Experimental section
General
NMR spectra were measured using a Bruker DPX-300 (300 MHz 1H-NMR and 75 MHz 13C-NMR) or DRX-400 (400 MHz 1H-NMR and 100 MHz 13C-NMR) instruments; deuterochloroform was used as solvent and tetramethylsilane as the internal standard; 31P-NMR spectra were measured with Bruker DPX-300 (121.5 MHz) using H3PO4/D2O as external standard. IR spectra were measured with a Perkin-Elmer 1600-FT or Nicolet 5ZDX spectrometers. TLC was performed on precoated silica gel 60 F254 plates (0.25 mm thick, Merck), and for column chromatography silica gel 60 70-230 mesh (Merck) was used. Reported yields refer to samples with the same purity as the samples used in the following step.
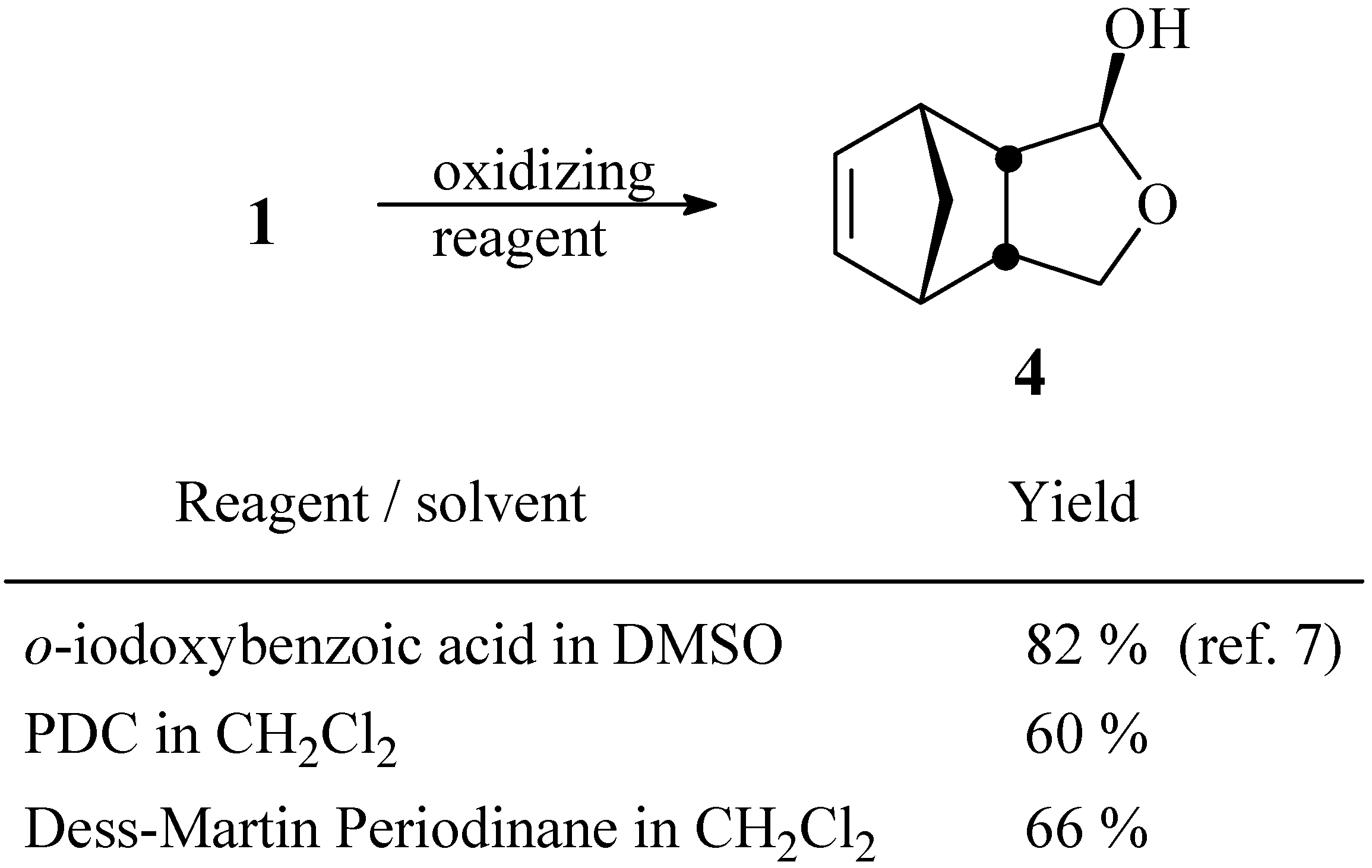
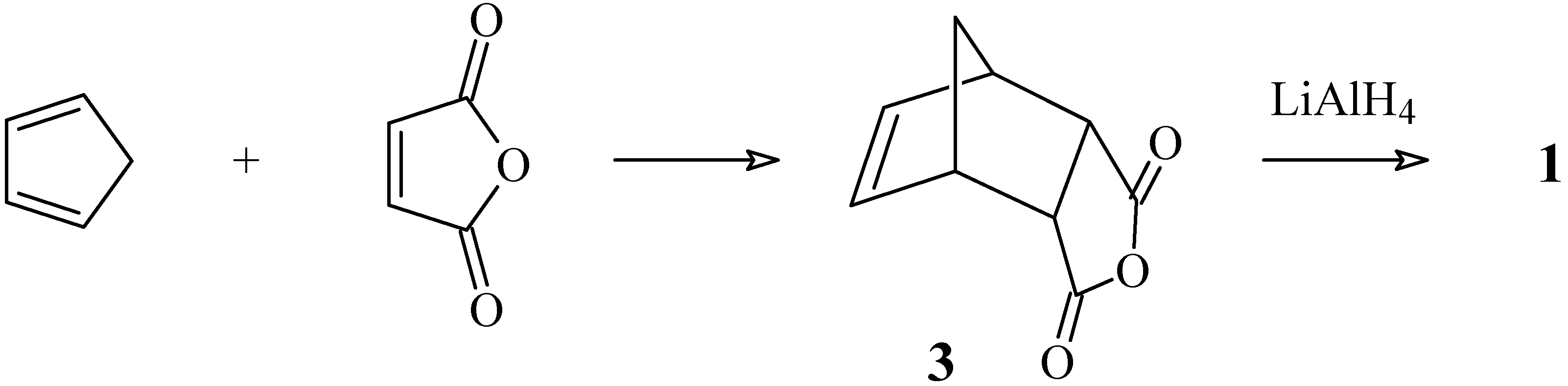
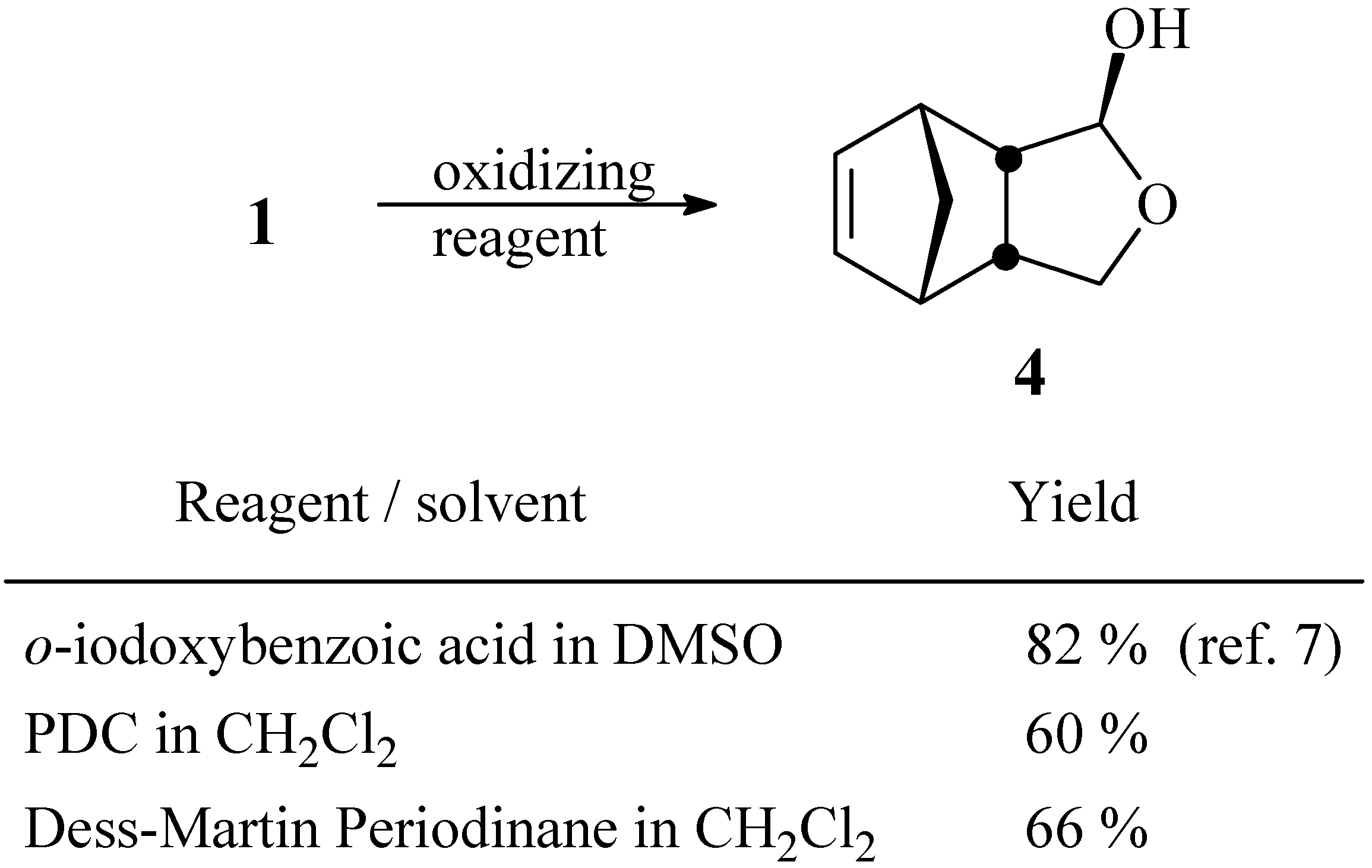
4-Oxatricyclo[5.2.1.02,6]dec-8-en-3-ol (4)
a) Oxidation with PDC. To a solution of pyridinium dichromate (3.0 g, 8.0 mmol) in anhydrous CH2Cl2 (30 mL), maintained under N2 atmosphere, was added a solution of compound 1 (640 mg, 4.1 mmol) in CH2Cl2 (10 mL). After stirring for 24 h at room temperature, the solution was filtered through a small column of MgSO4. The solvent was removed under vacuum and the residue was chromatographed in a column of silica gel using a 4:6 mixture of hexane / ethyl acetate. Yield 374 mg (60 %).
