2,3-Bifunctionalized Quinoxalines: Synthesis, DNA Interactions and Evaluation of Anticancer, Anti-tuberculosis and Antifungal Activity

Abstract
:Introduction
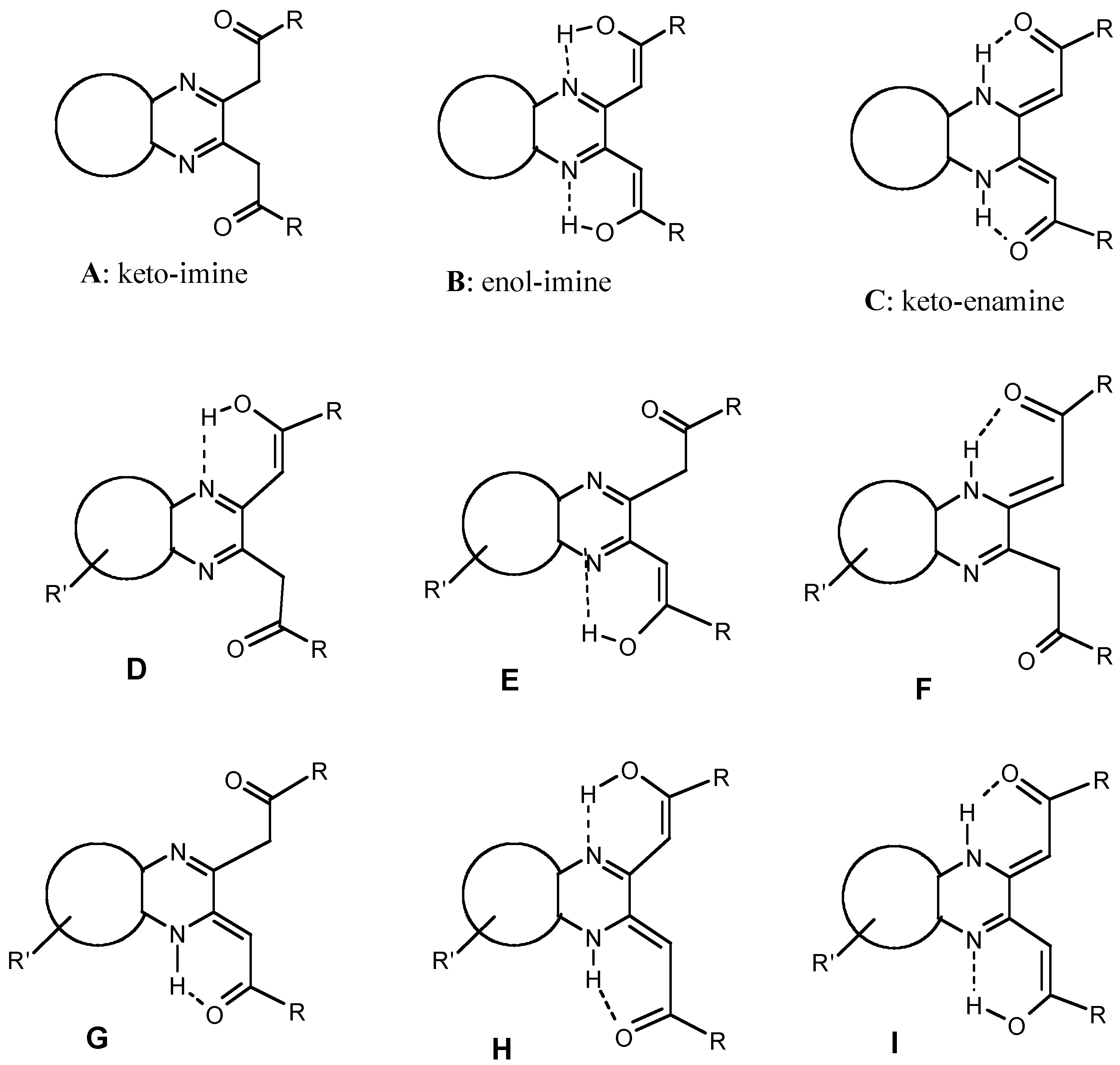
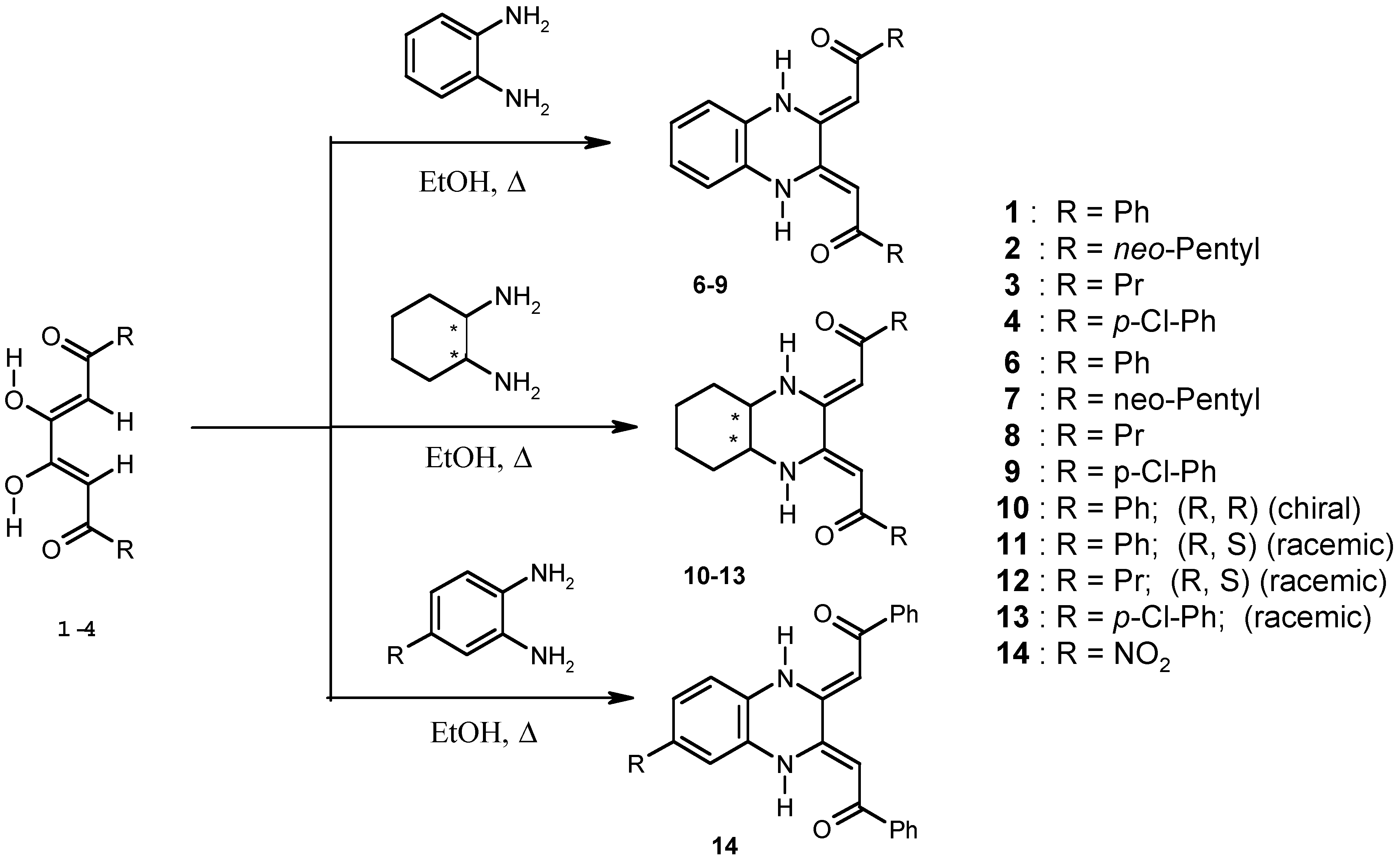
Results and Discussion
