Muscarinic Receptor Agonists and Antagonists

Abstract
:| Contents | |
| 1 | The Pharmacology of Muscarinic Receptors |
| 1.1 | Introduction |
| 1.2 | Muscarinic receptor subtypes |
| 2 | Pharmacological Effects of Agonists and Therapeutic Applications |
| 2.1 | Cardiovascular system |
| 2.2 | Gastointestinal tract |
| 2.3 | Other smooth muscle |
| 2.4 | Glandular secretions |
| 2.5 | The eye |
| 2.6 | Central nervous system |
| 3 | Pharmacological Effects of Antagonists and Therapeutic Applications |
| 3.1 | Introduction |
| 3.2 | Cardiovascular system |
| 3.3 | Gastrointestinal tract |
| 3.4 | Urinary bladder |
| 3.5 | Airways |
| 3.6 | The eye |
| 3.7 | Prostate gland |
| 4 | The Medicinal Chemistry of Muscarinic Receptor Agonists and Antagonists |
| 4.1 | Scope |
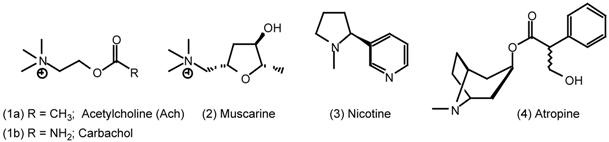
| 5 | Muscarinic Agonists |
| 5.1 | Arecoline |
| 5.2 | Heterocyclic and other derivatives of arecoline |
| 5.3 | The synthesis of 1-aza-bicyclo[2.2.1]heptanes (1-azanorbornanes) and 1-azabicyclo[2.2.2]octanes (quinuclidines) |
| 5.3.1 | Intramolecular nucleophilic displacement by amines |
| 5.3.2 | Dieckmann condensation |
| 5.3.3 | Ester surrogates |
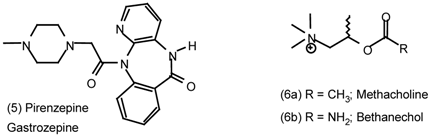
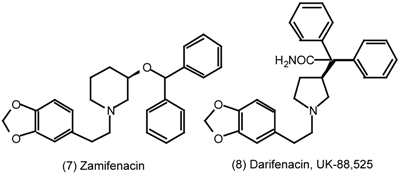
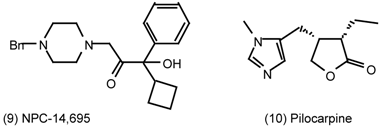
| 6 | Muscarinic Antagonists |
| 6.1 | Enantioselective synthesis of tertiary centres; tolterodine, rociverine, IQNP |
| 6.2 | Diels-Alder reactions |
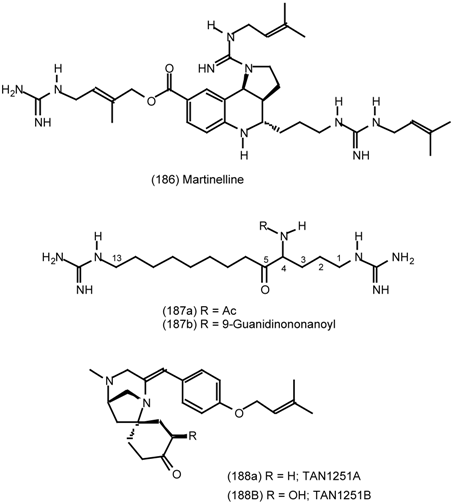
| 6.2.1 | Indole alkaloids; allosteric activators of muscarinic binding |
| 6.2.2 | Himbacine; a muscarinic antagonist with allosteric properties |
| 6.2.3 | Cyclostellettamines; macrocyclic muscarinic antagonists |
| 7 | Conclusions and Future Prospects |
| 8 | References |
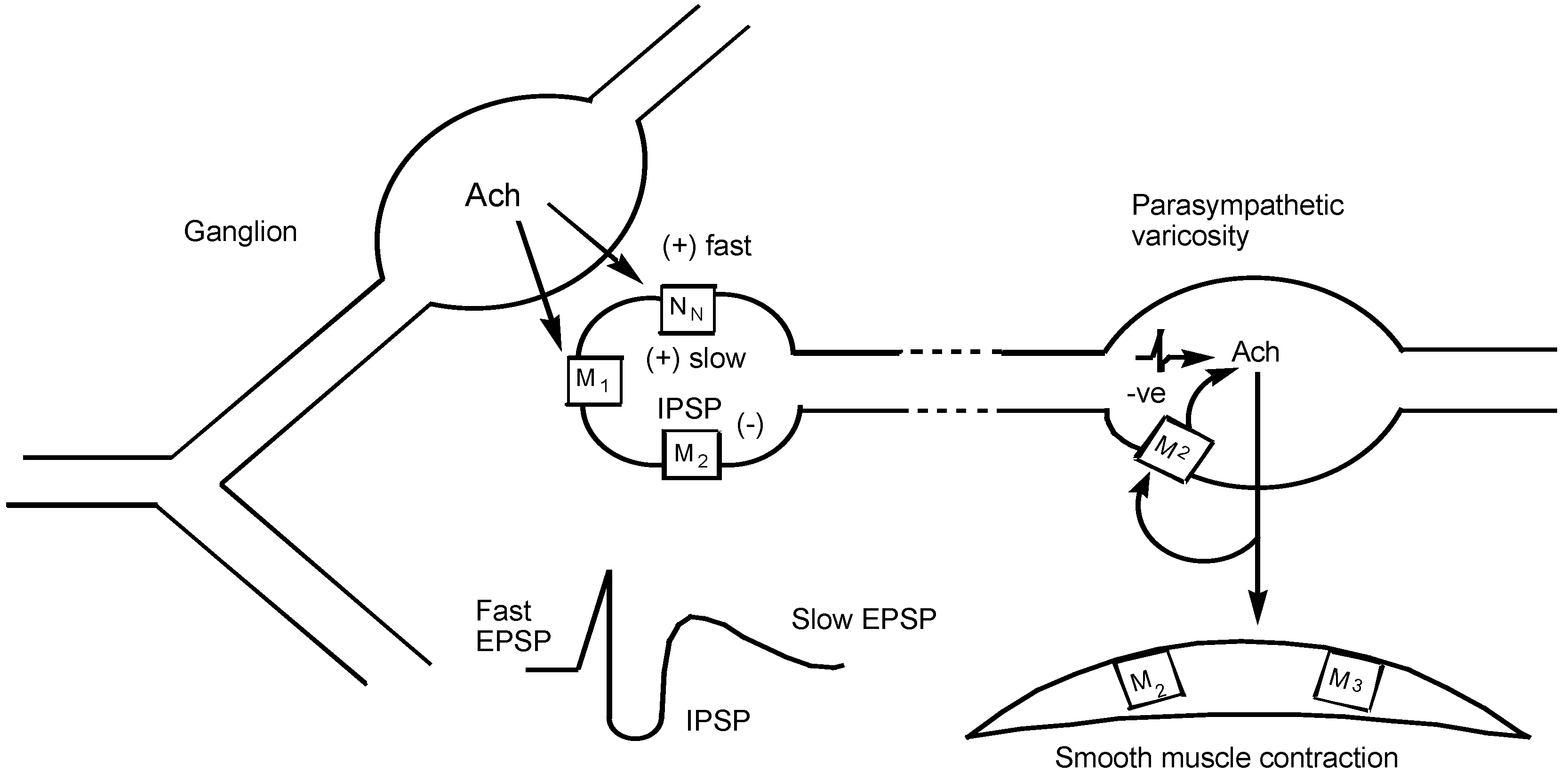
1 The Pharmacology of Muscarinic Receptors
1.1 Introduction
1.2 Muscarinic Receptor Subtypes

{kind=link}
| Receptor subtype | Second messenger | Organ/tissue | Response |
| M1 | IP3/DAG↑ | autonomic ganglion including myenteric plexus | depolarization, slow EPSP increased gastric acid secretion via vagus nerve |
| M2 | cAMP↓ and direct coupling to K+ channel | vas deferens (rabbit) brain (cerebral cortex) canine saphenous vein heart - | reduced twitch height and NA release binding sites contraction |
| atrium | reduced rate (negative chronotropy) | ||
| sinu-atrial node prejunctional at autonomic nerve endings | reduced rate (negative chronotropy) reduced NA or Ach release | ||
| ileum smooth muscle | predominant binding sites - uncertain function | ||
| M3 | IP3/DAG↑ | smooth muscle -gut urinary bladder trachea iris circular muscle blood vessels- | contraction minor binding sites |
