.11; H, 4.97; N, 4.32; O, 29.60%, found: C, 61.02; H, 5.05; N, 4.28; O, 29.70%.
1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-phthalimido-4-formyl]azetidin-2-one (7). A solution of compound 6 (2.00 g, 3.00 mmol) in dry CH2Cl2 (50 mL) was cooled to -78 °C. A stream of ozone was passed through the reaction mixture until a pale blue coloration was observed and then it was purged with nitrogen. A solution of dimethyl sulfide (3 mL) in CH2Cl2 (10 mL) was added dropwise at -78 °C. When the addition was complete, the cooling bath was removed and the solution was stirred at room temperature. The reaction mixture was washed with H2O (3 x 40 mL) and saturated brine (2 x 30 mL). The organic layer was separated and dried (Na2SO4). Evaporation of the solvent gave a syrup. The oily residue was heated under high vacuum for 20 hours at 50 °C to remove the benzaldehyde formed during the ozonolysis. Compound 7 was isolated as a solid and was recrystalized from ether-hexane in 97% yield; m.p. 88-90 °C; IR (cm–1): 1780 (β-lactam CO), 1765, 1730 (phthalimido CO), 1750 (CHO), 1740 (OCOCH3); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.95-4.20 (3H, m, H2, CH2OAc), 4.35 (1H, t, H3), 4.70 (1H, dd, J=5Hz, CH-CHO), 4.90 (1H, d, J=5Hz, CH-N(CO)2), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6), 7.60-7.95 (4H, m, C6H4(CO)2N), 10.00 (1H, d, CHO); Anal. Calc. for C26H26N2O13: C, 54.36; H, 4.56; N, 4.88; O, 36.20%, found: C, 54.30; H, 5.01; N, 4.87; O, 36.27%.
1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-phthalimido-4-hydroxymethyl] azetidin-2-one (8): To aldehyde 7 (1.73 g, 3.00 mmol) in tetrahydrofuran (80 mL) at 0 °C was added lithium tri (tert-butoxy) aluminum hydride (1.53 g, 6.00 mmol). After stirring under nitrogen for 3 hours (0 °C), the mixture was acidified with dilute HCl (2%) to pH 5, and 1 g of silica gel was added. The suspension was stirred for 10 minutes, filtered, and evaporated under reduced pressure. The residue was taken up