b) Oxidation with Dess-Martin periodinane. To a solution of diol
1 (61 mg, 0.37 mmol) in anhydrous CH
2Cl
2 (1.2 mL), maintained under N
2 atmosphere, was added a solution of Dess-Martin periodinane [
12] (351 mg, 0.80 mmol) in CH
2Cl
2 (3 mL). After stirring at room temperature for 20 min, the reaction mixture was diluted with ethyl ether (10 mL) and quenched with a 1.3 M aqueous solution of NaOH. After 10 min of vigorous stirring the organic phase was separated, washed with aqueous NaOH solution, water, and dried with MgSO
4. The solvent was removed under vacuum and the residue was purified by chromatography as described in (a). Yield 37 mg (66 %).
Spectroscopic data: 1H-NMR (CDCl3, 400 MHz) δ 6.20 (dd, 1 H, J1 = 5.6 Hz, J2 = 3.0 Hz), 6.09 (dd, 1 H, J1 = 5.8 Hz, J2 = 3.0 Hz), 4.98 (s, 1 H), 3.98 (dd, 1 H, J1 = 7.6 Hz, J2 = 8.8 Hz), 3.46 (dd, 1 H, J1 = 8.8 Hz, J2 = 2.0 Hz), 3.03 (m, 1 H), 2.95 (m, 1 H), 2.87 (m, 2 H), 2.63 (br. s, 1 H), 1.45 (dt, 1 H, J1 = 8.3 Hz, J2 = J3 = 1.6 Hz), 1.36 (d, 1 H, J = 8.3 Hz); 13C-NMR (CDCl3, 100 MHz), 134.7 (CH), 134.6 (CH), 100.4 (CH), 69.3 (CH2), 55.6 (CH), 51.9 (CH), 46.0 (CH), 45.9 (CH), 44.9 (CH).
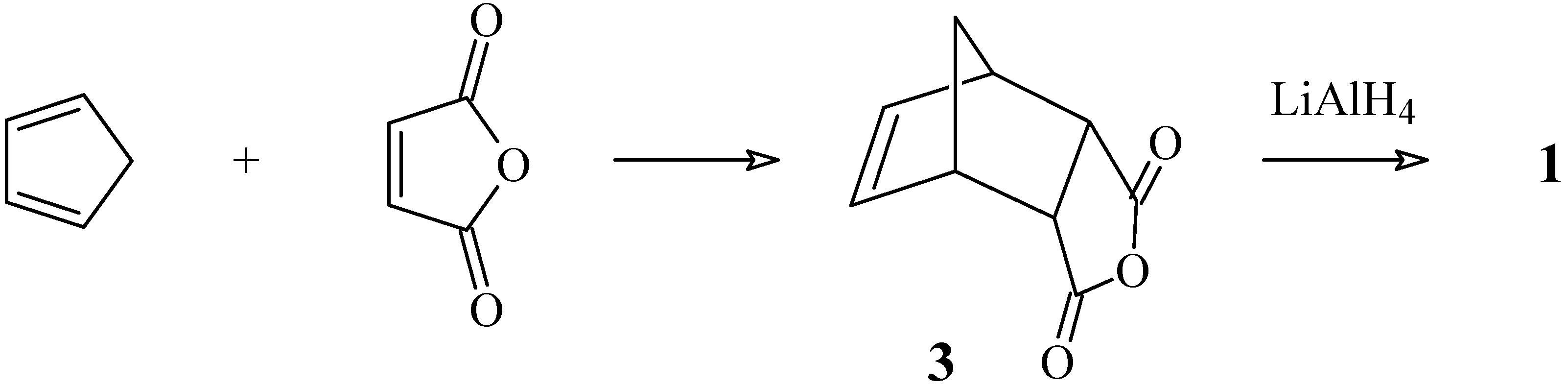
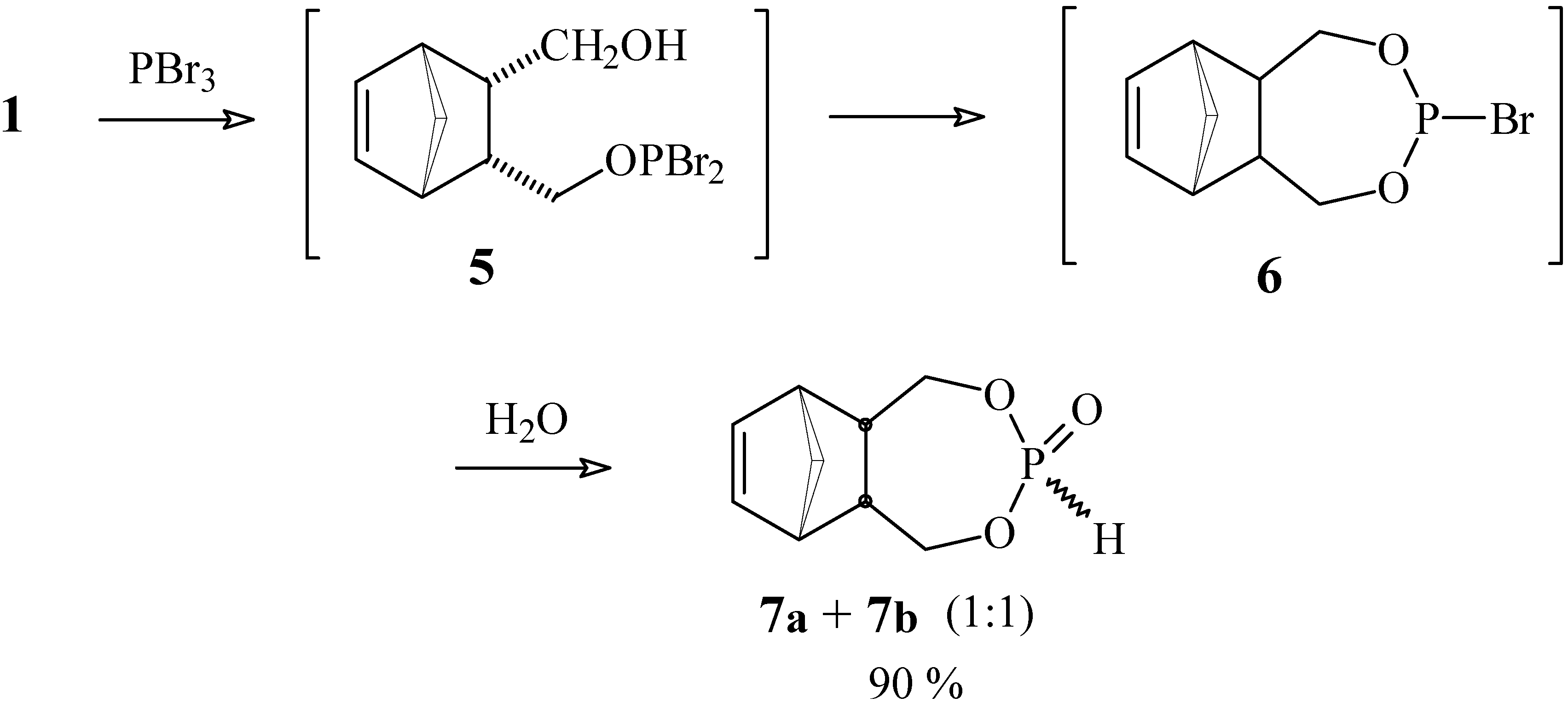
4,6-Dioxa-5-phosphatricyclo[7.2.1.02,8]dodec-10-en-5-one (7a and 7b).