| endothelium smooth muscle glands - | release of NO and vasodilatation contraction | ||
| oxyntic cells (gastric acid) salivary glands pancreatic β cells | increased acid secretion salivation insulin release | ||
| M4 | cAMP↓ | guinea-pig uterus | contraction |
| rabbit anococcygeus muscle | NO-dependent relaxation | ||
| rabbit lung | binding sites | ||
| M5 | IP3/DAG↑ | brain regions | binding sites after all others occluded |
| Abbreviations: Ach, acetylcholine; cAMP, cyclic adenosine 3',5'-monophosphate; DAG, 1,2-diacylglycerol; EPSP, slow depolarization; IP3, inositol 1,4,5-triphosphate; NA, noradrenaline; NO, nitric oxide. | |||
2 Pharmacological Effects of Agonists and Therapeutic Applications
2.1 Cardiovascular System
2.2 Gastrointestinal Tract
2.3 Other Smooth Muscle
2.4 Glandular Secretions
2.5 The Eye
2.6 Central Nervous System
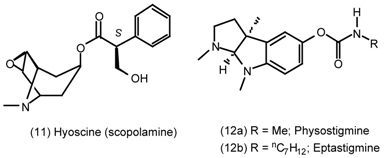
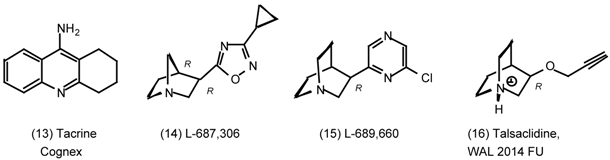
3. Pharmacological Effects of Antagonists and Therapeutic Applications
3.1 Introduction
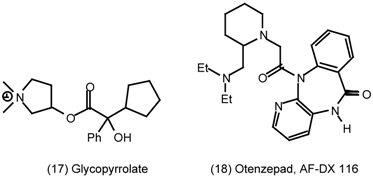
3.2 Cardiovascular System
3.3 Gastrointestinal Tract
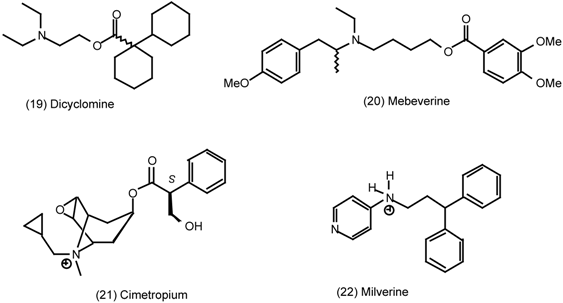
3.4 Urinary Bladder
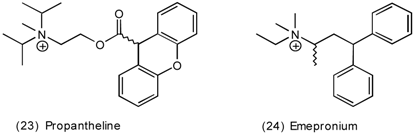
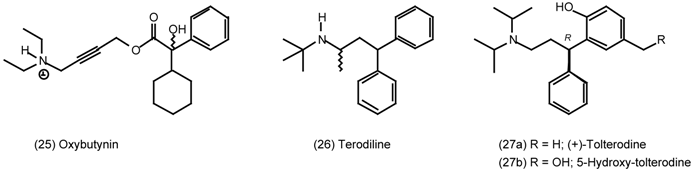
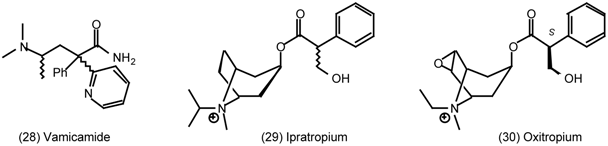
3.5 Airways
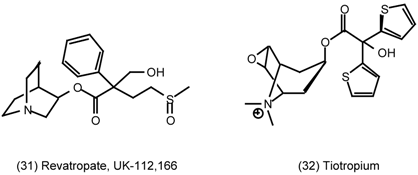
3.6 The Eye
3.7 Prostate Gland
4. The Medicinal Chemistry of Muscarinic Receptor Agonists and Antagonists
4.1 Scope
5 Muscarinic Agonists
5.1 Arecoline
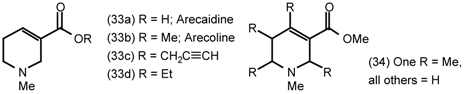
5.2 Heterocyclic and Other Derivatives of Arecoline

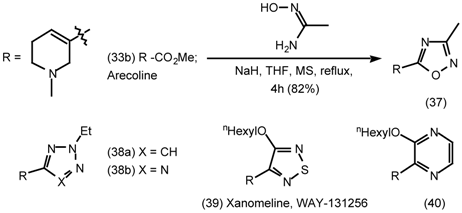
5.3 The Synthesis of 1-Aza-bicyclo[2.2.1]heptanes (1-Azanorbornanes) and 1-Aza-bicyclo- [2.2.2]-octanes (Quinuclidines)
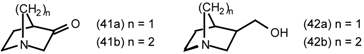
5.3.1 Intramolecular Nucleophilic Displacement by Amines
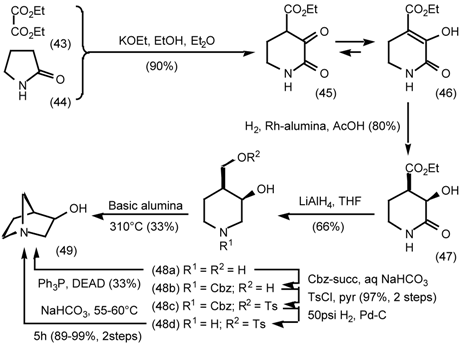
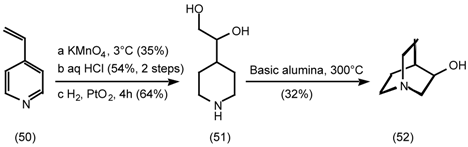
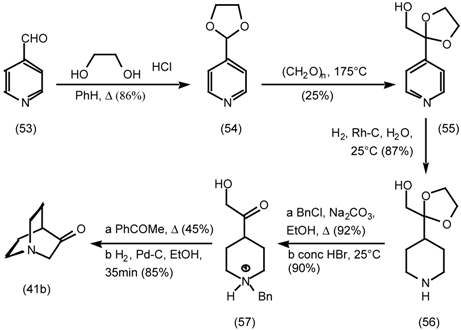
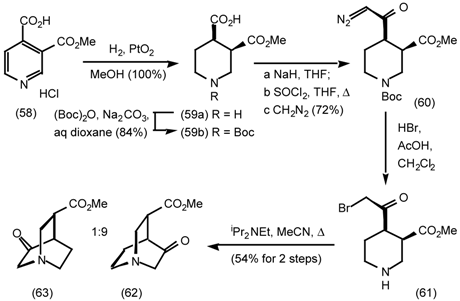
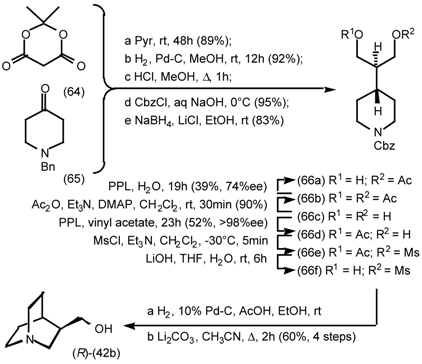
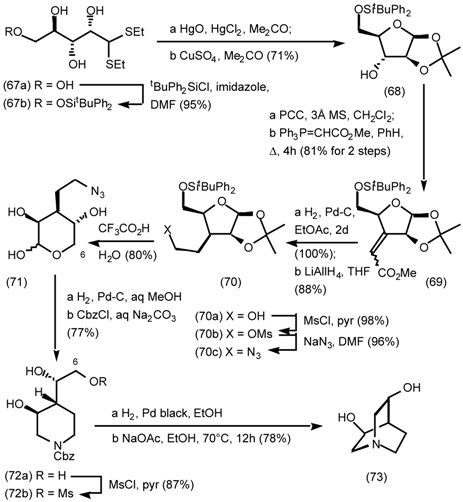
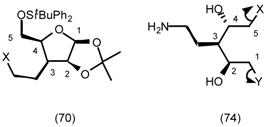
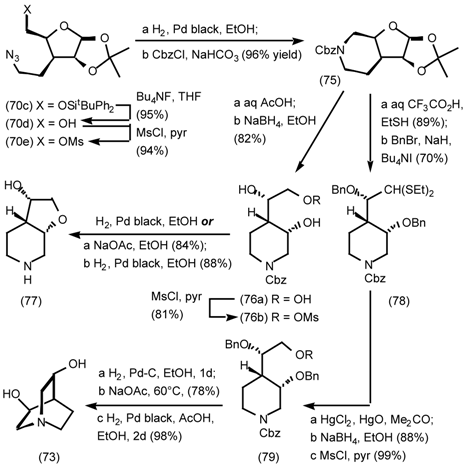
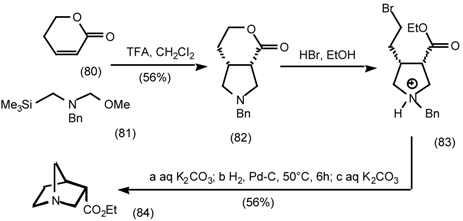
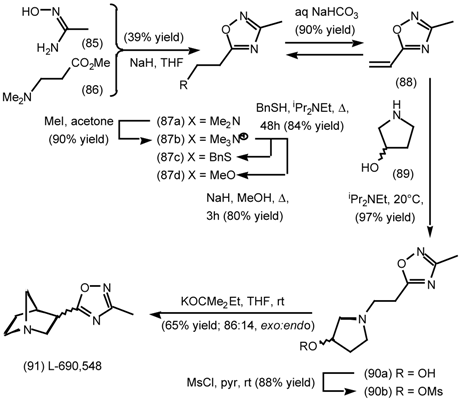
5.3.2 Dieckmann Condensation
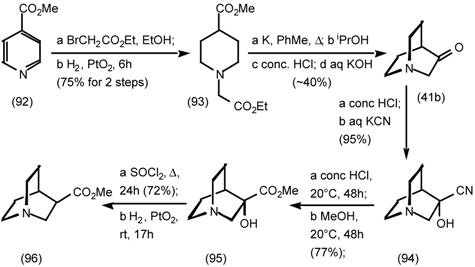
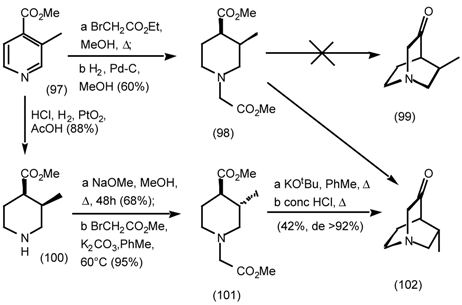
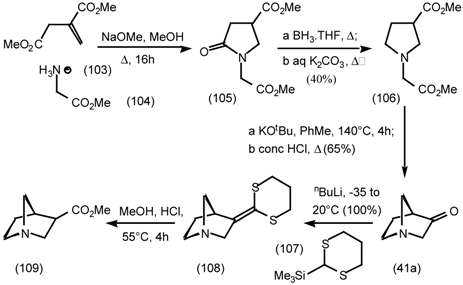
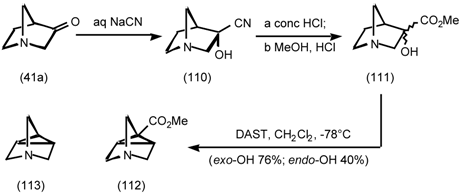
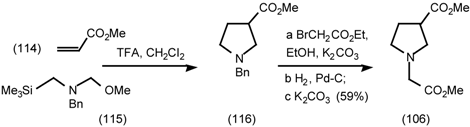
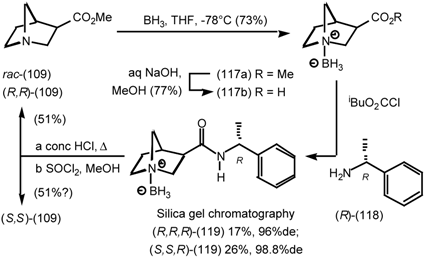
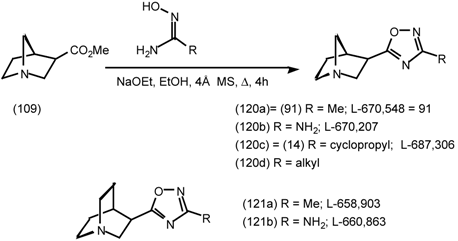
5.3.3 Ester Surrogates
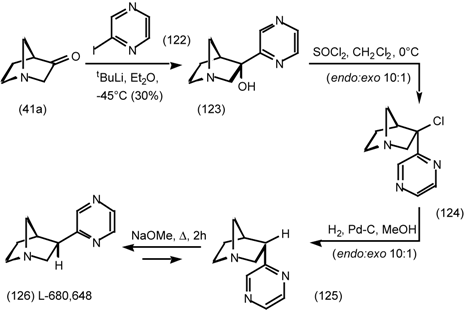
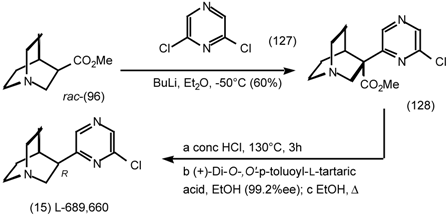
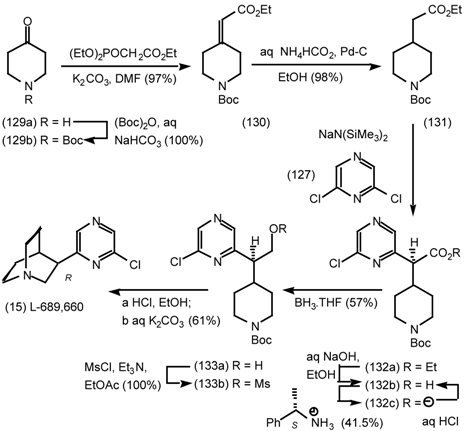
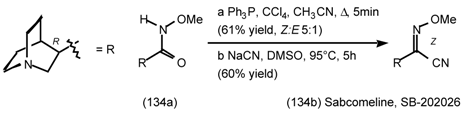