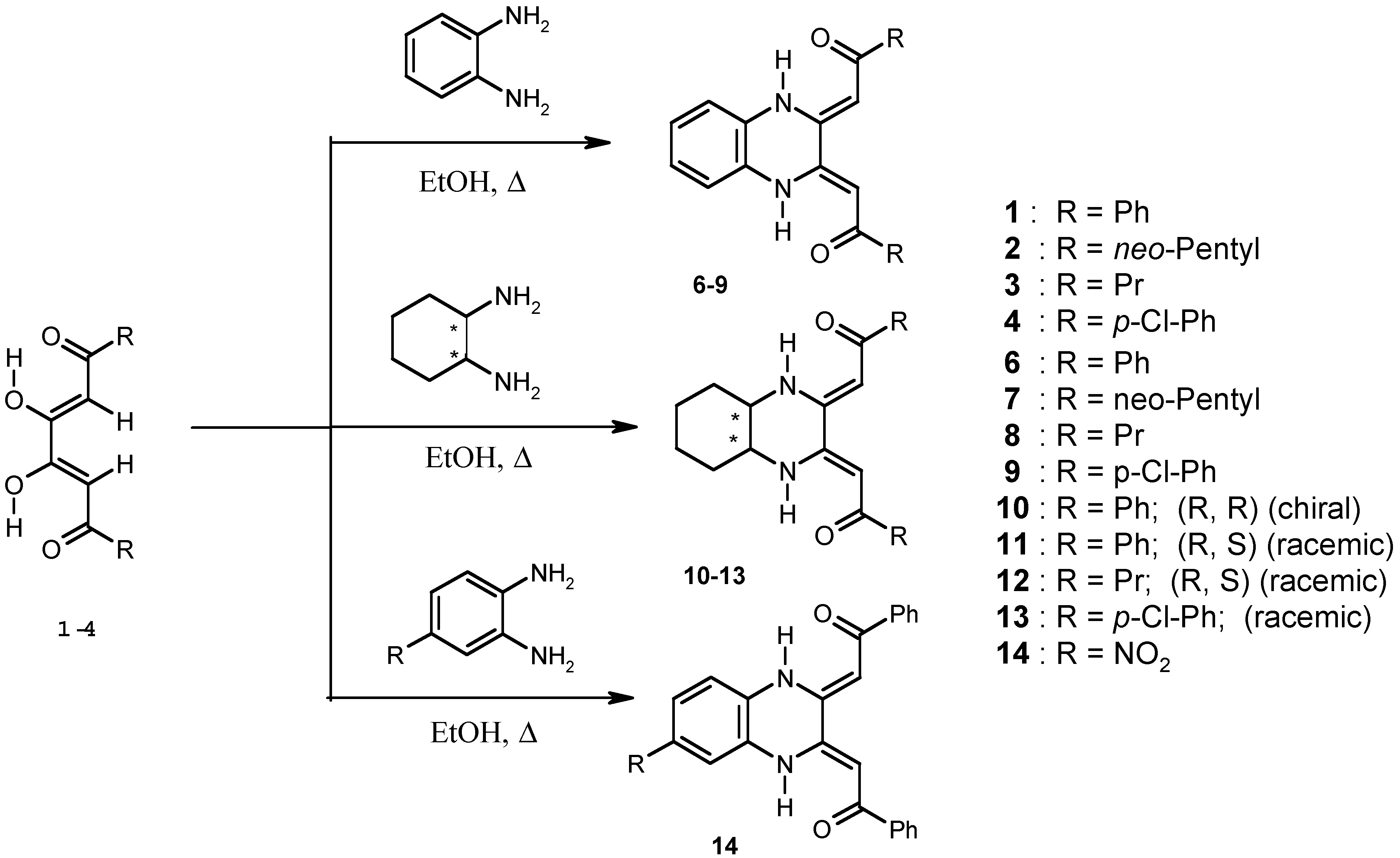
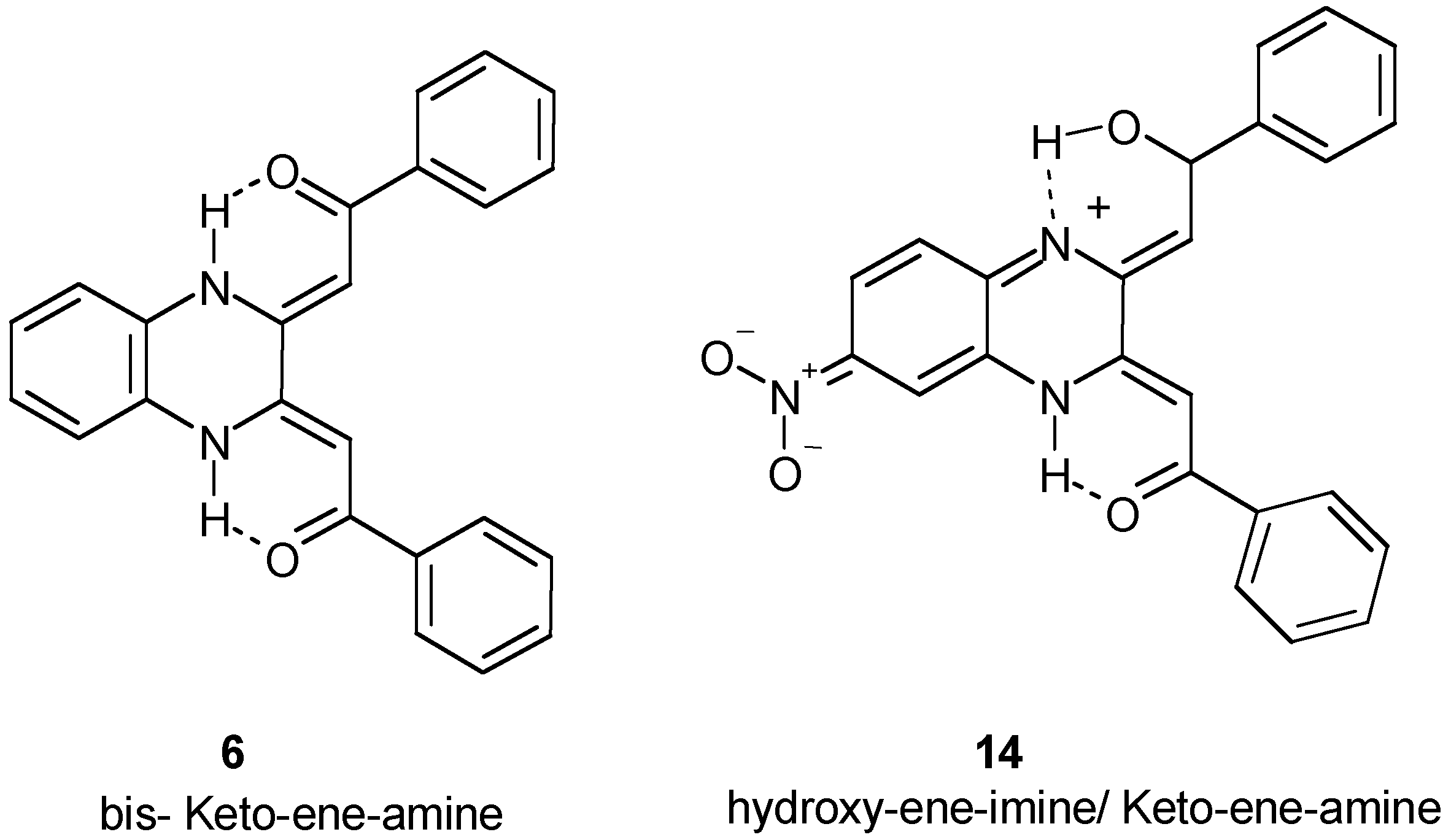
Chemistry
Biological Evaluation
Evaluation of Anti-fungal Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
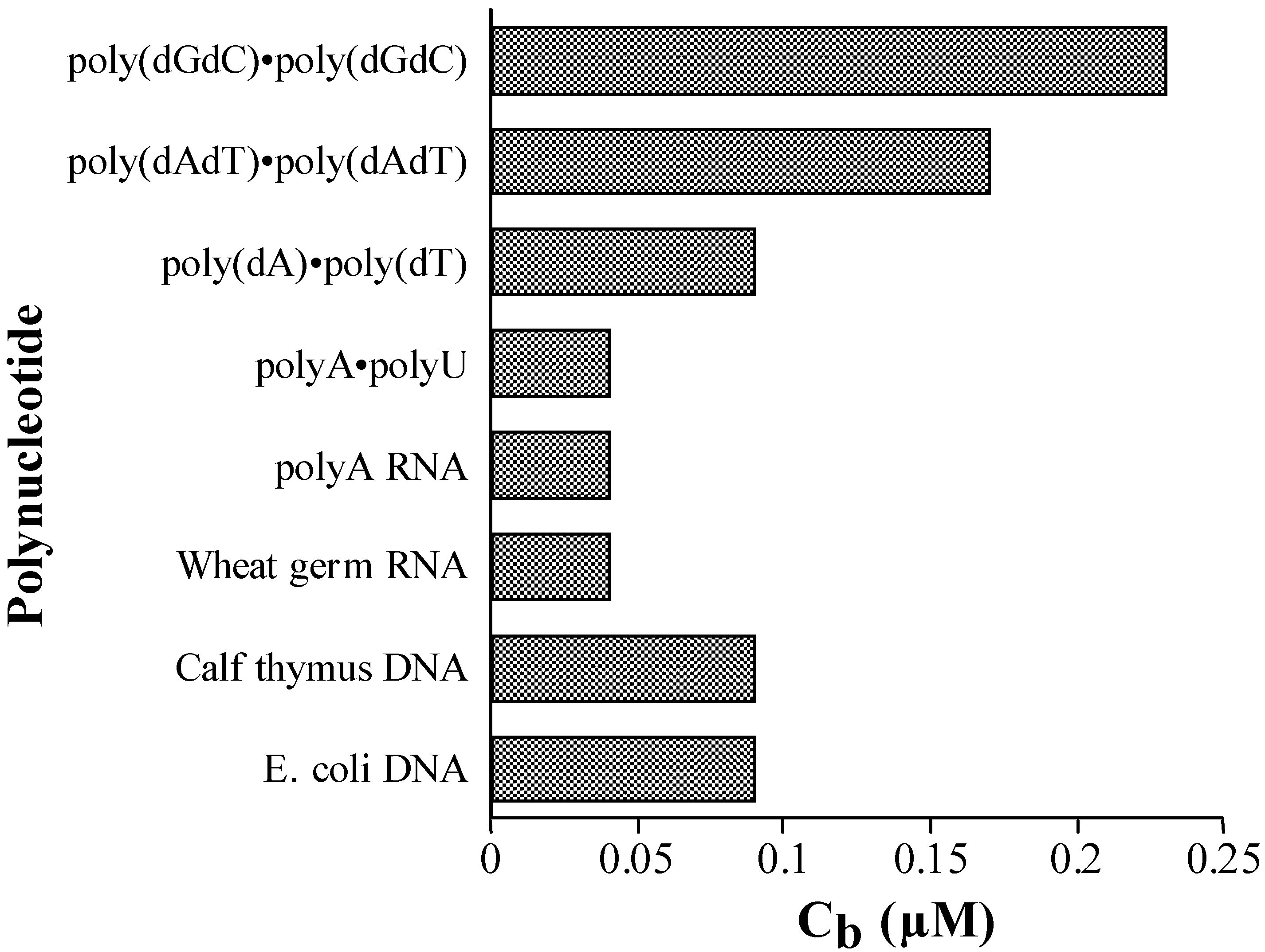{kind=link}
| Compound | Assay | Percent growth inhibition (Concentration, mg/L) | |||