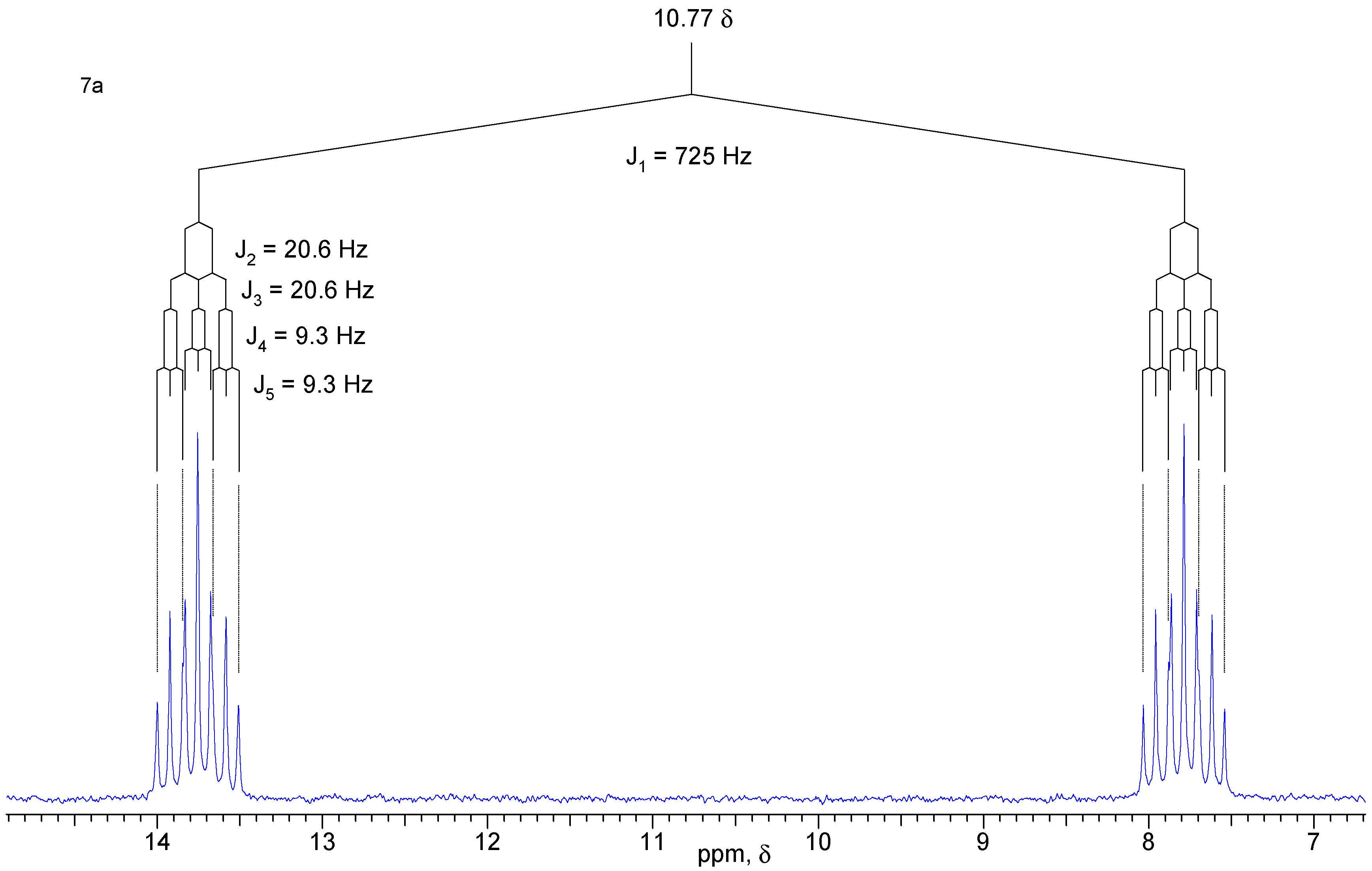
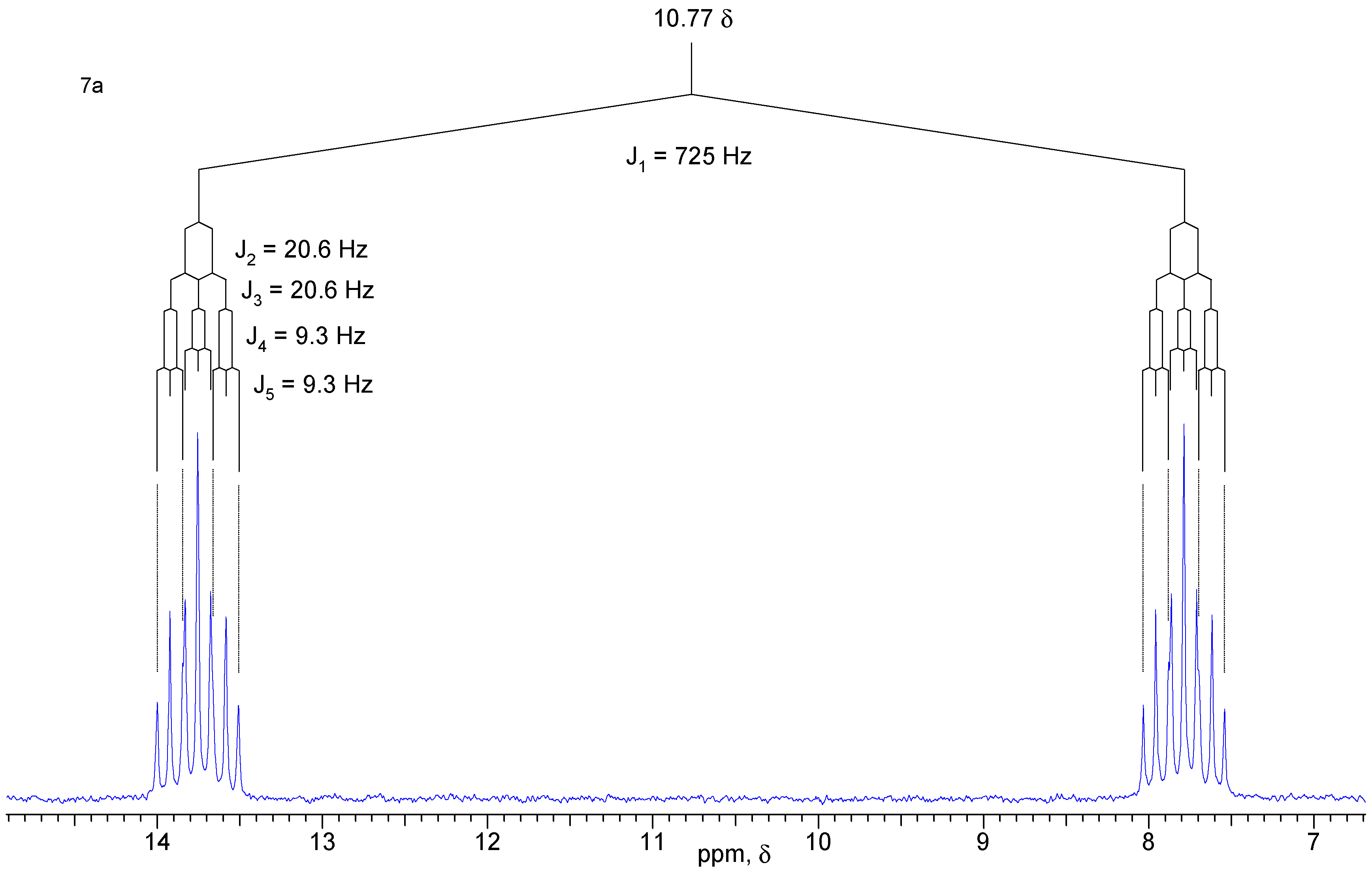
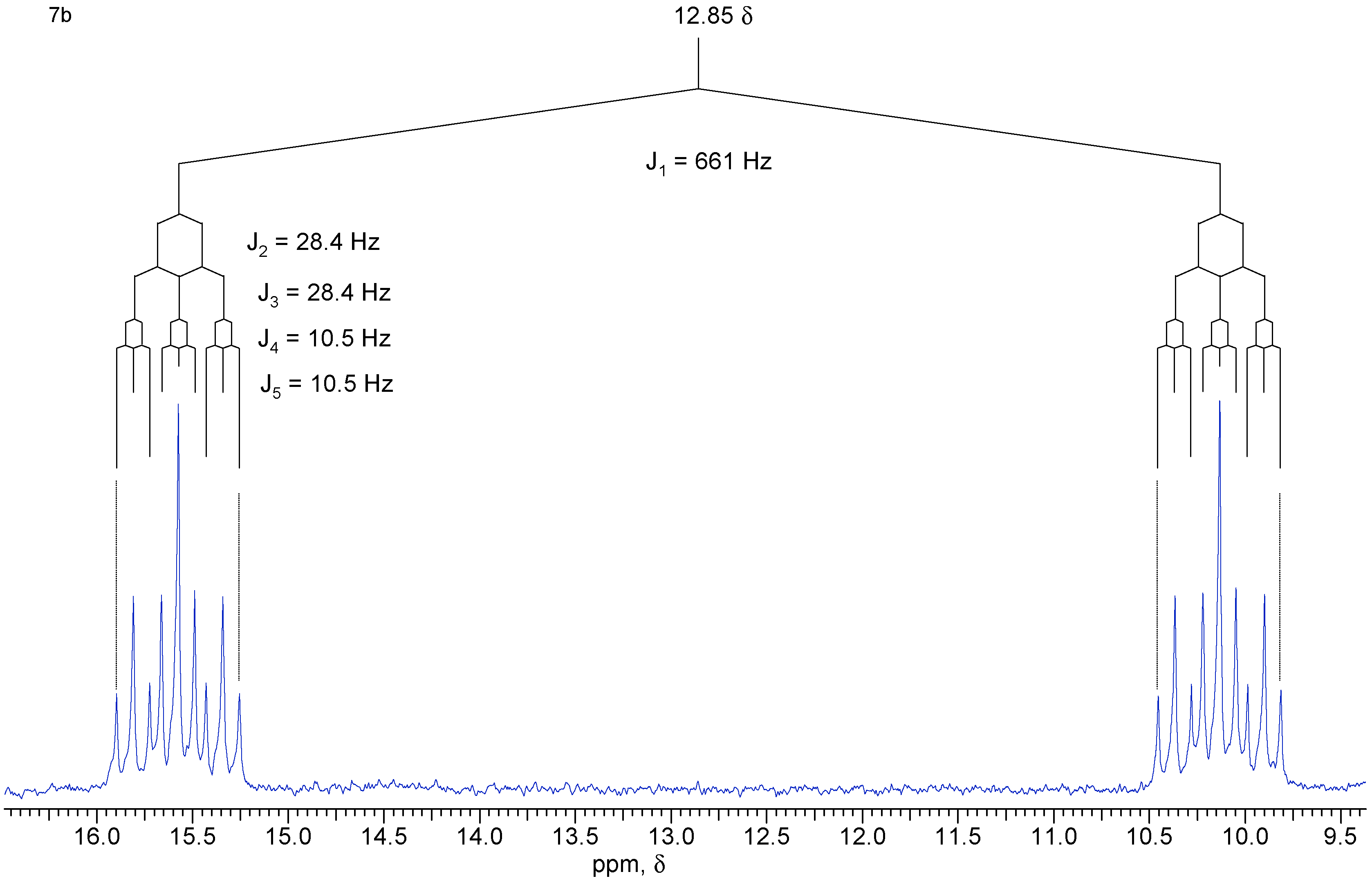
A solution of diol 1 (50 mg, 0.32 mmol) and PBr3 (26 mg, 0.96 mmol) in anhydrous ethyl ether (10 mL) was kept under N2 atmosphere at 5°C for 45 h. After quenching with chopped ice and extraction with ethyl ether, the organic phase was washed with water and saturated brine, and dried with MgSO4. The solvent was removed under vacuum to give 58 mg (90 %) of a mixture of isomers that were separated by chromatography in a silica gel column using a 3:7 mixture of hexane / ethyl acetate as eluent. Both isomers are white crystalline solids. Compound 7a: yield 29 mg (45 %); 1H-NMR (CDCl3, 300 MHz) δ 6.71 (d, 1 H, J [H-P] = 725 Hz), 6.13 (br. t, 2 H, J = 1.6 Hz), 4.16 (dt, 2 H, J1 = J2 = 11.7 Hz, J3 [H-P] = 9.3 Hz), 4.05 (ddd, 2H, J1 = 11.7 Hz, J2 [H-P] = 20.4 Hz, J3 = 3.6 Hz), 2.84 (m, 2 H), 2.81 (m, 2 H), 1.59 (dt, 1 H, J1 = 8.5 Hz, J2 = J3 = 1.7 Hz), 1.53 (br. d, 1 H, J = 8.