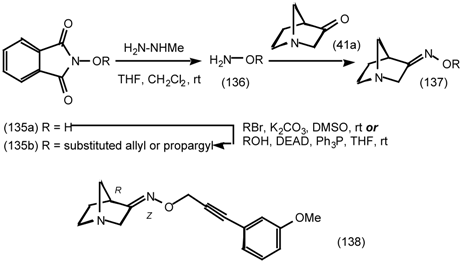
6 Muscarinic Antagonists
6.1 Enantioselective Synthesis of Tertiary Centres; Tolterodine, Rociverine, IQNP
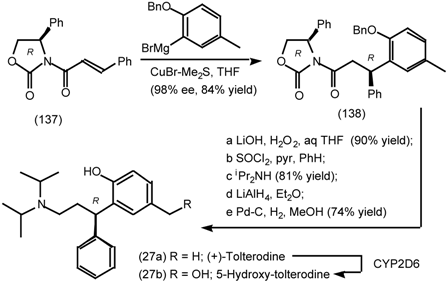
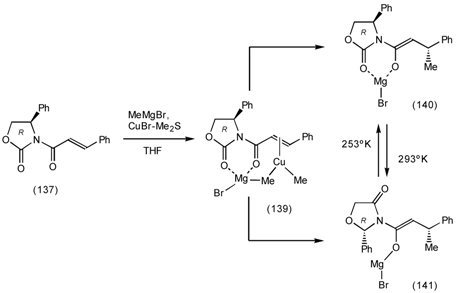
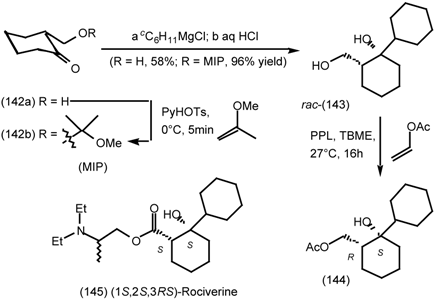
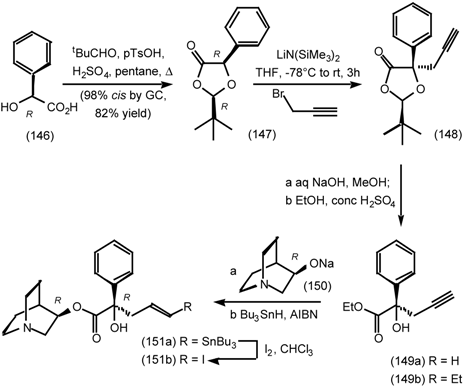
6.2 Diels-Alder Reactions
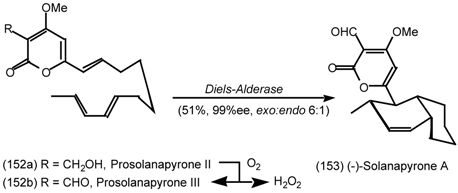
6.2.1 Indole Alkaloids; Allosteric Activators of Muscarinic Binding
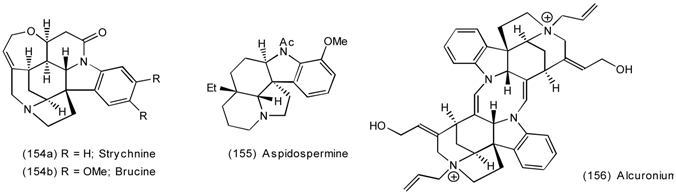
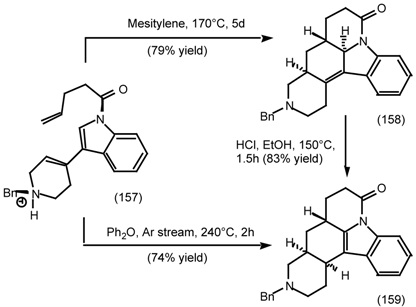
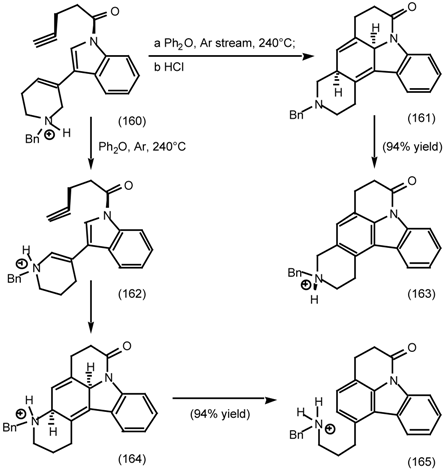
6.2.2 Himbacine; A Muscarinic Antagonist with Allosteric Properties
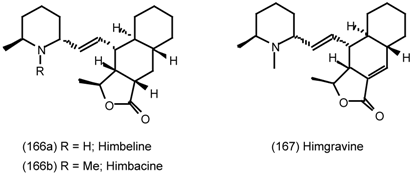
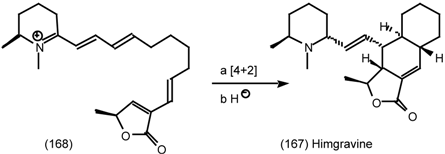
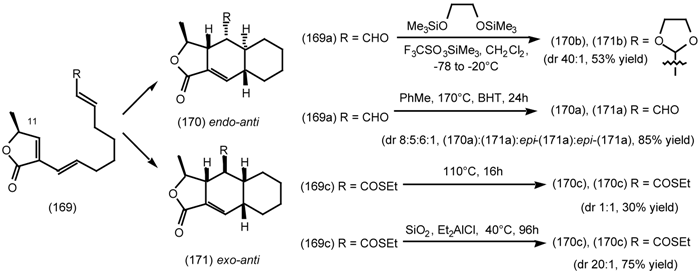
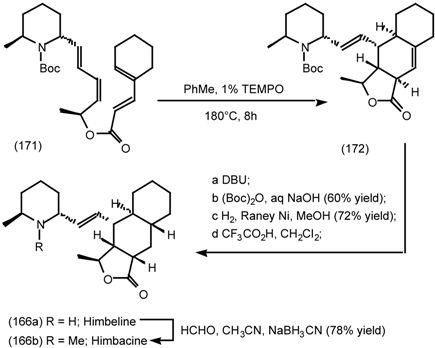
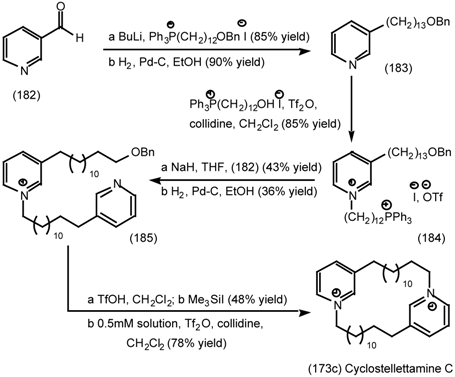
6.2.3 Cyclostellettamines; Macrocyclic Muscarinic Antagonists
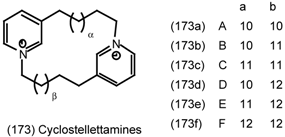
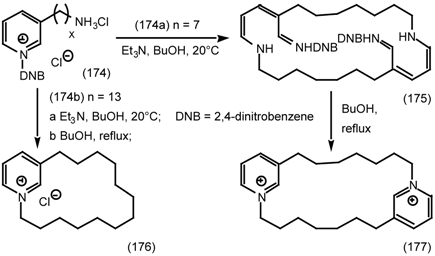
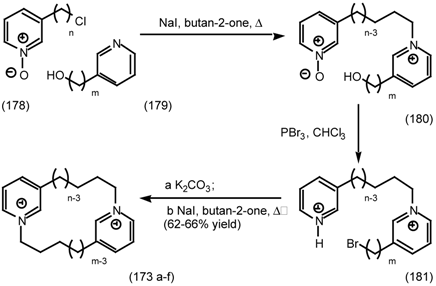
7 Conclusions and Future Prospects
Postscript added in proof, Developments in Drugs for Alzheimer’s Disease (23rd January 2001):
8 References
- Broadley, K. J. Autonomic Pharmacology; Taylor & Francis: London, 1996. [Google Scholar]
- Starke, K.; Gothert, M.; Kilbinger, H. Physiol. Rev. 1989, 69, 864–989. [PubMed]
- Newberry, N. R.; Priestley, T. Br. J. Pharmacol. 1987, 92, 817–826. [CrossRef] [PubMed]
- Hammer, R.; Berrie, C. P.; Birdsall, N. J. M.; Burgen, A. S. V.; Hulme, E. C. Nature 1980, 283, 90–92. [CrossRef] [PubMed]
- International Union of Pharmacology XVIII classification of muscarinic acetylcholine receptors, Caulfield, M. P.; Birdsall, N. J. M. Pharmacol. Rev. 1998, 50, 279–290.
- Reever, C. M.; Ferrari-DiLeo, G.; Flynn, D. D. Life Sci. 1997, 60, 1105–1112. [CrossRef]
- Hulme, E. C.; Birdsall, N. J. M.; Buckley, N. J. Annu. Rev. Pharmacol. Toxicol. 1990, 30, 633–673. [CrossRef] [PubMed]
- Caulfield, M. P. Pharmacol. Ther. 1993, 58, 319–379.
- Eglen, R. M.; Hegde, S. S.; Watson, N. Pharmacol. Rev. 1996, 48, 531–565. [PubMed]
- Urquhart, R. A.; Broadley, K. J. Gen. Pharmacol. 1992, 23, 619–626. [CrossRef]
- Sneddon, P.; Graham, A. J. Auton. Pharmacol. 1992, 12, 445–456. [CrossRef] [PubMed]
- Eglen, R. M.; Whiting, R. L. J. Auton. Pharmacol. 1990, 10, 233–245. [CrossRef] [PubMed]
- Kromer, W.; Eltze, M. J. Auton. Pharmacol. 1991, 11, 337–342. [CrossRef] [PubMed]
- Goyal, R. K. N. Engl. J. Med. 1989, 321, 1022–1029.
- Alabaster, V. A. Life Sci. 1997, 60, 1053–1060. [PubMed]
- Howell, R. E.; Laemont, K. D.; Kovalsky, M. P.; Lowe, V. C.; Waid, P. P.; Kinnier, W. J.; Noronha-Blob, L. J. Pharmacol. Exp. Ther. 1994, 270, 546–553. [PubMed]