in ethyl acetate (60 mL), washed with water (3 x 40 mL), brine (3 x 20 mL), dried (Na2SO4), and the solvent was evaporated. Recrystallization from n-hexane gave 8 as yellow crystals in 60% yield; m.p. 85-87 °C; IR (cm-1): 3490, 3460 (OH), 1780 (β-lactam CO), 1760, 1730 (phthalimido CO), 1740(OCOCH3); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.75 (2H, m, CH2OH), 3.95-4.20 (3H, m, H2,CH2OAc), 4.35 (1H, t, H3), 4.45 (1H, br, OH), 4.70 (1H, dd, J=5Hz, CH-CH2OH), 4.90 (1H, d, J=5Hz, CH-N(CO)2), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6), 7.60-7.95 (4H, m, C6H4(CO)2N); Anal. Calc. for C26H28N2O13: C, 54.17; H, 4.90; N, 4.86; O, 36.08%, found: C, 54.10, H, 5.00; N, 4.80; O, 36.18%.
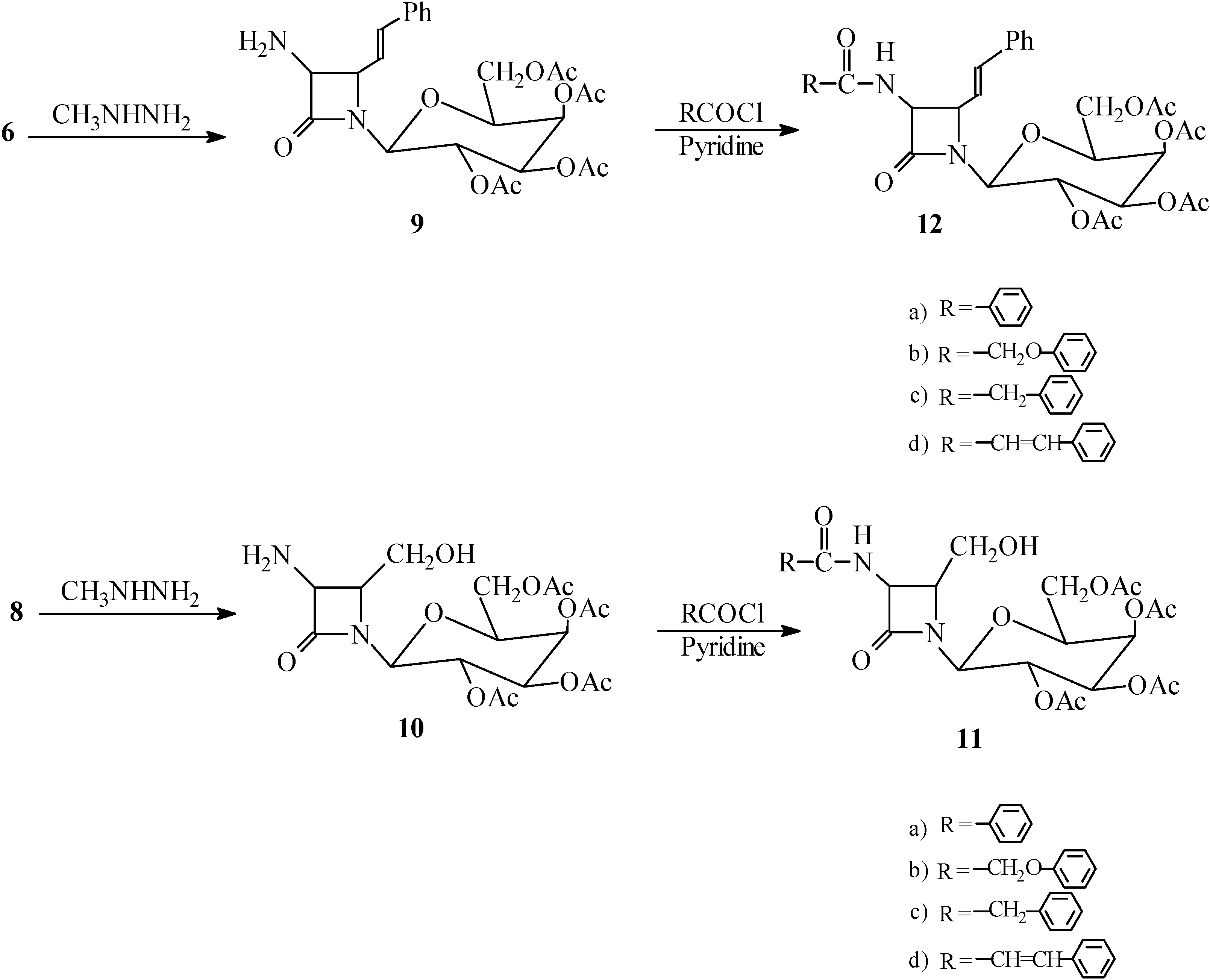
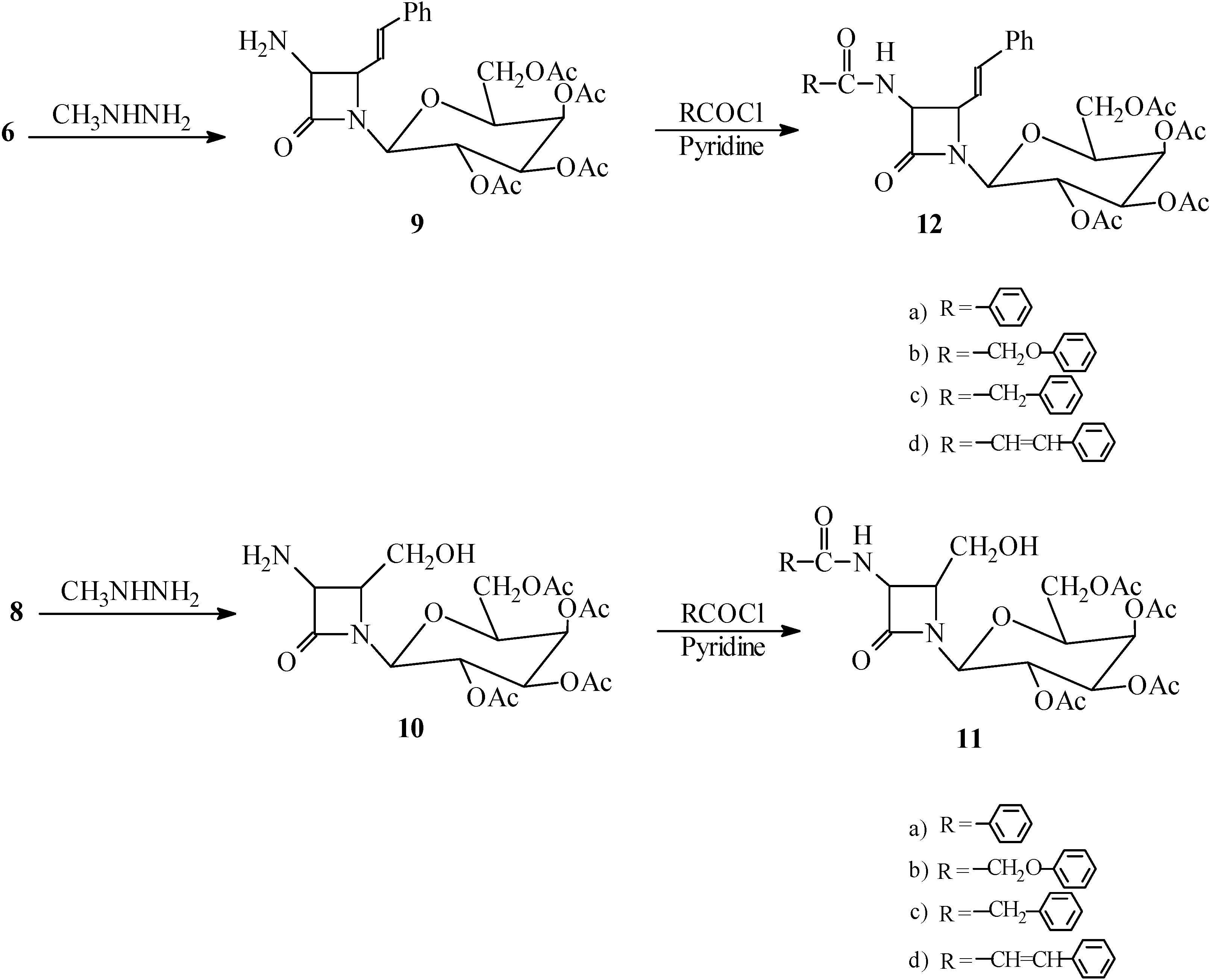
General procedure for dephthaloylation of protected β-lactams 6 and 8. The monocyclic β-lactam 6 (6.50 g, 10.00 mmol) (or 8) was dissolved in ethanol (25 mL) and methylhydrazine (0.046 g, 10.00 mmol) was added. After refluxing for 2 hours, the reaction mixture was stored overnight and then concentrated to dryness under vacuum. The methylphthalhydrazide residue was stirred for 2 hours with 25 mL of 5N HCl, and filtered. The aqueous and acid extracts were combined and 3 mL of concentrated HCl was added. After 2 hours, the aqueous solution was evaporated to give the product 1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-amino-4-styryl] azetidin-2-one (9, 3.64g, 70.0%) (or 1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-amino-4-hydroxymethyl]azetidin-2-one (10, 3.57g, 80.0%) as the corresponding hydrochloride salts.
(9) m.p.: 91-92 °C (free amine); IR (cm-1): 3250-3550 (br, N+H3), 1780 (β-lactam), 1740 (ester); 1-H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.95-4.20 (3H, m, H2,CH2OAc), 4.35 (1H, t, H3), 4.50 (1H, dd, J=5 and J=8Hz, CH-CH=CH), 4.90 (1H, d, J=5Hz, CH-NH2), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6), 6.20 (1H, dd, J=8 and J=16Hz, CH‑CH=CHPh), 6.40 (1H, d, CH=CHPh), 7.10-7.25 (5H, m, Ph); Anal. Calc. for C25H30N2O10: C, 57.91; H, 5.83; N, 5.40; O, 30.86%, found: C, 57.99; H, 5.70; N, 5.35; O, 30.91%.