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 134.9 (CH), 66.0 (CH2, d, JC-P = 6.9 Hz), 51.2 (CH2), 45.7 (CH), 44.7 (CH); 31P-NMR (CDCl3, 121.5 MHz) δ 10.77 (dtt, J1 = J2 = 9.3 Hz, J3 = J4 = 20.6 Hz, J5 = 725 Hz). Compound 7b: yield 29 mg (45 %) 1H-NMR (CDCl3, 300 MHz) δ 6.73 (d, 1H, J [H-P] = 660 Hz), 6.13 (t, 2H, J = 1.9 Hz), 4.14 (ddd, 2H, J1 [H-P] = 28.4 Hz, J2 = 12.0 Hz, J3 = 3.0 Hz), 3.68 (dt, 2H, J1 = J2 = 12.0 Hz, J3 [H-P] = 10.7 Hz), 2.88 (m, 2H), 2.83 (m, 2H), 1.59 (dt, 1H, J1 = 8.5 Hz, J2 = J3 = 1.7 Hz), 1.50 (br. d, 1H, J = 8.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 135.0 (CH), 65.5 (CH2, d, JC-P = 5.1 Hz), 50.6 (CH2), 45.7 (CH), 42.8 (CH); 31P-NMR (CDCl3, 121.5 MHz) δ 12.85 (dtt, J1 = J2 = 10.5 Hz, J3 = J4 = 28.4 Hz, J5 = 660 Hz); IR (KBr) νmax 2966, 2389, 1267, 1047, 797 cm-1.