- Kaufman, P. L.; Weidman, T.; Robinson, J. R. Pharmacology of the Eye. Handbook of Experimental Pharmacology; Sears, M. L., Ed.; Spriger-Verlag: Berlin, 1984; pp. 149–191. [Google Scholar]
- Growdon, J. H. Life Sci. 1997, 60, 993–998. [PubMed]
- Babic, T.; Smith, R. J.; Petravic, D. Neurologia Croatica 1997, 46, 55–72.
- Wen, G. Y. J. Food Drug. Anal 1998, 6, 465–476.
- Huff, F. J.; Mickel, S. F.; Corkin, S.; Growdon, J. H. Drug Devel. Res. 1988, 12, 271–278.
- Yankner, B. A.; Duffy, K. L.; Kirschner, D. A. Science 1990, 250, 279–282. [CrossRef] [PubMed]
- Hung, A. Y.; Haass, C.; Nitsch, R. M.; Qiu, W. Q.; Citron, M.; Wurtman, R. J.; Growdon, J. H.; Selkoe, D.J. J. Biol. Chem. 1993, 268, 22959–22962. [PubMed]
- Muller, D. M.; Mendla, K.; Farber, S. A.; Nitsch, R. M. Life Sci. 1997, 60, 985–991. [PubMed]
- Avery, E. E.; Baker, L. D.; Asthana, S. Drugs & Aging 1997, 11, 450–459.
- Unni, L. K. CNS Drugs 1998, 10, 447–460.
- Weinstock, M. Neurodegeneration 1995, 4, 349–356. [PubMed]
- Giacobini, E. Neurochem. Int. 1998, 32, 413–419.
- Hunter, A. J.; Murray, T. K.; Jones, J. A.; Cross, A. J.; Green, A. R. Br. J. Pharmacol. 1989, 98, 79–86. [CrossRef] [PubMed]
- Nordberg, A.; Svensson, A. L. Drug Safety 1998, 19, 465–480. [CrossRef] [PubMed]
- Doods, H. N.; Quirion, R.; Mihm, G.; Engel, W.; Rudolf, K.; Entzeroth, M.; Schiavi, G. B.; Ladinsky, H.; Bechtel, W. D.; Ensinger, H. A.; Mendla, K. D.; Eberlein, W. Life Sci. 1993, 52, 497–503. [CrossRef]
- Baker, R.; Showell, G. A.; Street, L. J.; Saunders, J.; Hoogsteen, K.; Freedman, S. B.; Hargreaves, J. J. Chem. Soc., Chem. Commun. 1992, 817–819. [CrossRef]
- Street, L. J.; Baker, R.; Book, T.; Reeve, A. J.; Saunders, J.; Willson, T.; Marwood, R. S.; Patel, S.; Freedman, S. B. J. Med. Chem. 1992, 35, 295–305. [CrossRef] [PubMed]
- Baker, R.; Street, L. J.; Reeve, A. J.; Saunders, J. J. Chem. Soc., Chem. Commun. 1991, 760–762. [CrossRef]
- Hargreaves, R. J.; McKnight, A. T.; Scholey, K.; Newberry, N. R.; Street, L. J.; Hutson, P. H.; Semark, J. E.; Harley, E. A.; Patel, S.; Freedman, S. B. Br. J. Pharmacol. 1992, 107, 494–501. [CrossRef] [PubMed]
- Freedman, S. B.; Dawson, G. R.; Iversen, L. L.; Baker, R.; Hargreaves, R. J. Life Sci. 1993, 52, 489–495. [CrossRef]
- Ensinger, H. A.; Bechtel, W. D.; Birke, F. W.; Mendla, K. D.; Mierau, J.; Speck, G.; Troger, W. Drug Devel. Res. 1997, 40, 144–157. [CrossRef]
- Adamus, W. S.; Leonard, J. P.; Troger, W. Life Sci. 1995, 56, 883–890. [CrossRef]
- Schulte, B.; Volz-Zang, C.; Mutschler, E.; Horne, C.; Palm, D.; Wellstein, A.; Pitschner, H.F. Clin. Pharmacol. Ther. 1991, 50, 372–378. [CrossRef] [PubMed]
- Eglen, R. M.; Watson, N. Pharmacol. Toxicol. 1996, 78, 59–68. [CrossRef] [PubMed]
- Anerson, K.-E. Drugs 1988, 35, 477–494.