| (C1) | (C2) | (C3) | Assessment | ||
| 6 | Bayoud | 9 (20) | 7 (40) | 22 (80) | Inactive |
| 7 | Bayoud | 9 (60) | 15 (120) | 15 (180) | Inactive |
| 8 | Bayoud | 17 (60) | 17 (120) | 19 (180) | Inactive |
| 9 | Bayoud | 21 (60) | 32 (120) | 35 (180) | Inactive |
| 11 | Bayoud | 15 (34) | 31 (67) | 33 (134) | Inactive |
| 14 | Bayoud | 29 (18) | 31 (36) | 51 (72) | Active |
Evaluation of Anti-tuberculosis Activity in vitro
| Compound | Assay | MIC (μg/mL) | % Inhibition | Activity |
| 6 | Alamar | > 6.25 | 0 | - |
| 7 | Alamar | > 6.25 | 7 | - |
| 9 | Alamar | > 6.25 | 9 | - |
| 10 | Alamar | > 6.25 | 0 | - |
| 11 | Alamar | > 6.25 | 0 | - |
| 14 | Alamar | > 6.25 | 7 | - |
Evaluation of Anti-tumour Activity in vitro
| Compound | Concentration | Growth percentages | |||
| (Lung) NCI H460 | (Breast) MCF7 | (CNS) SF-268 | Assessment | ||