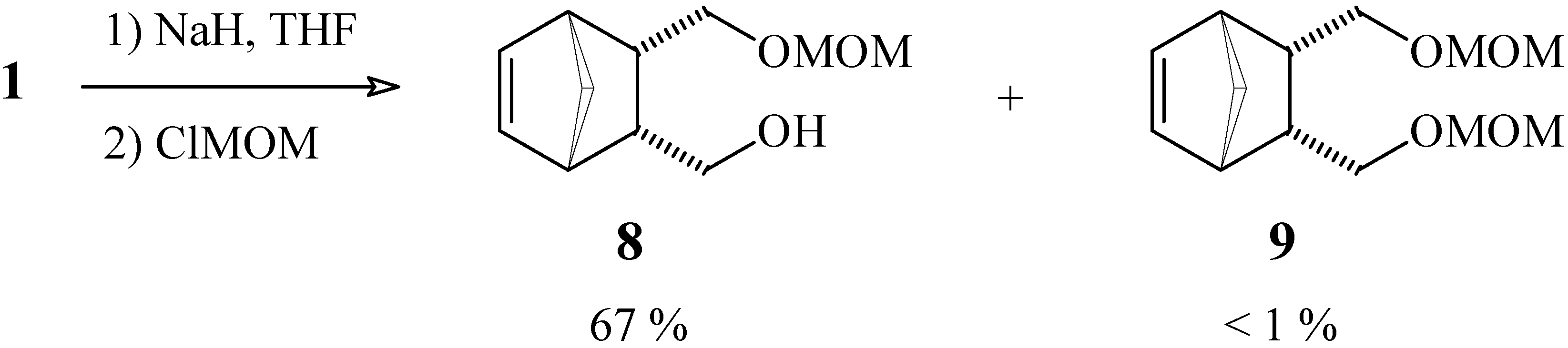
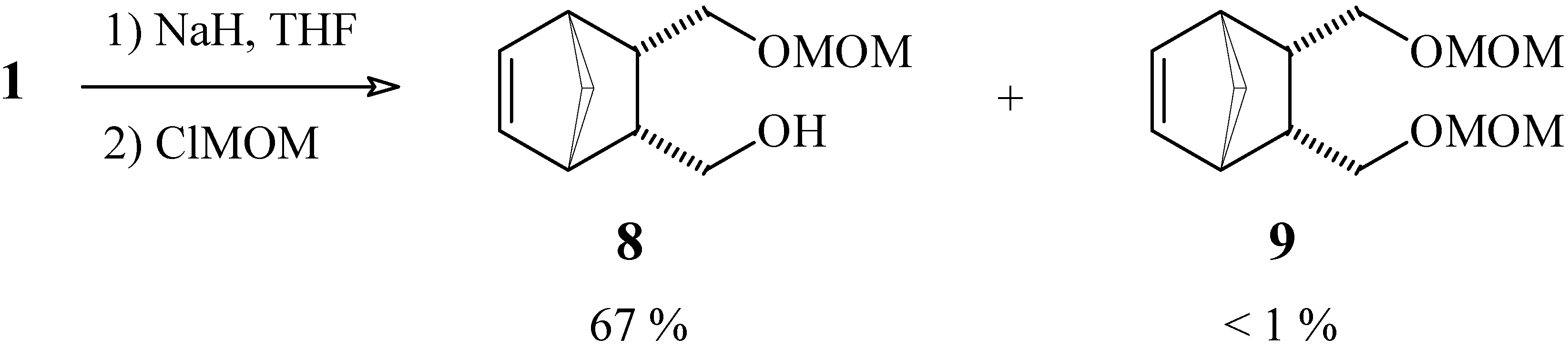
{3-[(Methoxymethoxy)methyl]bicyclo[2.2.1]hept-5-en-2-yl}methan-1-ol (8) and (methoxy-methoxy)-{3-[(methoxymethoxy)methyl]bicyclo[2.2.1]hept-5-en-2-yl}methane (9).
To a suspension of NaH (29 mg of a 60 % suspension in mineral oil, corresponding to 1.2 mmol), previously washed with anhydrous hexane, in anhydrous THF (10 mL), maintained at 0°C under N2 atmosphere, was added a solution of diol 1 (103 mg, 0.66 mmol) in THF (1 mL). After 5 min the ice bath was removed and the reaction mixture was stirred for 1 h. Chloromethyl methyl ether (80 mg, 1.0 mmol) was then added and the mixture was stirred for a further 1 h period. After quenching with chopped ice, the product was extracted with ethyl ether, the organic phase was dried with MgSO4 and the solvent was removed under vacuum. The crude product was purified by column chromatography in silica gel, using a 7:3 mixture of hexane and ethyl acetate as eluent. Compound 8: yield 88 mg (67 %); 1H-NMR (CDCl3, 300 MHz) δ 6.10 (dd, 1H, J1 = 5.5 Hz, J2 = 2.6 Hz), 6.06 (dd, 1H, J1 = 5.5 Hz, J2 = 2.6 Hz), 4.60 (s, 2H), 3.42 (m, 4H), 3.37 (s, 3H), 2.85 (m, 2H), 2.55 (m, 2H), 1.45 (dt, 1H, J1 = 8.3 Hz, J2 = J3 = 1.9 Hz), 1.37 (dt, 1H, J1 = 8.3 Hz, J2 = J3 = 1.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 135.1 (CH), 134.8 (CH), 96.6 (CH2), 68.9 (CH2), 63.0 (CH2), 55.6 (CH3), 49.7 (CH2), 46.43 (CH), 46.38 (CH), 45.4 (CH), 41.9 (CH); IR (KBr) νmax 3421, 2934, 1383, 1157, 1111, 1045 cm-1. Compound 9: yield 1 mg (< 1 %); this sample was identical (by 1H-NMR) to other samples obtained in higher yields by other methods, from which come some of the spectral data that follow: 1H-NMR (CDCl3, 300 MHz) δ 6.14 (t, 2H, J = 1.9 Hz), 4.58 (d, 2H, J = 8.4 Hz), 4.56 (d, 2H, J = 8.4 Hz), 3.38 (m, 2H), 3.35 (s, 6H), 3.13 (t, 2H, J = 9.1 Hz), 2.95 (m, 2H), 2.48 (m, 2H), 1.50 (dt, 1H, J1 = 8.3 Hz, J2 = J3 = 1.9 Hz), 1.35 (dt, 1H, J1 = 8.3 Hz, J2 = J3 = 1.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 135.4 (CH), 96.5 (CH2), 67.9 (CH2), 55.2 (CH3), 49.0 (CH2), 45.6 (CH), 41.5 (CH); IR (KBr) νmax 2991, 1158, 1111, 1055 cm-1.
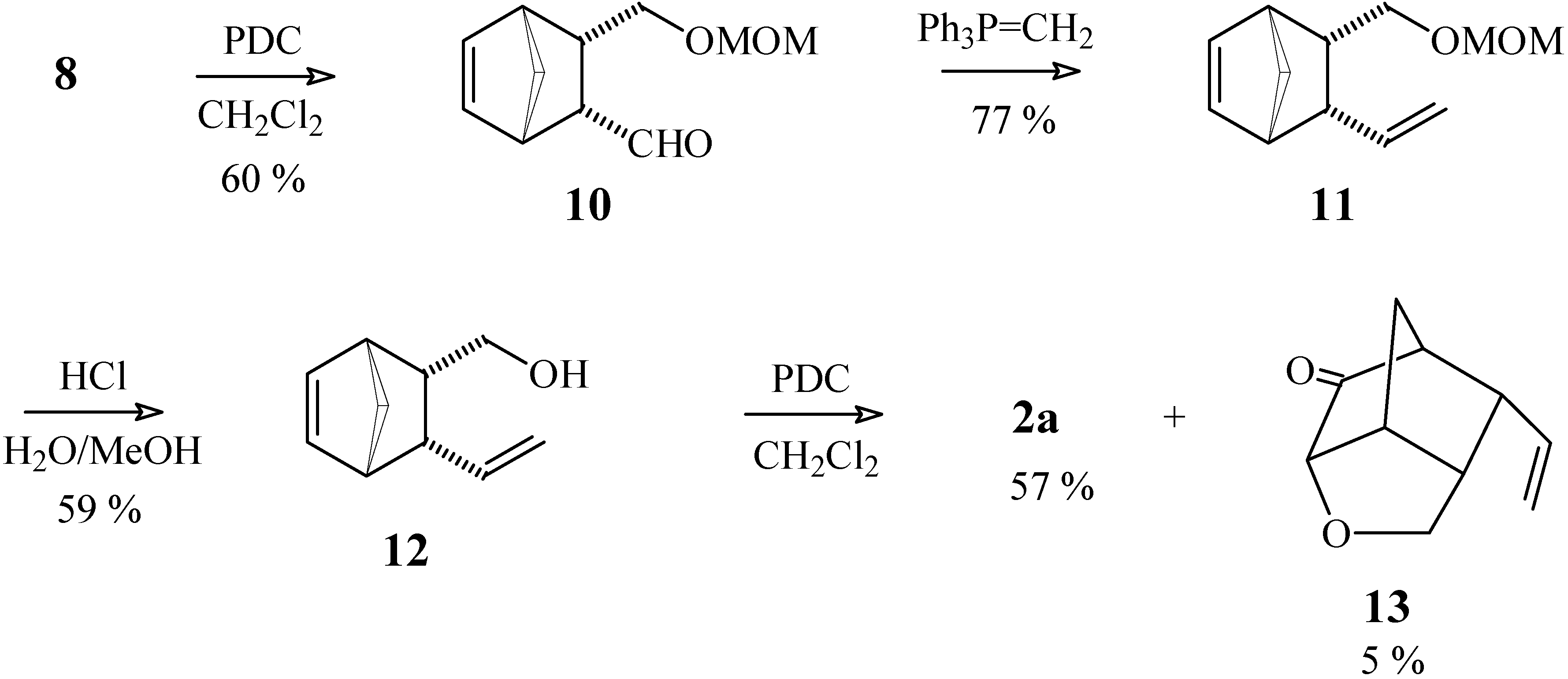
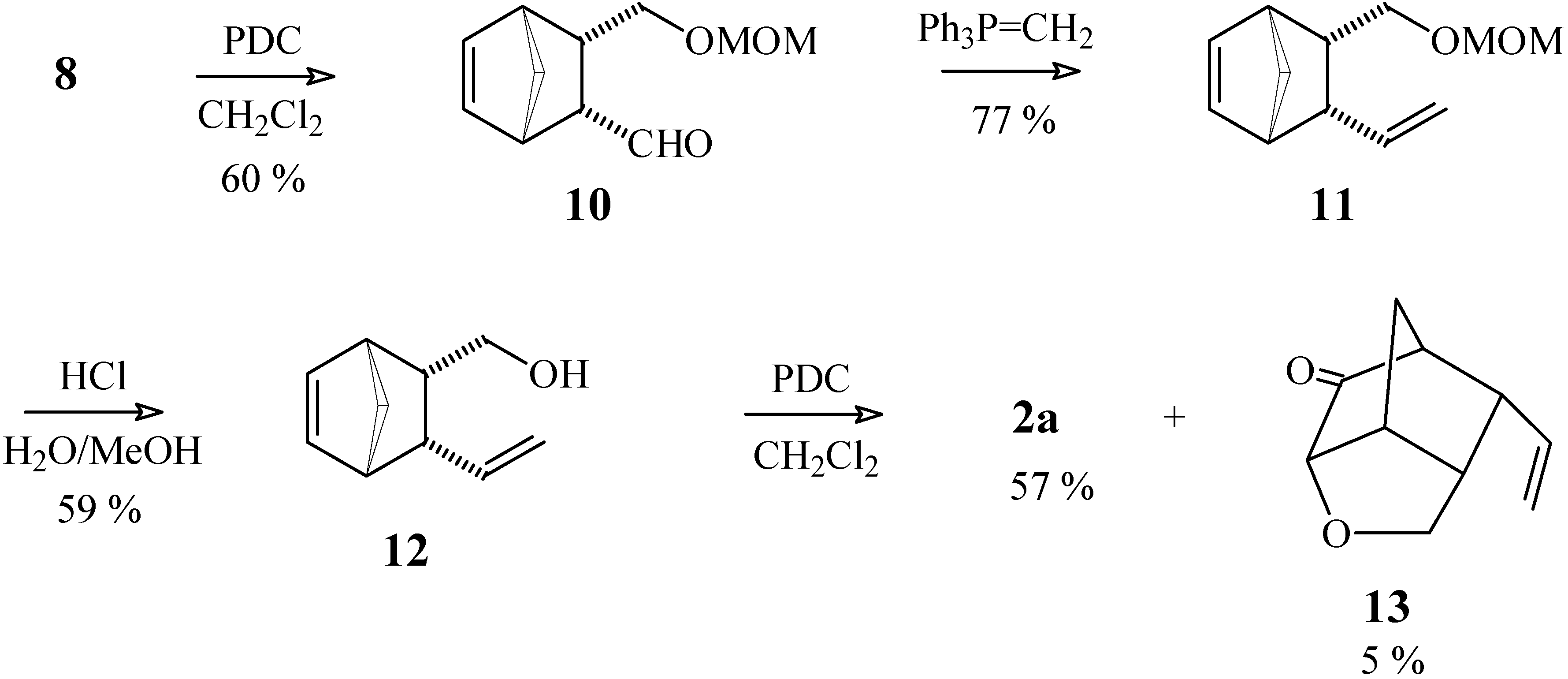
3-[(Methoxymethoxy)methyl]bicyclo[2.2.1]hept-5-ene-2-carbaldehyde (10).