- Langtry, H. D.; McTavish, D. Drugs 1990, 40, 748–761. [CrossRef] [PubMed]
- Nilvebrannt, L.; Hallen, B.; Larsson, G. Life Sci. 1997, 60, 1129–1136.
- Oyasu, H.; Yamamoto, T.; Sato, N.; Ozaki, R.; Mukai, T.; Ozaki, T.; Nishii, T.; Sato, H.; Fujisawa, T.; Tozuka, Z.; Koibuchi, Y.; Honbo, T.; Esumi, K.; Ohtsuka, M.; Shimomura, K. Arzneimittelforsch./Drug Res. 1994, 44, 1242–1249.
- Barnes, P. J. Life Sci. 1993, 52, 521–527. [PubMed]
- Gross, N.J. Asthma: Basic Mechanisms and Clinical Management, 2nd Edn; Barnes, P. J., Rodger, I. W., Thomson, N. C., Eds.; Academic Press: London, 1992; pp. 555–566. [Google Scholar]
- Maesen, F. P. V.; Smeets, J. J.; Costongs, M. A. L.; Cornelissen, P. J. G.; Wald, F. D. M. Eur. Resp. J. 1993, 6, 1031–1036.
- Dail, W.G. Nervous Control of the Urogenital System. In The Autonomic Nervous System; Maggi, C.A., Ed.; Harwood: Switzerland, 1993; vol. 3, pp. 69–101. [Google Scholar]
- Ruggieri, M. R.; Colton, M. D.; Wang, P.; Wang, J.; Smyth, R. J.; Pontari, M. A.; Luthin, G. R. J. Pharmacol. Exp. Ther. 1995, 274, 976–982. [PubMed]
- Caine, M.; Raz, S.; Zeigler, M. Br. J. Urol. 1975, 47, 193–202. [CrossRef] [PubMed]
- Lau, W. A. K.; Pennefather, J. N.; Mitchelson, F. Proc. Aust. Soc. Clin. Exp. Pharmacol. Toxicol. 1996, 110.
- Holdsworth, D. K.; Jones, R. A.; Self, R. Phytochemistry 1998, 48, 581–582. [CrossRef]
- Wang, C. K.; Lee, W. H.; Peng, C. H. Agric. Biol. Chem. 1997, 45, 1185–1188. [CrossRef]
- Ref 1, p341.
- Jeng, J. H.; Hahn, L. J.; Lin, B. R.; Hsieh, C. C.; Chan, C. P.; Chang, M. C. J. Oral. Pathol. Med. 1999, 28, 64–71. [CrossRef] [PubMed]
- Sharan, R. N. Cancer J. 1996, 9, 13–19.
- Raffaele, K. C.; Berardi, A.; Morris, P. P.; Asthana, S.; Haxby, J. V.; Schapiro, M. B.; Rapoport, S. I.; Soncrant, T. T. Prog. Neuro-Psychopharmacol. & Biol. Psychiatry 1991, 15, 643–648.
- Raffaele, K. C.; Berardi, A.; Asthana, S.; Morris, P.; Haxby, J. V.; Soncrant, T. T. Psychopharmacology Bull. 1991, 27, 315–319.
- Raffaele, K. C.; Asthana, S.; Berardi, A.; Haxby, J. V.; Morris, P. P.; Schapiro, M. B.; Soncrant, T. T. Neuropsychopharmacology 1996, 15, 163–170. [CrossRef]
- Soncrant, T. T.; Raffaele, K. C.; Asthana, S.; Berardi, A.; Morris, P. P.; Haxby, J. V. Psychopharmacology 1993, 112, 421–427. [CrossRef] [PubMed]
- Asthana, S.; Raffaele, K. C.; Greig, N. H.; Berardi, A.; Morris, P. P.; Schapiro, M. B.; Rapoport, S. I.; Blackman, M. R.; Soncrant, T. T. Psychoneuroendocrinology 1995, 20, 623–636. [CrossRef]
- Asthana, S.; Greig, N. H.; Holloway, H. W.; Raffaele, K. C.; Berardi, A.; Schapiro, M. B.; Rapoport, S. I.; Soncrant, T. T. Clin. Pharmacol. & Therap. 1996, 60, 276–282.
- Law, M. Y. L.; Pershing, L. K.; Roberts, L. K. Int. J. Pharmaceutics 1992, 88, 251–260. [CrossRef]
- Kurihara-Bergstrom, T.; Good, W. R.; Feisullin, S.; Signor, C. J. Control. Release 1991, 15, 271–278. [CrossRef]
- Hussain, M. A.; Mollica, J. A. J. Pharm. Sci. 1991, 80, 750751.
- Patterson, T. A.; Kosh, J. W. Gen. Pharmacol. 1993, 24, 641–647. [CrossRef]
- Moos, W. H.; Bergmeier, S. G.; Coughenour, L. L.; Davis, R. E.; Hershenson, F. M.; Kester, J. A.; McKee, J. S.; Marriott, J. G.; Schwarz, R. D.; Tecle, H.; Thomas, A. J. J. Pharm. Sci. 1992, 81, 1015–1019. [CrossRef] [PubMed]
- Moser, U.; Gubitz, C.; Galvan, M.; Immelsehr, A.; Lambrecht, G.; Mutschler, E. Drug Res. 1995, 45-1, 449–455.
- Toja, E.; Bonett, C.; Butti, A.; Hunt, P.; Fortin, M.; Barzaghi, F.; Formento, M. L.; Maggioni, A.; Nencioni, A.; Galliani, G. Eur. J. Med. Chem. 1991, 26, 853–868. [CrossRef]
- Toja, E.; Bonett, C.; Butti, A.; Hunt, P.; Fortin, M.; Barzaghi, F.; Formento, M. L.; Maggioni, A.; Nencioni, A.; Galliani, G. Eur. J. Med. Chem. 1992, 27, 519–526. [CrossRef]
- Showell, G. A.; Gibbons, T. L.; Kneen, C. O.; MacLeod, A. M.; Merchant, K.; Saunders, J.; Freedman, S. B.; Patel, S.; Baker, R. J. Med. Chem. 1991, 34, 1086–1094. [CrossRef] [PubMed]
- Moltzen, E. K.; Pedersen, H.; Bogeso, K. P.; Meier, E.; Frederiksen, K.; Sanchez, C.; Lembol, H.L. J. Med. Chem. 1997, 37, 4085–4099. [CrossRef]
- Bymaster, F. P.; Whitesitt, C. A.; Shannon, H. E.; DeLapp, N.; Ward, J. S.; Calligaro, D. O.; Shipley, L. A.; BuelkeSam, J. L.; Bodick, N. C.; Farde, L.; Sheardown, M. J.; Olesen, P. H.; Hansen, K. T.; Suzdak, P. D.; Swedberg, M. D. B.; Sauerberg, P.; Mitch, C. H. Drug Dev. Res. 1997, 40, 158–170. [CrossRef]
- Christopoulos, A.; Pierce, T. L.; Sorman, J. L.; El Fakahany, E. E. Mol. Pharmacol. 1998, 53, 1120–1130. [PubMed]
- Christopoulos, A.; El Fakahany, E. E. Eur. J. Pharmacol. 1997, 334, R3–R4. [CrossRef]
- DeLapp, N.; Wu, S.; Belagaje, R.; Johnstone, E.; Little, S.; Shannon, H.; Bymaster, F.; Calligaro, D.; Mitch, C.; Whitesitt, C.; Ward, J.; Sheardown, M.; Fink-Jensen, A.; Jeppesen, L.; Thomsen, C.; Sauerberg, P. Biochem. Biophys. Res. Commun. 1998, 244, 156–160. [CrossRef] [PubMed]
- Bodick, N. C.; Offen, W. W.; Levey, A. I.; Cutler, N. R.; Gauthier, S. G.; Satlin, A.; Shannon, H. E.; Tollefson, G. D.; Rasmussen, K.; Bymaster, F. P.; Hurley, D. J.; Potter, W. Z.; Paul, S. M. Archiv. Neurol. 1997, 54, 465–473. [CrossRef]
- Bodick, N. C.; Offen, W. W.; Shannon, H. E.; Satterwhite, J.; Lucas, R.; Van Lier, R.; Paul, S. M. Alzheimer Disease Assoc. Disorders 1997, 11, S16–S22.