(10) m.p.: 110-112 °C (free amine); IR (cm-1): 3200-3550 (NH3 and OH), 1780 (β-lactam CO), 1740(OCOCH3); 1H-NMR (CDCl3) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.75 (2H, m, CH2OH), 3.95-4.20 (3H, m, H2,CH2OAc), 4.35 (1H, t, H3), 4.45 (1H, br, OH), 4.70 (1H, dd, J=5Hz, CH-CH2OH), 4.90 (1H, d, J=5Hz, CH-NH2), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6); Anal Calc. for C18H26N2O11 : C, 48.43; H, 5.87; N, 6.28; O, 39.42%, found: C, 48.64; H, 5.80; N, 6.18; O, 39.50
General procedure for acylation of free-amino β-lactams 11a-d and 12a-d. Pyridine (1.50 g, 18.96 mmol) was added to a solution of 9 (3.12 g, 6.00 mmol), followed by dropwise addition of phenylacetyl chloride (0.93 g 6.00 mmol) in CH2Cl2 (20 mL). The solution was stirred for 2h. at 25 °C. Then it was washed with 10% HCl, 10% NaHCO3 and water, dried (MgSO4), and the solvent was evaporated to give the impure amide which was recrystallized from ethanol to give 1-[(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-3-phenylacetamido-4-styryl] azetidin-2-one ) 12c (2.00 g, 64.00%). M.p. 85-87 °C. IR (cm–1): 3410 (NH), 1770 (β‑lactam), 1740 (ester), 1685 (amide); 1H-NMR (DMSO-d6) δ (ppm): 1.95-2.15 (12H, s, 4COCH3), 3.45 (2H, s, PhCH2CO), 3.95-4.20 (3H, m, H2, CH2OAc), 4.35 (1H, t, H3), 4.50 (1H, dd, J=5 and J=8Hz, CH-CH=CH), 4.90 (1H, dd, J=5 and J=8Hz, CH-NH), 5.05-5.2 (2H, m, H4, H5), 5.95 (1H, d, H6), 6.20 (1H, dd, J=8 and J= 16Hz, CH-CH=CHPh), 6.40 (1H, d, CH=CHPh), 6.65 (1H, d, J=8Hz, NH), 7.00-7.30 (10H, m, 2Ph); Anal. Calc. for C33H36N2O11: C, 62.26; H, 5.70; N, 4.40; O, 27.64%, found: C, 62.20; H, 5.75; N, 4.42; O, 27.70%.
Compounds 11a-d, 12a-b and 12d were treated identically and their spectra were similar except for the expected variations due to the amide side chains.


{kind=link}
{kind=link}