| 7 | 0.1 mM | 98 | 86 | 105 | Inactive |
| 9 | 0.1 mM | 85 | 76 | 95 | Inactive |
| 10 | 0.1 mM | 89 | 84 | 100 | Inactive |
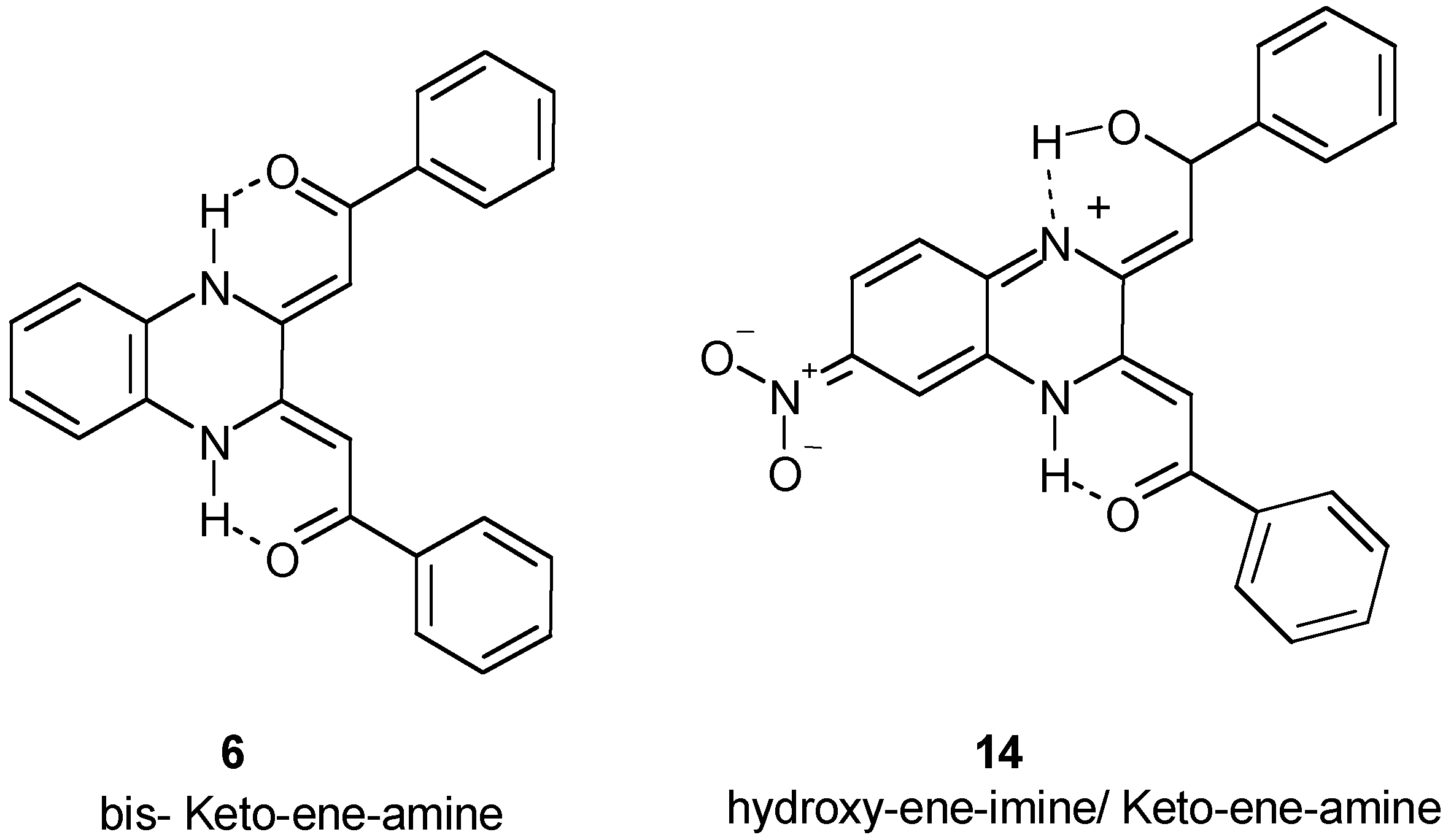
Investigation of binding to DNA

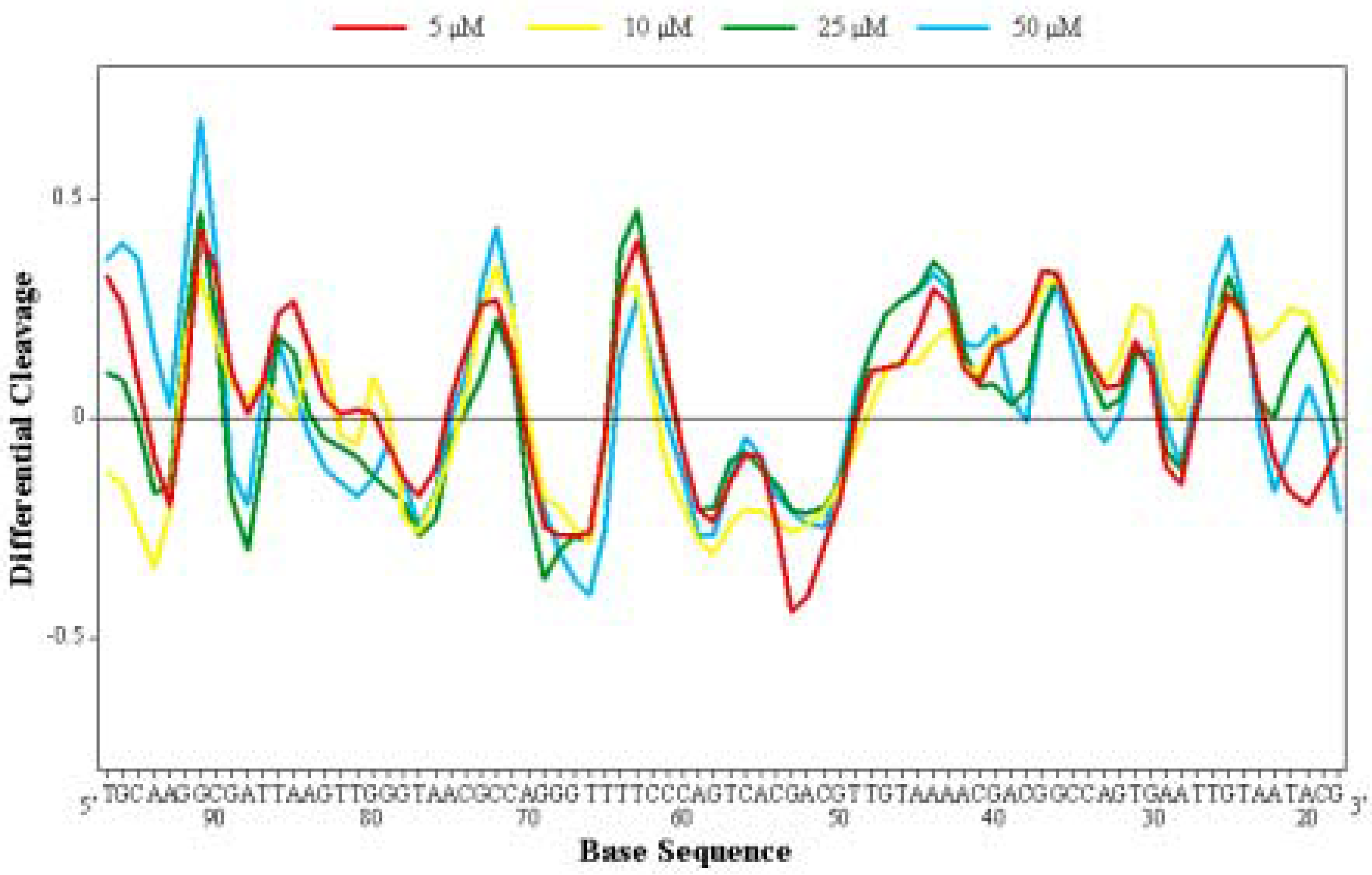
Differential Cleavage Plot