- Ward, J. S.; Merritt, L.; Stratford, R.; Brown, T. J.; Wijayaratne, R.; Szewczyk, S. P.; Calligaro, D. O.; Bymaster, F. P.; Shannon, H. E.; Mitch, C. H.; Johnson, D.W.; Wikel, J. H.; Olesen, M. J.; Sheardown, M. J.; Swedberg, M. D. B.; Sauerberg, P. Life Sci. 1997, 60, 6.
- Ward, J. S.; Merritt, L.; Calligaro, D. O.; Bymaster, F. P.; Shannon, H. E.; Sawyer, B. D.; Mitch, C. H.; Deeter, J. B.; Peters, S. C.; Sheardown, M. J.; Olesen, P.H.; Swedberg, M. D. B.; Sauerberg, P. J. Med. Chem. 1995, 38, 2469–3481.
- Ward, J. S.; Merritt, L.; Klimkowski, V. J.; Lamb, M. L.; Mitch, C. H.; Bymaster, F. P.; Sawyer, B.; Shannon, H. E.; Olesen, P. H.; Honore, T.; Sheardown, M. J.; Sauerberg, P. J. Med. Chem. 1992, 35, 4011–4019. [CrossRef] [PubMed]
- Hass, K.; Wieland, A. Chem. Ber. 1960, 93, 1686–1692.
- Spry, D. O.; Aaron, H. S. J. Org. Chem. 1969, 34, 3674–3676. [CrossRef]
- Barrett, S. D.; Jaen, J. C.; Caprathe, B. W.; Thomas, A. J.; Tecle, H. Org. Prep. Proc. Int. 1997, 29, 330–335. [CrossRef]
- Aaron, H. S.; Owens, O. O.; Rosenstock, P. D.; Leonard, S.; Elkin, S.; Miller, J. I. J. Org. Chem. 1965, 30, 1331–1333. [CrossRef]
- Nielson, A. T.; Platt, E. N. J. Heteocyclic Chem. 1969, 6, 891–900.
- Nielson, A. T. J. Heterocyclic Chem. 1970, 7, 231–234.
- Snow, R. J.; Baker, R.; Herbert, R. H.; Hunt, I. J.; Merchant, K. J.; Saunders, J. J. Chem. Soc., Perkin Trans. 1 1991, 409–420. [CrossRef]
- Review; Kazlauskas, R.; Bornscheuer, U. Biotransformations; Volume 8a in the series Biotechnology; Kelly, D. R., Ed.; Wiley-VCH: Weinheim, 1998; pp. 37–192. [Google Scholar]
- Guanti, G.; Banfi, L.; Brusco, S.; Narisano, E. Tetrahedron: Asym. 1994, 5, 537–540. [CrossRef]
- Hanessian, S. Total Synthesis of Natural Products: The Chiron Approach; Pergamon: Oxford, 1983; 291p. [Google Scholar]
- Bols, M. Carbohydrate Building Blocks; Wiley: New York, 1996. [Google Scholar]
- Fraser-Reid, B. O. Acc. Chem. Res. 1996, 29, 57–66. [CrossRef]
- Nicolaou, K. C.; Veale, C. A.; Hwang, C. K.; Hutchinson, J.; Prasad, C. V. C.; Ogilvie, W. W. Angew. Chem. Int. Ed. Engl. 1991, 30, 299–303. [CrossRef]
- Mori, K. Tetrahedron 1989, 45, 3233–3298.
- Vazqueztato, M. P.; Seijas, J. A.; Fleet, G. W. J.; Mathews, C. J.; Hemmings, P. R.; Brown, D. Tetrahedron 1995, 51, 959–974.
- Fleet, G. W. J.; Mathews, C. J.; Seijas, J. A.; Tato, M. P. V.; Brown, D. J. J. Chem. Soc., Perkin Trans I 1989, 1065–1066.
- Fleet, G. W. J.; James, K.; Lunn, R. J. Tetrahedron Lett. 1986, 27, 3053–3056. [CrossRef]
- Fleet, G. W. J.; James, K.; Lunn, R. J., C. J.; Matthews, C. J. Tetrahedron Lett. 1986, 27, 3057–3058. [CrossRef]
- Fleet, G. W. J.; Mathews, C. J.; Seijas, J. A.; Tato, M. P. V.; Brown, D. J. J. Chem. Soc., Perkin Trans I 1989, 1067–1068.
- Terao, Y.; Kotake, H.; Imai, N.; Achiwa, K. Chem. Pharm. Bull. 1985, 33, 2762–2766. [CrossRef]
- Orlek, B. S.; Wadsworth, H.; Wyman, P.; Hadley, M. S. Tetrahedron Lett. 1991, 32, 1241–1244. [CrossRef]
- Cotterell, I. F.; Hands, D.; Kennedy, D. J.; Kerensa, J. P.; Wright, S. H. B.; Hoogsteen, K. J. Chem. Soc., Perkin Trans I 1990, 1091–1097.
- Orlek, B. S.; Wadsworth, H.; Wyman, P.; Hadley, M. S. Tetrahedron Lett. 1991, 32, 1245–1246. [CrossRef]
- Jenkins, S. M.; Wadsworth, H. J.; Bromidge, S.; Orlek, B. S.; Wyman, P. A.; Riley, G. J.; Hawkins, J. J. Med. Chem. 1992, 35, 2392–2406. [CrossRef] [PubMed]
- Diana, G. D.; Volkots, D. L.; Nitz, T. J.; Bailey, T. R.; Long, M. A.; Vescio, N.; Aldous, S.; Pevear, D. C.; Dutko, F. J. J. Med. Chem. 1994, 37, 2421–2436. [CrossRef] [PubMed]
- Andersen, K. E.; Lundt, B. F.; Jorgensen, A. S.; Braestrup, C. Eur. J. Med. Chem. 1996, 31, 417–425. [CrossRef]
- Street, L. J.; Baker, R.; Book, T.; Kneen, C. O.; Macleod, A. M.; Merchant, K. J.; Showell, G. A.; Saunders, J.; Herbert, R. H.; Freedman, S. B.; Harley, E. A. J. Med. Chem. 1990, 33, 2690–2697. [CrossRef] [PubMed]
- Macor, J. E.; Ordway, T.; Smith, R. L.; Verhoest, P. R.; Mack, R. A. J. Org. Chem. 1996, 61, 3228–3229. [CrossRef]
- Saunders, J.; MacLeod, A. M.; Merchant, K.; Showell, G. A.; Snow, R. J.; Street, L. J.; Baker, R. J. Chem. Soc., Chem. Commun. 1988, 1618–1619. [CrossRef]
- Sternbach, L. H.; Kaiser, S. J. Am. Chem. Soc. 1952, 74, 2215-18 & 2219-2221.
- Daeniker, H. U.; Grob, C. A. Org. Syn. Coll. V 1973, 989–993.
- Grob, C.A.; Renk, E. Helv. Chim. Acta. 1954, 37, 1672-1680 & 1681-1688 & 1689-1698.