To a solution of pyridinium dichromate (113 mg, 0.30 mmol) in anhydrous CH2Cl2 (3 mL), maintained under N2 atmosphere, was added a solution of compound 8 (39 mg, 0.19 mmol) in CH2Cl2 (3 mL). After stirring for 24 h at room temperature, the product was extracted and purified as described in experiment 1a. Yield 22 mg (60 %). 1H-NMR δ (CDCl3, 300 MHz) 9.41 (d, 1H, J = 3.9 Hz), 6.33 (dd, 1H, J1 = 5.7 Hz, J2 = 2.7 Hz), 6.20 (dd, 1H, J1 = 5.7 Hz, J2 = 3.0 Hz), 4.54 (s, 3H), 3.38 (d, 2H, J = 7.8 Hz), 3.32 (s, 3H), 3.10 (m, 1H), 3.01 (m, 1H), 2.97 (m, 1H), 2.83 (m, 1H), 1.55 (dt, 1H, J1 = 8.7 Hz, J2 = J3 = 2.0 Hz), 1.40 (dt, 1H, J1 = 8.7 Hz, J2 = J3 = 1.5 Hz); 13C-NMR δ (CDCl3, 75 MHz) 204.6 (C=O), 135.03 (CH), 134.95 (CH), 96.1 (CH2), 67.9 (CH2), 54.9 (CH or CH3), 54.3 (CH3 or CH), 49.1 (CH2), 45.7 (CH2), 44.9 (CH), 44.8 (CH); IR (KBr) νmax 2935, 2740, 1713, 1391 cm-1.
Methoxy[(3-vinylbicyclo[2.2.1]hept-5-en-2-yl)methoxy]methane (11)
To a mixture of the phosphonium salt Ph3PCH3Br (207 mg, 0.58 mmol) and THF (5 mL), cooled to 0°C under a N2 atmosphere, was added a solution of n-BuLi (0.35 mL of a 1.17 M solution in hexane, 0.67 mmol). After stirring for 15 min at 0°C, a solution of compound 10 (56 mg, 0.29 mmol) in THF (1 mL) was added. The ice bath was removed and the reaction mixture was stirred at room temperature for 22 h. After quenching with chopped ice and extracting with ethyl ether, the resulting organic solution was washed with water and saturated brine, and dried with MgSO4. The solvent was removed under vacuum and the residue was purified by column chromatography (silica gel), eluting with a 7:3 mixture of hexane / ethyl acetate. Yield 43 mg (77 %). 1H-NMR δ (CDCl3, 300 MHz) 6.18 (t, 2H, J = 1.1 Hz), 5.30 (dt, 1H, J1 = 16.9 Hz, J2 = J3 = 9.9 Hz), 5.07 (ddd, 1H, J1 = 16.9 Hz, J2 = 2.5 Hz, J3 = 0.6 Hz), 4.90 (ddd, 1H, J1 = 9.9 Hz, J2 = 2.6 Hz, J3 = 0.4 Hz), 4.58 (d, 1H, J = 6.4 Hz), 4.55 (d, 1H, J = 6.4 Hz), 3.34 (s, 3H), 3.29 (dd, 1H, J1 = 9.6 Hz, J2 = 5.6 Hz), 3.04 (dd, 1H, J1 = 9.6 Hz, J2 = 10.5 Hz), 2.98 (m, 1H), 2.86 (dt, 1H, J1 = J2 = 9.9 Hz, J3 = 3.4 Hz), 2.79 (m, 1H), 2.46 (dddd, 1H, J1 = 10.5 Hz, J2 = 9.9 Hz, J3 = 5.6 Hz, J4 = 3.4 Hz), 1.49 (dt, 1H, J1 = 8.1 Hz, J2 = J3 = 1.5 Hz), 1.35 (dt, 1H, J1 = 8.1 Hz, J2 = J3 = 1.5 Hz); 13C-NMR δ (CDCl3, 75 MHz) 139.6 (CH), 135.8 (CH), 135.3 (CH), 115.9 (CH2), 96.5 (CH2), 69.0 (CH2), 55.1 (CH3), 49.0 (CH2), 48.2 (CH), 46.9 (CH), 45.2 (CH), 43.6 (CH); IR (KBr) νmax 2924, 1634, 1453, 1151, 1041, 912, 732 cm-1.