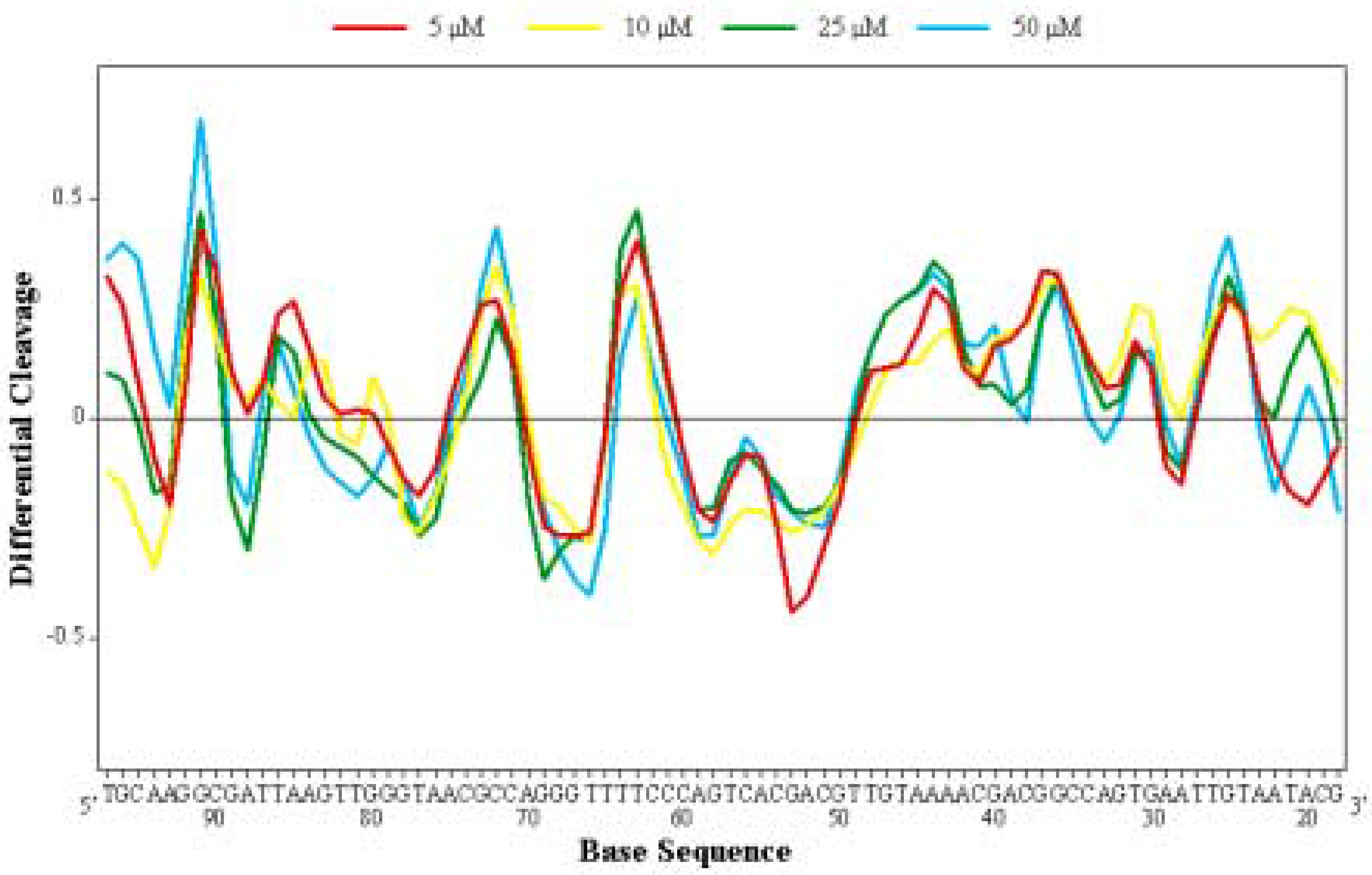
Competition Dialysis
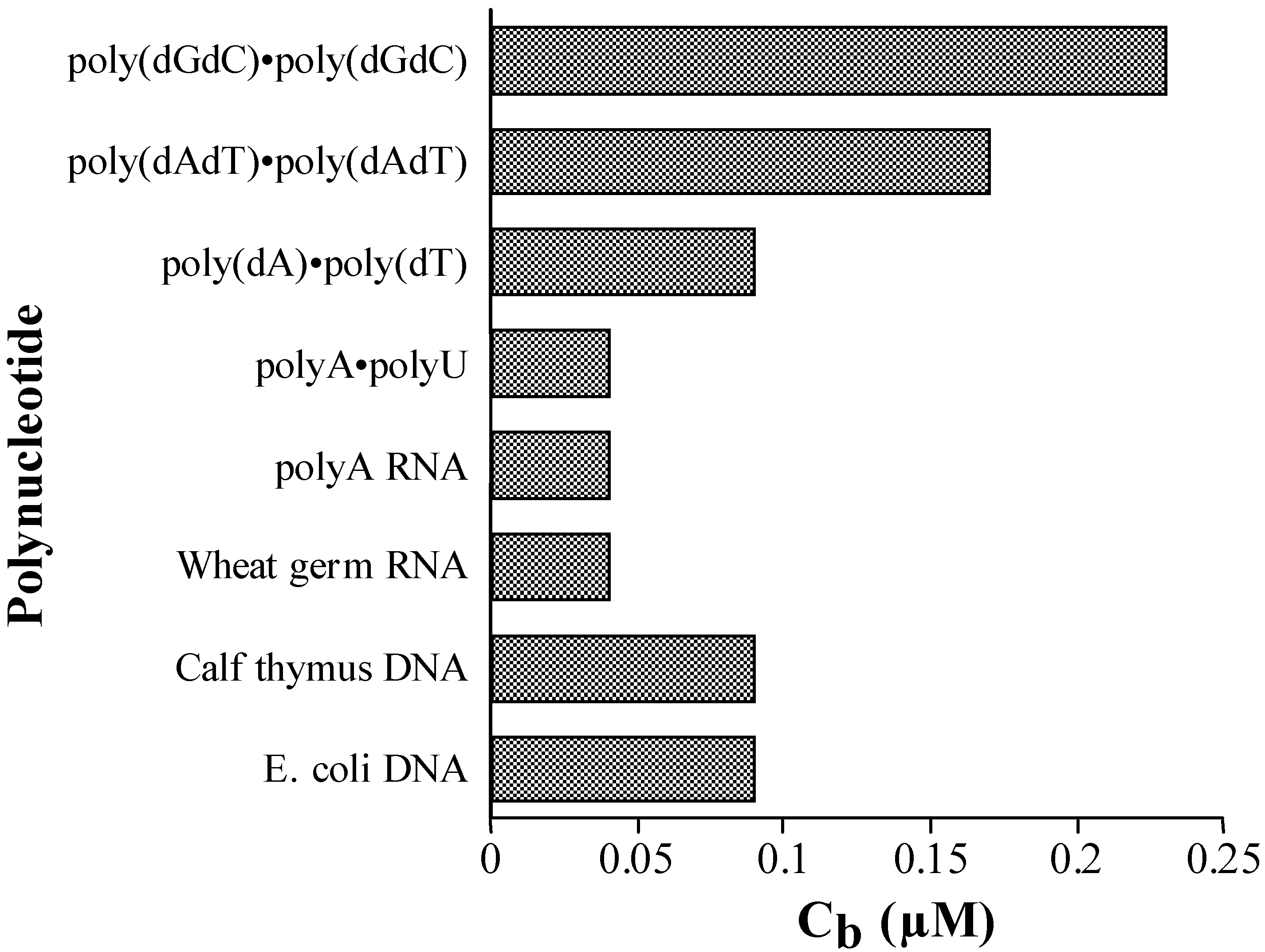
Conclusions
Experimental
General
Biological Evaluation
Synthesis of Compounds 1-4 and 6-14
Acknowledgments
References
- Bailly, C.; Waring, M.J. Biochem. J. 1998, 330, 81–87.