- Snow, R. J.; Baker, R.; Herbert, R. H.; Hunt, I. J.; Merchant, K. J.; Saunders, J. J. Chem. Soc., Perkin Trans. 1 1991, 409–420. [CrossRef]
- Saunders, J.; MacLeod, A. M.; Merchant, K.; Showell, G. A.; Snow, R. J.; Street, L. J.; Baker, R. J. Chem. Soc., Chem. Commun. 1988, 1618–1619. [CrossRef]
- Street, L. J.; Baker, R.; Book, T.; Kneen, C. O.; Macleod, A. M.; Merchant, K. J.; Showell, G. A.; Saunders, J.; Herbert, R. H.; Freedman, S. B.; Harley, E. A. J. Med. Chem. 1990, 33, 2690–2697. [CrossRef] [PubMed]
- MacLeod, A. M.; Herbert, R.; Hoogsteen, K. J. Chem. Soc., Chem. Commun. 1990, 100–102. [CrossRef]
- Jenkins, S. M.; Wadsworth, H. J.; Bromidge, S.; Orlek, B. S.; Wyman, P. A.; Riley, G. J.; Hawkins, J. J. Med. Chem. 1992, 35, 2392–2406. [CrossRef] [PubMed]
- Boelsterli, J.; Eggnauer, U.; Pombo-Villar, E.; Weber, H.-P.; Walkinshaw, M. Helv. Chim. Acta. 1992, 75, 507–512. [CrossRef]
- Showell, G. A.; Baker, R.; Davis, J.; Hargreaves, R.; Freedman, S. B.; Hoogsteen, K.; Patel, S.; Snow, R. J. J. Med. Chem. 1992, 35, 911–916. [CrossRef] [PubMed]
- Freedman, S. B.; Harley, E. A.; Marwood, R. S.; Patel, S. Eur. J. Pharmacol. 1990, 187, 193–199. [CrossRef]
- Freedman, S. B.; Harley, E. A.; Patel, S.; Newberry, N.; Gilbert, A. T.; McKnight, A.; Tang, J. K.; Macquire, J. J.; Mudunkotuwa, N. T.; Baker, R.; Street, L. J.; MacLeod, A. M.; Saunders, J.; Iversen, L. L. Br. J. Pharamacol. 1990, 101, 575–580. [CrossRef]
- Baker, R.; Showell, G. A.; Street, L. J.; Saunders, J.; Hoogsteen, K.; Freedman, S. B.; Hargreaves, R. J. J. Chem. Soc., Chem. Commun. 1992, 817–819. [CrossRef]
- Freedman, S. B.; Patel, S.; Harley, E. A.; Iversen, L. L.; Baker, R.; Showell, G. A.; Saunders, J.; McKnight, A.; Newberry, N.; Scholey, K.; Hargreaves, R. Eur. J. Pharamacol. 1992, 215, 135–136. [CrossRef]
- Baker, R.; Street, L. J.; Reeves, A. J.; Saunders, J. Chem. Soc., Chem. Commun. 1991, 760–762. [CrossRef]
- Street, L. J.; Baker, R.; Book, T.; Reeve, A. J.; Saunders, J.; Willson, T.; Marwood, R. S.; Patel, S.; Freedman, S. B. J. Med. Chem. 1992, 35, 295–305. [CrossRef] [PubMed]
- Ashwood, M. S.; Gibson, A. W.; Houghton, P. G.; Humphrey, G. R.; Roberts, D. G.; Wright, S. H. B. J. Chem. Soc., Perk. Trans. I. 1995, 641–644. [CrossRef]
- Hargreaves, R. J.; McKnight, A. T.; Scholey, K.; Newberry, N. R.; Street, L. J.; Hutson, P. H.; Semark, J. E.; Harley, E. A.; Patel, S.; Freedman, S. B. Br. J. Pharmacol. 1992, 107, 494–501. [CrossRef] [PubMed]
- Freedman, S. B.; Dawson, G. R.; Iverson, L. L.; Baker, R.; Hargreaves, R. J. Life Sci. 1993, 52, 489–495. [CrossRef]
- Loudon, J. M.; Bromidge, S. M.; Brown, F.; Clark, M. S. G.; Hatcher, J. P.; Hawkins, J.; Riley, G. J.; Noy, G.; Orlek, B. S. J. Pharmacol. Exper. Therap. 1997, 283, 1059–1068.
- Bromidge, S. M.; Cassidy, F.; Clark, M. S. G.; Eggleston, D. S.; Oriek, B. S. J. Chem. Soc., Chem. Commun. 1994, 2189–2190. [CrossRef]
- Bromidge, S. M.; Brown, F.; Cassidy, F.; Clark, M. S. G.; Dabbs, S.; Hadley, M. S.; Hawkins, J.; Loudon, J. M.; Naylor, C. B.; Orlek, B. S.; Riley, G. J. J. Med. Chem. 1997, 40, 4265–4280. [CrossRef] [PubMed]
- Harries, M. H.; Samson, N. A.; Cilia, J.; Hunter, A. J. Br. J. Pharmacol. 1998, 124, 409–415. [CrossRef] [PubMed]
- Hatcher, J. P.; Loudon, J. M.; Hagan, J. J.; Clark, M. S. G. Psychopharmacology 1998, 138, 275–282. [CrossRef] [PubMed]
- Grochowski, E.; Jurczak, J. Synthesis 1976, 682–684. for recent examples see Maier, L.; Sporri, H. Phosph. Sulfur & Related Element 1992, 70, 39-48 & 49-57.
- Tecle, H.; Jaen, J. C.; Augelli-Szafran, C.; Barrett, S. D.; Caprathe, B. W.; Lauffer, D. J.; Mirzadegan, T.; Moos, W. H.; Moreland, D. W.; Pavia, M. R.; Schwarz, R. D.; Thomas, A. J.; Davis, R. E. Bioorg. Med. Chem. Lett. 1995, 5, 631–636. [CrossRef]
- Tecle, H.; Lauffer, D. J.; Davis, R. E.; Mirzadegan, T.; Moreland, D. W.; Schwarz, R. D.; Thomas, A. J.; Raby, C.; Eubanks, D.; Brann, M. R.; Jaen, J. C. Bioorg. Med. Chem. Lett. 1995, 5, 637–642. [CrossRef]
- Jaen, J. C.; Barrett, S.; Brann, M.; Callahan, M. J.; Davis, R.; Doyle, P.; Eubanks, D.; Lauffer, D.; Lauffer, L.; Lipiniski, W.; Moreland, D. W.; Nelson, C. B.; Raby, C.; Schwarz, R.; Spencer, C.; Tecle, H. Life Sci. 1995, 56, 845–852. [CrossRef]
- Tecle, H.; Barrett, S.D.; Lauffer, D. J.; Augelli-Szafran, C.; Brann, M. R.; Callahan, M. J.; Caprathe, B. W.; Davis, R. E.; Doyle, P. D.; Eubanks, D.; Lipiniski, W.; Mirzadegan, T.; Moos, W. H.; Moreland, D. W.; Nelson, C. B.; Pavia, M. R.; Raby, C.; Schwarz, R. D.; Spencer, C. J.; Thomas, A. J.; Jaen, J. C. J. Med. Chem. 1998, 41, 2524–2536. [PubMed]
- Hills, C. J.; Winter, S. A.; Balfour, J. A. Drugs 1998, 55, 813–820. [CrossRef] [PubMed]
- Gillberg, P.G.; Sundquist, S.; Nilvebrant, L. Eur. J. Pharmacol. 1998, 349, 285–292. [CrossRef]
- Postlind, H.; Danielson, A.; Lindgren, A.; Andersson, S. H. G. Drug Metab. Disp. 1998, 26, 289–293.
- Andersson, S. H. G.; Lindgren, A.; Postlind, H. Drug Metab. Disp. 1998, 26, 528–535.
- Nilvebrant, L.; Gillberg, P. G.; Sparf, B. Pharmacol. & Toxicol. 1997, 81, 169–172.
- Nilvebrant, L.; Hallen, B.; Larsson, G. Life Sci. 1997, 60, 1129–1136. [CrossRef]
- Brynne, N.; Dalen, P.; Alvan, G.; Bertilsson, L.; Gabrielsson, J. Clin. Pharmacol. Therap. 1998, 63, 529–539. [CrossRef]
- Swart, R.; Koivisto, P.; Markides, K. E. J. Chromatog. A. 1998, 828, 209–218. [CrossRef]
- Palmer, L.; Andersson, L.; Andersson, T.; Stenberg, U. J. Pharm. Biomed. Anal. 1997, 16, 155–165. [PubMed]
- For a review of asymmetric conjugate additions see Rossiter, B.E.; Swingle, N. M. Chem. Rev. 1992, 92, 771–806.
- For a review of the use of chiral oxazolidinones in asymmetric synthesis see Ager, D. J.; Prakash, I.; Schaad, D. R. Aldrichimica Acta 1997, 30, 3–12.
- Andersson, P. G.; Schink, H. E.; Osterlund, K. J. Org. Chem. 1998, 63, 8067–8070. [CrossRef]
- Leu, B. S.; Li, G. G.; Lung, F. D.; Hruby, V. J. J. Org. Chem. 1995, 60, 5509–5514.
- Barbier, P.; Renzetti, A. R.; Turbanti, L.; Di Bugno, C.; Fornai, F.; Vaglini, F.; Maggio, R.; Corsini, G. U. Eur J. Pharm. 1995, 290, 125–132. [CrossRef]
- Review; Kazlauskas, R.; Bornscheuer, U. Biotransformations; Volume 8a in the series Biotechnology; Kelly, D. R., Ed.; Wiley-VCH: Weinheim, 1998; pp. 37–192. [Google Scholar]
- Di Bussolo, V.; Catelani, G.; Mastrorilli, E.; Di Bugno, C.; Giorgi, R. Tetrahedron: Asym. 1996, 7, 3585–3592. [CrossRef]
- Di Bugno, C.; Colombani, S.M.; Dapporto, P.; Garzelli, G.; Paoli, P.; Subissi, A. Turbanti Chirality 1997, 9, 713–721.
- Frey, K. A.; Minoshima, S.; Kuhl, D. E. Quart. J. Nucl. Med. 1998, 42, 166–178.
- Morgan, A. E.; Brodie, J. D.; Dewey, S. L. Quart. J. Nucl. Med. 1998, 42, 151–157.
- Maziere, M. Pharmacol. Therap. 1995, 66, 83–101. [CrossRef]
- Mullan, B. P.; O'Connor, M. K.; Hung, J. C. Neurosurgery Clinics of North Am. 1996, 7, 617–652.
- Devous, M. D. J. Neuroimaging 1995, 5, S2–S13. [CrossRef]
- Waldemar, G. Cerebrovascular Brain Metab. Rev. 1995, 7, 89–130.