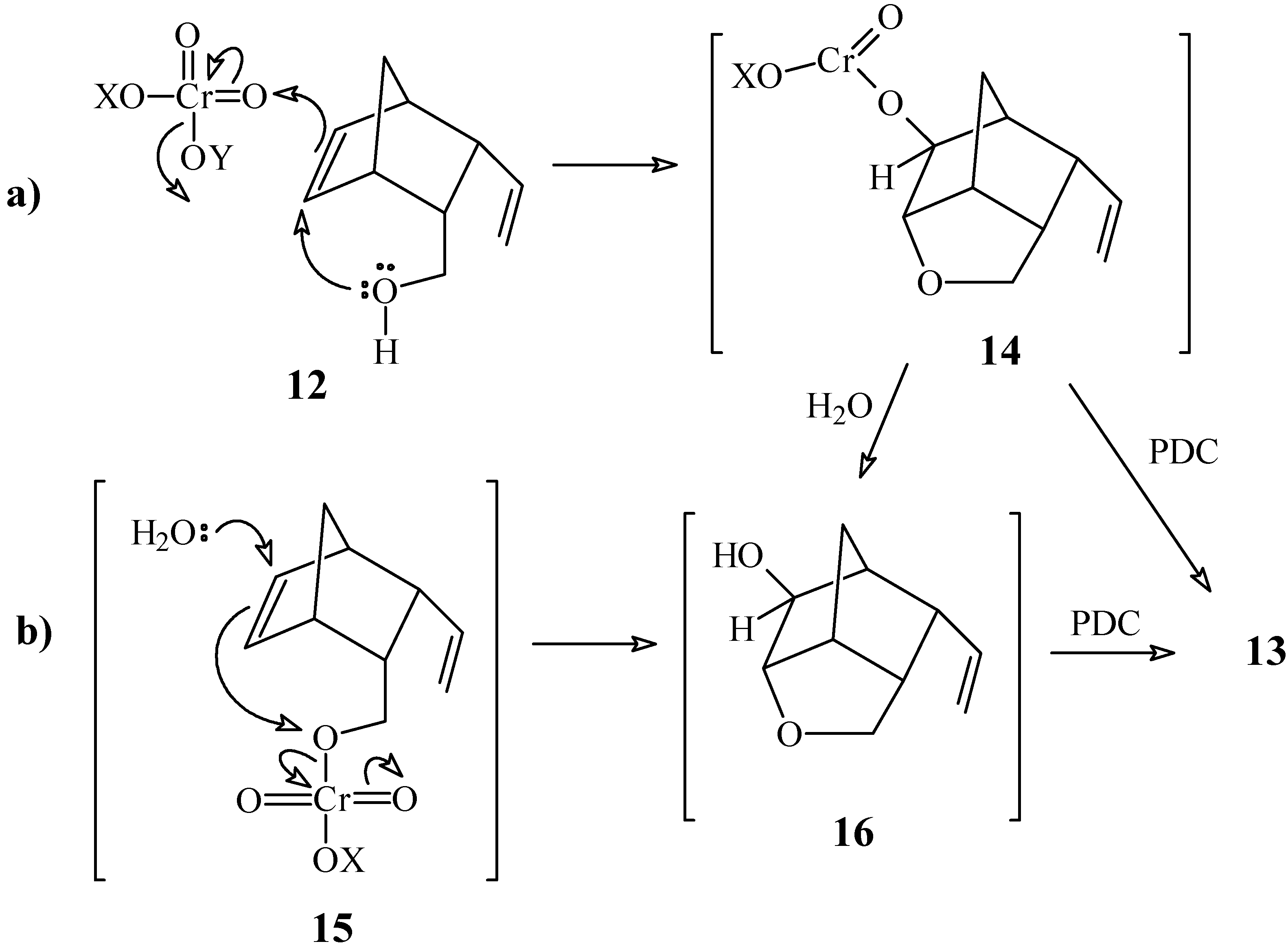
(3-Vinylbicyclo[2.2.1]hept-5-en-2-yl)methan-1-ol (12).
To a solution of compound 11 (2.41 g, 12.4 mmol) in MeOH (70 mL) were added a few drops of concentrated HCl. The reaction mixture was heated to 60°C for 3 h (following the disappearance of the starting material by TLC) and then most of the MeOH was removed under vacuum. The product was extracted with ethyl ether, the organic solution was washed with NaHCO3 solution and dried with MgSO4. The solvent was removed under vacuum and the residue was purified by chromatography on a silica gel column, using a 7:3 mixture of hexane / ethyl acetate as eluent. Yield 1.10 g (59 %). 1H-NMR δ (CDCl3, 300 MHz) 6.17 (m, 2H), 5.34 (dt, 1H, J1 = 17.0 Hz, J2 = J3 = 10 Hz), 5.11 (dd, 1H, J1 = 17.0 Hz, J2 = 2.4 Hz), 4.94 (dd, 1H, J1 = 10 Hz, J2 = 2.4 Hz), 3.37 (dd, 1H, J1 = 10 Hz, J2 = 6.6 Hz), 3.15 (dt, 1H, J1 = J2 = 10 Hz, J3 = 3.5 Hz), 3.02 (br. s, 1H), 2.94 (m, 1H), 2.85 (dt, 1H, J1 = J2 = 10 Hz, J3 = 3.4 Hz), 2.78 (m, 1H), 2.40 (ddt, 1H, J1 = J2 = 10 Hz, J3 = 6.6 Hz, J4 = 3.4 Hz), 1.49 (dt, 1H, J1 = 8.4 Hz, J2 = J3 = 1.8 Hz), 1.35 (d, 1H, J = 8.4 Hz); 13C-NMR δ (CDCl3, 75 MHz) 139.9 (CH), 135.6 (CH), 135.3 (CH), 116.1 (CH2), 63.4 (CH2), 49.4 (CH2), 48.9 (CH), 46.8 (CH), 46.6 (CH), 44.9 (CH); IR (KBr) νmax 3329, 2966, 1636, 1451, 1342, 910, 732 cm-1.
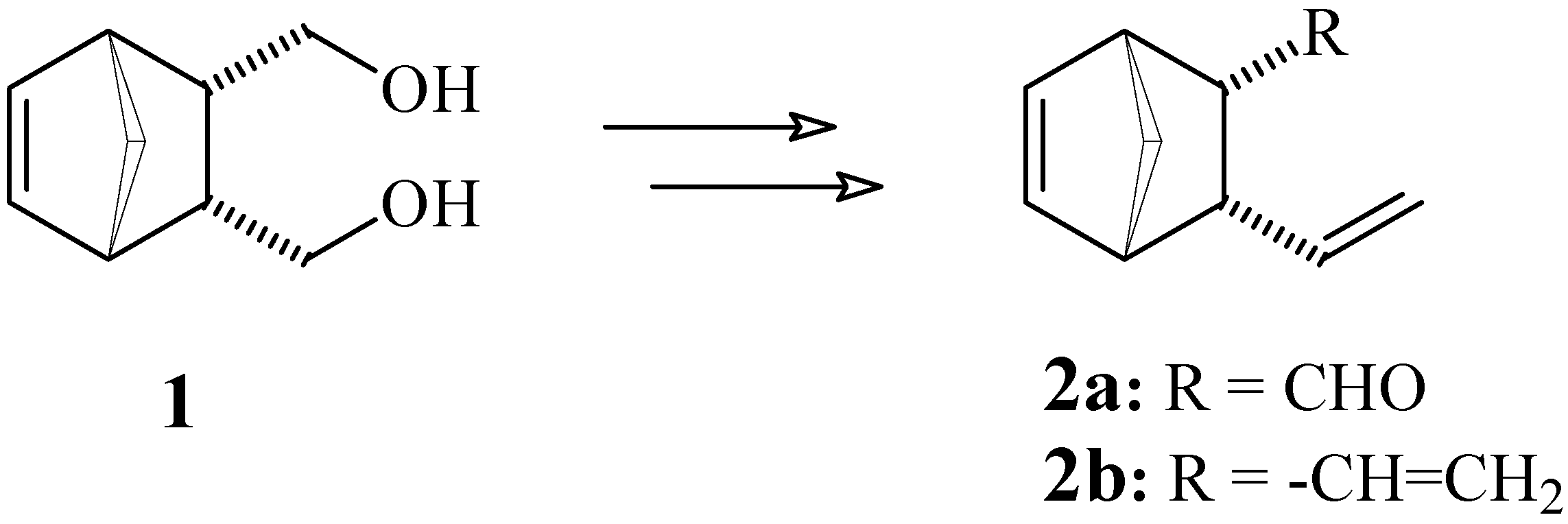
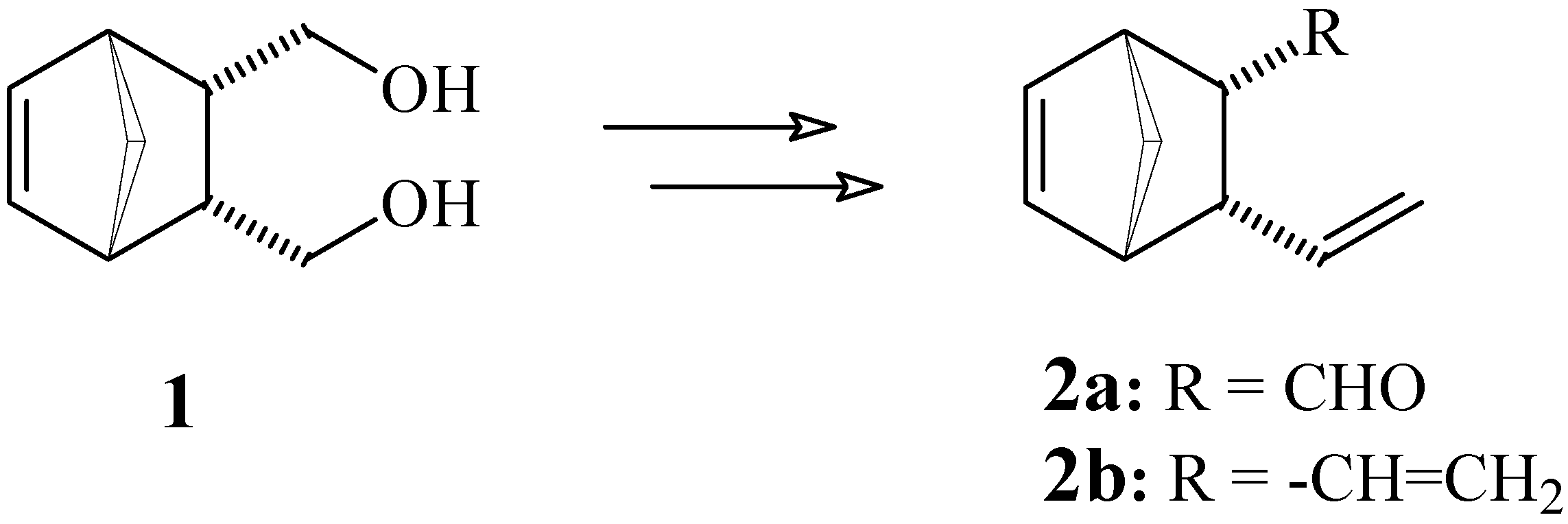
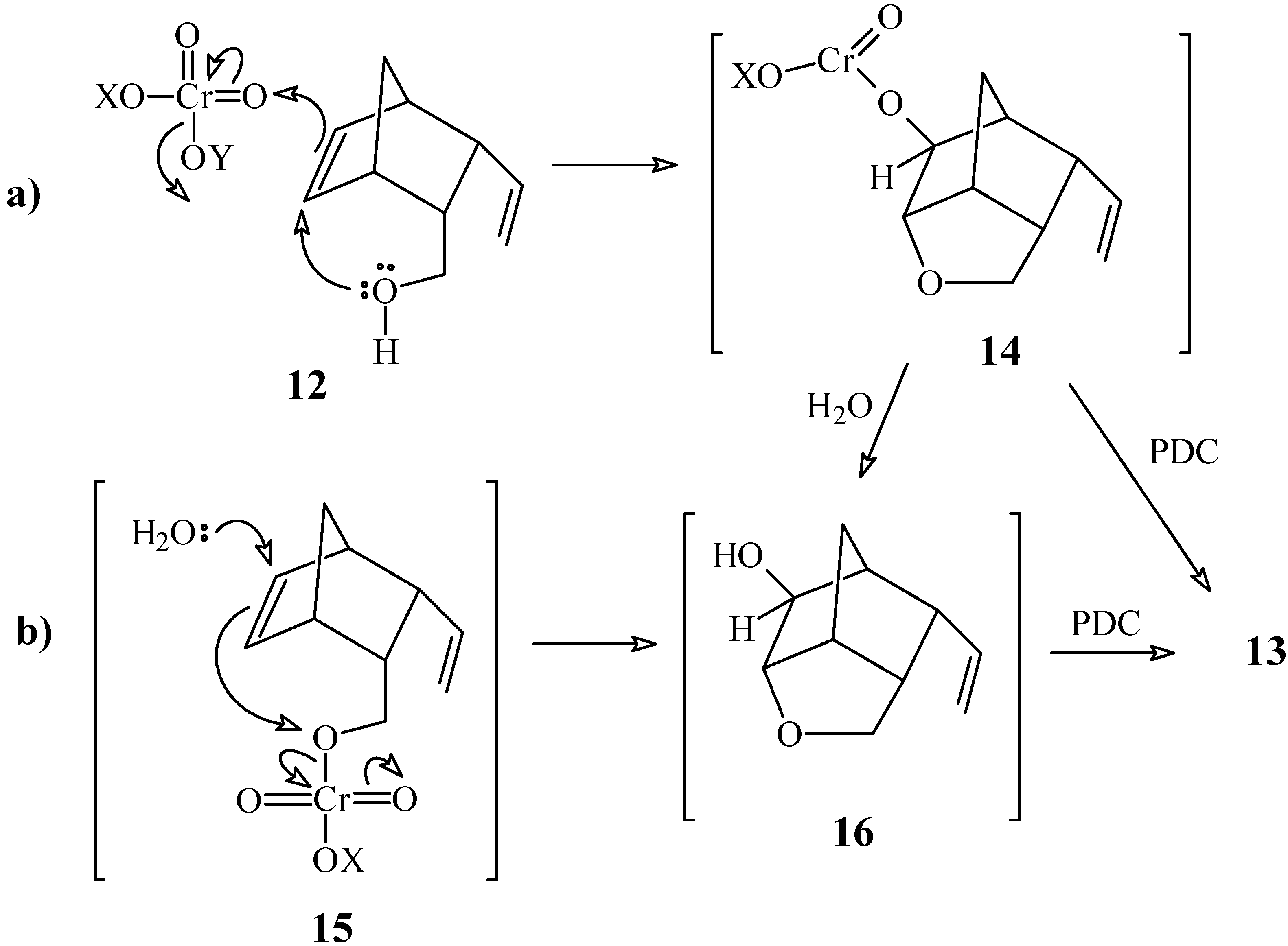
3-Vinylbicyclo[2.2.1]hept-5-ene-2-carbaldehyde (2a) and 4-Oxa-9-vinyltricyclo[4.2.1.03,7]-nonan- 2-one (13)