- Addess, K.J.; Feigon, J. Nucleic Acids Res. 1994, 22, 5484–5491.
- Branka, J.E.; Vallette, G.; Jarry, A.; Laboisse, C.L. Biochem. J. 1997, 323, 521–524.
- Balzarini, J.; Karisson, A.; Meichsner, C.; Riess, A.Ps.G.; De Clercq, E.; Kleim, J.P. J. Virol. 1994, 68, 7986–7992.
- Stilwell, W.G.; Turesky, R.J.; Sinha, R.; Skiper, P.L. Tannenbaum S.R. Cancer Lett. 1999, 143, 145–148. [Google Scholar]
- Nallas, G.N.A.; Brewer, K.J. Inorg. Chim. Acta 1996, 253, 7–13.
- Milkevitch, M.; Brauns, E.; Brewer, K.J. Inorg. Chem. 1996, 35, 1737.
- Molnar, S.M.; Nallas, G.; Bridgewater, J.S.; Brewer, K.J. J. Am. Chem. Soc. 1994, 116, 5206–5210.
- El-Bendary, E.R.; El-Ashmawy, M.B.; Barghash, A.M.; Shehata, I.A.; El-Kerdawy, M.M. Boll. Chim. Farm. 1996, 135, 617–620.
- Keeble, J.; Al-Swayeh, O.A.; Moore, P.K. Br. J. Pharmacol. 2001, 133, 1023–1028.
- Lin, S.-K. Molecules 1996, 1, 37–40.
- Finar, I.L. J. Chem. Soc. 1955, 1205–1209.
- Wolfe, J.F.; Portlock, D.E.; Feuerbach, D.J. J. Org. Chem. 1974, 39, 2006–2009.
- Kaiser, E.M.; Petty, J.D. J. Organomet. Chem. 1976, 108, 139–143.
- Lee, B.L.; Yamamoto, T. Macromolecules 1999, 32, 1375–1382.
- Touzani, R.; Ben-Hadda, T.; Elkadiri, S.; Ramdani, A.; Maury, O.; Le Bozec, H.; Toupet, L.; Dixneuf, P.H. New J. Chem. 2001, 25, 391–395.
- Gale, E.F.; Cundliffe, E.; Reynolds, P.E.; Richmond, M.H.; Waring, M.J. The Molecular Basis of Antibiotic Action, 2nd ed.; John Wiley & Sons: London, 1981; pp. 258–401. [Google Scholar]
- Waring, M.J.; Ponder, B.A.J. The Search for New Anticancer Drugs; Kluwer: Dordrecht, 1992. [Google Scholar]
- Waring, M.J. Molecular Aspects of Anticancer Drug-DNA Interactions; Neidle, S., Waring, M.J., Eds.; Macmillan: London, 1993; Vol. 1, pp. 213–242. [Google Scholar]
- Gordon, T.R.; Martyn, R.D. Annu. Rev. Phytopathol. 1997, 35, 111–128.
- Kotchevar, A.T.; Ghosh, P.; Uckun, F.M. J. Phys. Chem. B 1998, 102, 10925–10930.
- Bailly, C.; Crow, S.; Minnock, A.; Waring, M.J. J. Mol. Biol. 1999, 291, 561–573.
- Bailly, C.; Waring, M.J. Nucleic Acids Res. 1998, 26, 4309–4314.
- Bailly, C.; Mollegaard, N.E.; Nielsen, P.E.; Waring, M.J. EMBO J. 1995, 14, 2121–2131.
- Ren, J.; Chaires, J.B. Biochemistry 1999, 38, 16067–16075.
- Ren, J.; Chaires, J.B. Methods in Enzymol 2001, 340, 99–108.
- Collins, L.; Franzblau, S.G. Antimicrob. Agents Chemother 1997, 41, 1004–1009.
- Sample Availability: Not Available.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Waring, M.J.; Ben-Hadda, T.; Kotchevar, A.T.; Ramdani, A.; Touzani, R.; Elkadiri, S.; Hakkou, A.; Bouakka, M.; Ellis, T. 2,3-Bifunctionalized Quinoxalines: Synthesis, DNA Interactions and Evaluation of Anticancer, Anti-tuberculosis and Antifungal Activity. Molecules 2002, 7, 641-656. https://doi.org/10.3390/70800641
Waring MJ, Ben-Hadda T, Kotchevar AT, Ramdani A, Touzani R, Elkadiri S, Hakkou A, Bouakka M, Ellis T. 2,3-Bifunctionalized Quinoxalines: Synthesis, DNA Interactions and Evaluation of Anticancer, Anti-tuberculosis and Antifungal Activity. Molecules. 2002; 7(8):641-656. https://doi.org/10.3390/70800641
Chicago/Turabian StyleWaring, Michael J., Taibi Ben-Hadda, Ann T. Kotchevar, Abdelkrim Ramdani, Rachid Touzani, Sghir Elkadiri, Abdelkader Hakkou, Mohamed Bouakka, and Tom Ellis. 2002. "2,3-Bifunctionalized Quinoxalines: Synthesis, DNA Interactions and Evaluation of Anticancer, Anti-tuberculosis and Antifungal Activity" Molecules 7, no. 8: 641-656. https://doi.org/10.3390/70800641