- Knapp, F. F.; McPherson, D. W.; Luo, H.; Zeeburg, B. Anticancer Res. 1997, 17, 1559–1572. [PubMed]
- Kiesewetter, D. O.; Carson, R. E.; Jagoda, E. M.; Herscovitch, P.; Eckelman, W. C. Life Sci. 1998, 64, 511–518. [CrossRef]
- Lee., K. S.; He, X. S.; Jones, D. W.; Coppola, R.; Gorey, J. G.; Knable, M. B.; de Costa, B. R.; Rice, K. C.; Weinberger, D. R. J. Nucl. Med. 1996, 37, 2021–2024. [PubMed]
- Seebach, D.; Naef, R.; Calderari, G. Tetrahedron 1984, 40, 1313–1324. [CrossRef]
- McPherson, D. W.; Knapp, F. F., Jr. J. Org. Chem. 1996, 61, 8335–8336. [CrossRef] [PubMed]
- McPherson, D. W.; Lambert, C. R.; Jahn, K.; Sood, V.; McRee, R. C.; Zeeberg, B.; Reba, R. C.; Knapp, F. F., Jr. J. Med. Chem. 1995, 38, 3908–3917. [CrossRef] [PubMed]
- Cohen, V. I.; Zeeberg, B. R.; Boulay, S. F.; Sood, V. K.; Rayeq, M. R.; Danesh, R. A.; McPherson, D. W.; Reba, R. C. J. Mol. Neuroscience 1998, 11, 1–9. [CrossRef]
- Ichihara, A.; Oikawa, H. Curr. Org. Chem. 1998, 2, 365–394.
- Katayama, K.; Kobayashi, T.; Oikawa, Honma, M. H.; Ichihara, A. Biochim. Biophys. Acta 1998, 1384, 387–395. [CrossRef]
- Oikawa, H.; Kobayashi, T.; Katayama, K.; Suzuki, Y.; Ichihara, A. J. Org. Chem. 1998, 63, 8748–8756. [CrossRef]
- Oikawa, H.; Yagi, K.; Watanabe, K.; Honma, M.; Ichihara, A. J. Chem. Soc., Chem. Commun. 1997, 97–98. [CrossRef]
- Ichihara, A.; Oikawa, H. Biosci. Biotech. Bioch. 1997, 61, 12–18.
- Jakubik, J.; Bacakova, L.; El Fakahany, E. E.; Tucek, S. Mol. Pharmacol. 1997, 52, 172–179. [PubMed]
- Dolezal, V.; Tucek, S. Br. J. Pharmacol. 1998, 124, 1213–1218.
- Lazareno, S.; Gharagozloo, P.; Kuonen, D.; Popham, A.; Birdsall, N. J. M. Mol. Pharmacol. 1998, 53, 573–589, correction 1999, 55, 194.
- Gharagozloo, P.; Miyauchi, M.; Birdsall, B.; Birdsall, N. J. M. J. Org. Chem. 1998, 63, 1974–1980. [CrossRef]
- Lazareno, S.; Birdsall, B.; Fukazawa, T.; Gharagozloo, p.; Hashimoto, T.; Kuwano, H.; Popham, A.; Sugimoto, M.; Birdsall, N. J. M. Life Sci. 1999, 64, 519–526.
- Dorje, F.; Wess, J.; Lambrecht, G.; Tacke, R.; Mutschler, E.; Brann, M. R. J. Pharmacol. & Exper. Therap. 1991, 256, 727–733.
- Kilbinger, H.; Von Bardeleben, R. S.; Siefken, H.; Wolf, D. Life Sci. 1995, 56, 981–987. [CrossRef]
- Matsui, H.; Lazareno, S.; Birdsall, N. J. M. Mol. Pharmacol. 1995, 47, 88–98. [PubMed]
- Kozikowski, A. P.; Fauq, A. H.; Miller, J. H.; McKinney, M. Bioorg. Med. Chem. Lett. 1992, 2, 797–802. [CrossRef]
- Malaska, M. J.; Fauq, A. H.; Kozikowski, A. P.; Aagaard, P. J.; McKinney, M. Bioorg. Med. Chem. Lett. 1995, 5, 61–66. [CrossRef]
- Malaska, M. J.; Fauq, A. H.; Kozikowski, A. P.; Aagaard, P. J.; McKinney, M. Bioorg. Med. Chem. Lett. 1993, 3, 1247–1252. [CrossRef]
- Baldwin, J. E.; Chesworth, R.; Parker, J. S.; Russell, A. T. Tetrahedron Lett. 1995, 36, 9551–9554. [CrossRef]
- Debaecke, G.; Declercq, P. J. Tetrahedron Lett. 1995, 36, 7515–7518.
- Hofman, S.; DeBaecke, G.; Kenda, B.; DeClerg, P. J. Synthesis 1998, 479–489. [CrossRef]
- Hart, D. J.; Wu, W. L.; Kozikowski, A. P. J. Am. Chem. Soc. 1995, 117, 9369–9370. [CrossRef]
- Hart, D. J.; Li, J.; Wu, W. L.; Kozikowski, A. P. J. Org. Chem. 1997, 62, 5023–5033. [CrossRef]
- Chackalamannil, S.; Davies, R. J.; Asberom, T.; Doller, D.; Leone, D. J. Am. Chem. Soc. 1996, 118, 9812–9813. [CrossRef]
- Andersen, R. J.; Van Soest, R. W. M.; Kong, F. Alkaloids; Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon Press/Elsevier Science: Oxford, UK, 1996; pp. 301–355. [Google Scholar]
- Tsuda, M.; Kobayashi, J. Heterocycles 1997, 46, 765–794.
- Fusetani, N.; Asai, N.; Matsunaga, S.; Honda, K.; Yasumuro, K. Tetrahedron Lett. 1994, 35, 3967–3970. [CrossRef]
- Baldwin, J. E.; Whitehead, R. C. Tetrahedron Lett. 1992, 33, 2059–2062. [CrossRef]
- Baldwin, J. E.; Claridge, T. D. W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead, R. C.; Broughtflower, R. J.; Mutton, I. M.; Upton, R. J. Angew. Chem. Int. Ed. Engl. 1998, 37, 2661–2663. [CrossRef]
- Kaiser, A.; Billot, X.; Gateau-Olesker, A.; Marazano, C.; Das, B. C. J. Am. Chem. Soc. 1998, 120, 8026–8034. [CrossRef]
- Baldwin, J. E.; Spring, D. R.; Atkinson, C. E.; Lee, V. Tetrahedron 1998, 54, 13655–13680. [CrossRef]
- Wanner, M. J.; Koomen, G. J. Eur. J. Org. Chem. 1998, 889–895. [CrossRef]
- Anan, H.; Seki, N.; Noshiro, O.; Honda, K.; Yasumuro, K.; Ozasa, T.; Fusetani, N. Tetrahedron 1996, 52, 10849–10860. [CrossRef]
- Adem, A.; Karlsson, E. Life Sci. 1997, 60, 1069–1076. [CrossRef]
- Liang, J. S.; Carsi-Gabrenas, J.; Krajewski, J. L.; McCaffrey, J. M.; Purkerson, S. L.; Santiago, M.P.; Strauss, W. L.; Valentine, H. H.; Potter, L. T. Toxicon 1996, 34, 1257–1267. [CrossRef]
- Jerusalinsky, D.; Kornisiuk, E.; Alfaro, P.; Quillfeldt, J.; Alonso, M.; Verde, E. R.; Cervenansky, C.; Harvey, A. Neuroreport 1998, 9, 1407–1411. [CrossRef]
- Potter, L. T.; Hanchett-Valentine, H.; Liang, J.-S.; Max, S. I.; Purkerson, S. L.; Silberberg, H. A.; Strauss, W. L. Life Sci. 1993, 52, 433–440. [CrossRef]
- Adem, A.; Jolkkonen, M.; Bogdanovic, N.; Islam, A.; Karlsson, E. Brain. Res. Bull. 1997, 44, 597–601. [CrossRef]
- Witherup, K. M.; Ransom, R. W.; Graham, A. C.; Bernard, A. M.; Salvatore, M. J.; Lumma, W.C.; Anderson, P. S.; Pitzenberger, S. M.; Varga, S. L. J. Am. Chem. Soc. 1995, 117, 6682–6685. [CrossRef]
- Nagumo, S.; Nishida, A.; Yamazaki, C.; Murashige, K.; Kawahara, N. Tetrahedron Lett. 1998, 39, 4493–4496. [CrossRef]
- Sample Availability: Not applicable.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
Broadley, K.J.; Kelly, D.R. Muscarinic Receptor Agonists and Antagonists. Molecules 2001, 6, 142-193. https://doi.org/10.3390/60300142
Broadley KJ, Kelly DR. Muscarinic Receptor Agonists and Antagonists. Molecules. 2001; 6(3):142-193. https://doi.org/10.3390/60300142
Chicago/Turabian StyleBroadley, Kenneth J., and David R. Kelly. 2001. "Muscarinic Receptor Agonists and Antagonists" Molecules 6, no. 3: 142-193. https://doi.org/10.3390/60300142
APA StyleBroadley, K. J., & Kelly, D. R. (2001). Muscarinic Receptor Agonists and Antagonists. Molecules, 6(3), 142-193. https://doi.org/10.3390/60300142