To a solution of pyridinium dichromate (230 mg, 0.59 mmol) in anhydrous CH2Cl2 (4 mL) maintained under N2 atmosphere, was added a solution of compound 12 (62 mg, 0.41 mmol) in CH2Cl2 (2 mL). After stirring for 24 h at room temperature, the product was extracted and purified as described in experiment 1a, using a 7:3 mixture of hexane / ethyl acetate as eluent. Compound 2a: yield 35 mg (57 %). 1H-NMR δ (CDCl3, 300 MHz) 9.35 (d, 1H, J = 3.4 Hz), 6.36 (dd, 1H, J1 = 5.9 Hz, J2 = 2.8 Hz), 6.24 (dd, 1H, J1 = 5.9 Hz, J2 = 2.8 Hz), 5.41 (dt, 1H, J1 = 17.0 Hz, J2 = J3 = 10.0 Hz), 5.21 (ddd, 1H, J1 = 17.0 Hz, J2 = 2.0 Hz, J3 = 0.6 Hz), 5.02 (ddd, 1H, J1 = 10.0 Hz, J2 = 2.0 Hz, J3 = 0.6 Hz), 3.24 (dt, 1H, J1 = J2 = 10.0 Hz, J3 = 3.4 Hz), 3.12 (m, 1H), 3.02 (dt, 1H, J1 = 10.0 Hz, J2 = J3 = 3.4 Hz), 2.92 (m, 1H), 1.54 (dt, 1H, J1 = 8.6 Hz, J2 = J3 = 1.9 Hz), 1.41 (m, 1H); 13C-NMR δ (CDCl3, 75 MHz) 205.5 (C=O), 138.6 (CH), 135.9 (CH), 135.5 (CH), 117.0 (CH2), 57.4 (CH), 49.4 (CH2), 49.0 (CH), 48.5 (CH), 45.4 (CH); IR (KBr) νmax 2972, 2729, 1716, 1636, 1453, 1392, 912, 737 cm-1. Compound 13: yield 3 mg (5 %). 1H-NMR δ (CDCl3, 300 MHz) 5.59 (ddd, 1H, J1 = 17.4, J2 = 10.0 Hz, J3 = 8.3 Hz), 5.19 (ddd, 1H, J1 = 17.4 Hz, J2 = 1.5 Hz, J3 = 1.0 Hz), 5.15 (dd, 1H, J1 = 10.0, J2 = 1.5 Hz), 3.85 (m, 3H), 3.09 (m, 1H), 2.78 (m, 2H), 2.47 (m, 1H), 1.91 (d, 1H, J = 11.5 Hz), 1.78 (d, 1H, J = 11.5 Hz); 13C-NMR (CDCl3, 75 MHz) δ 211.9 (C=O), 134.9 (CH), 118.3 (CH2), 80.8 (CH), 70.4 (CH2), 50.7 (CH), 44.6 (CH), 44.1 (CH), 41.2 (CH), 29.1 (CH2); IR (KBr) νmax 2974, 1753, 1639, 1178, 1129, 1040 cm-1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}