High-level Computational Study of the Site-, Facial- and Stereoselectivities for the Diels-Alder Reaction Between o-Benzoquinone and Norbornadiene

Abstract
:Introduction.
Computational Methodology
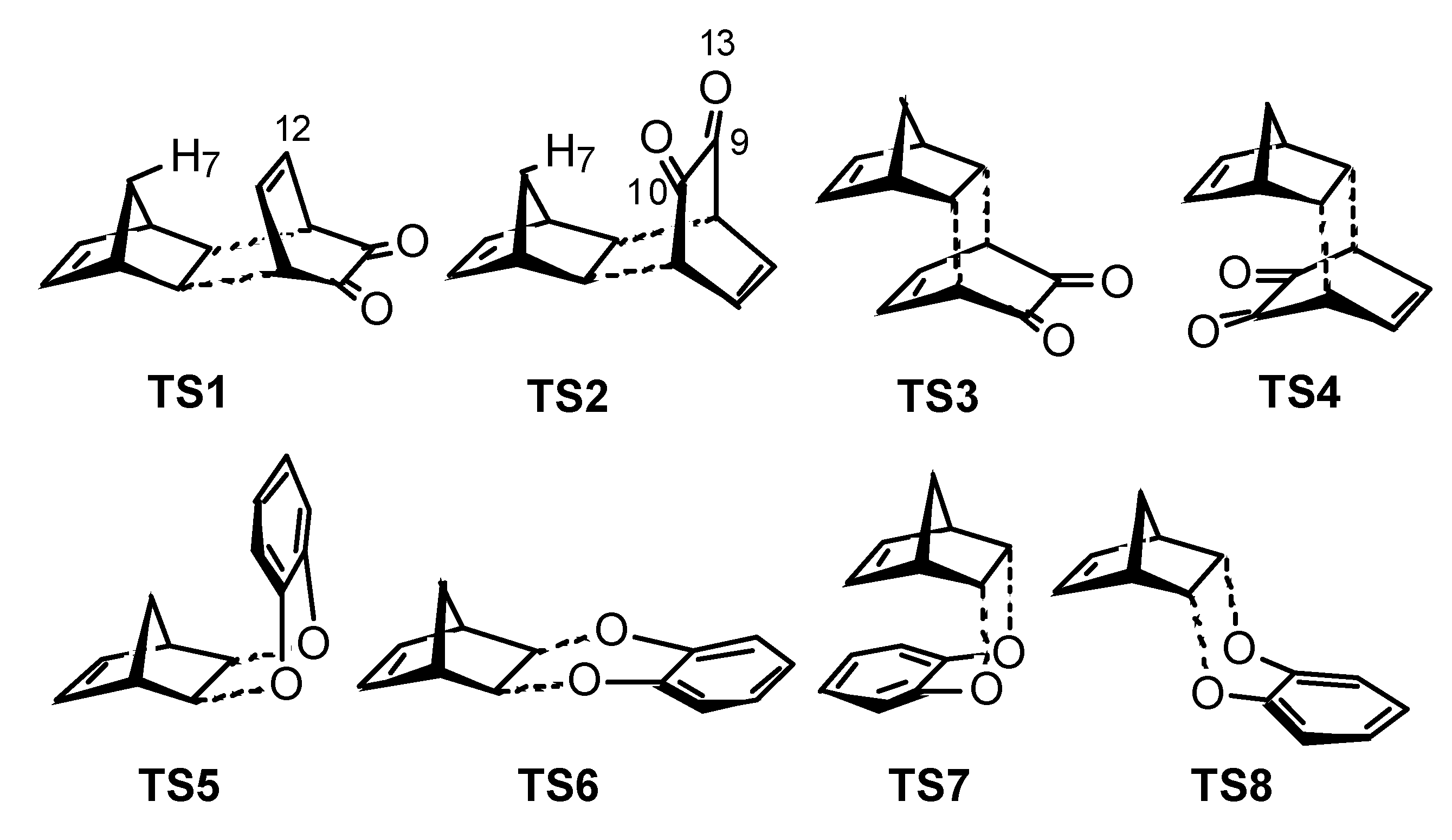
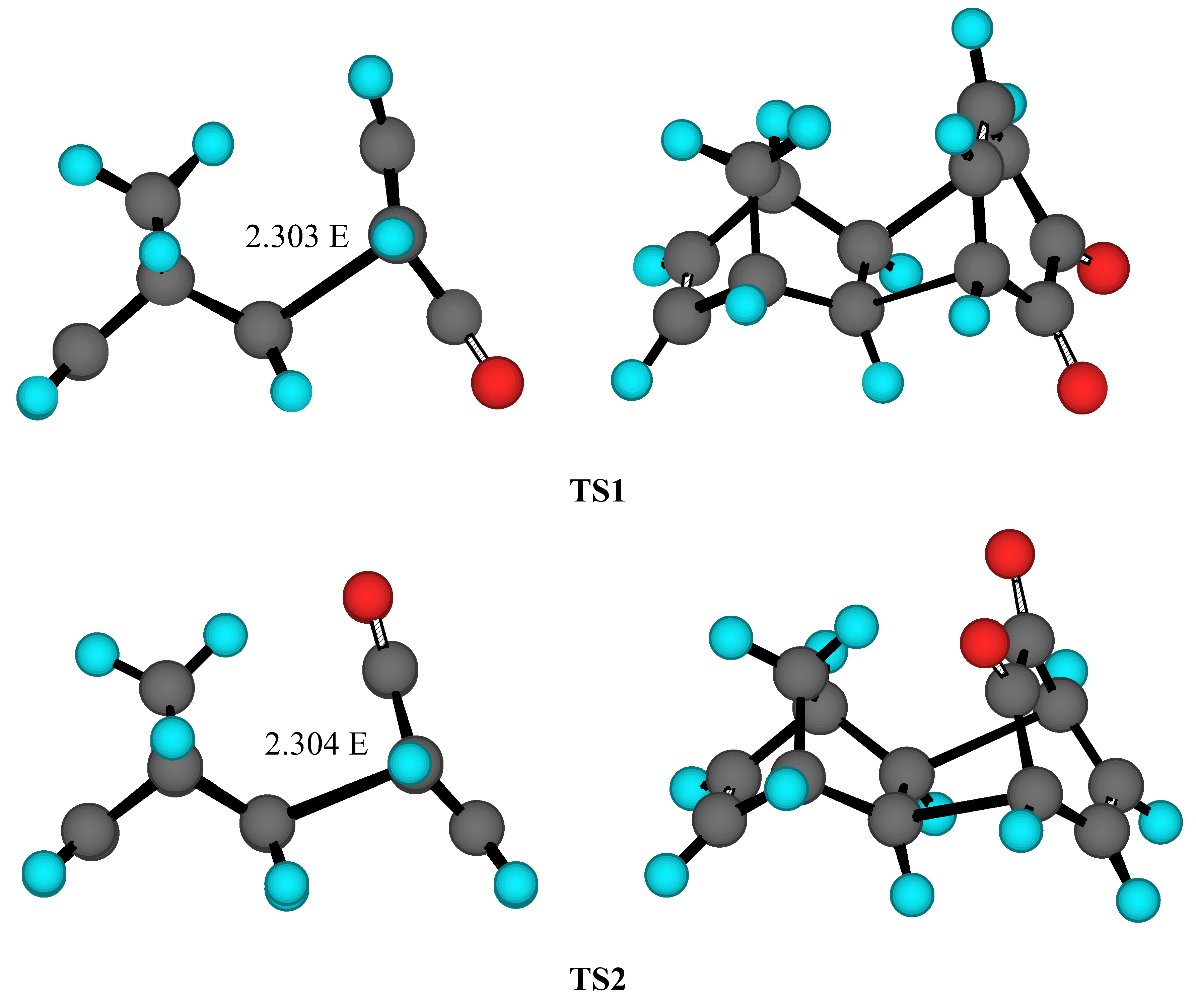
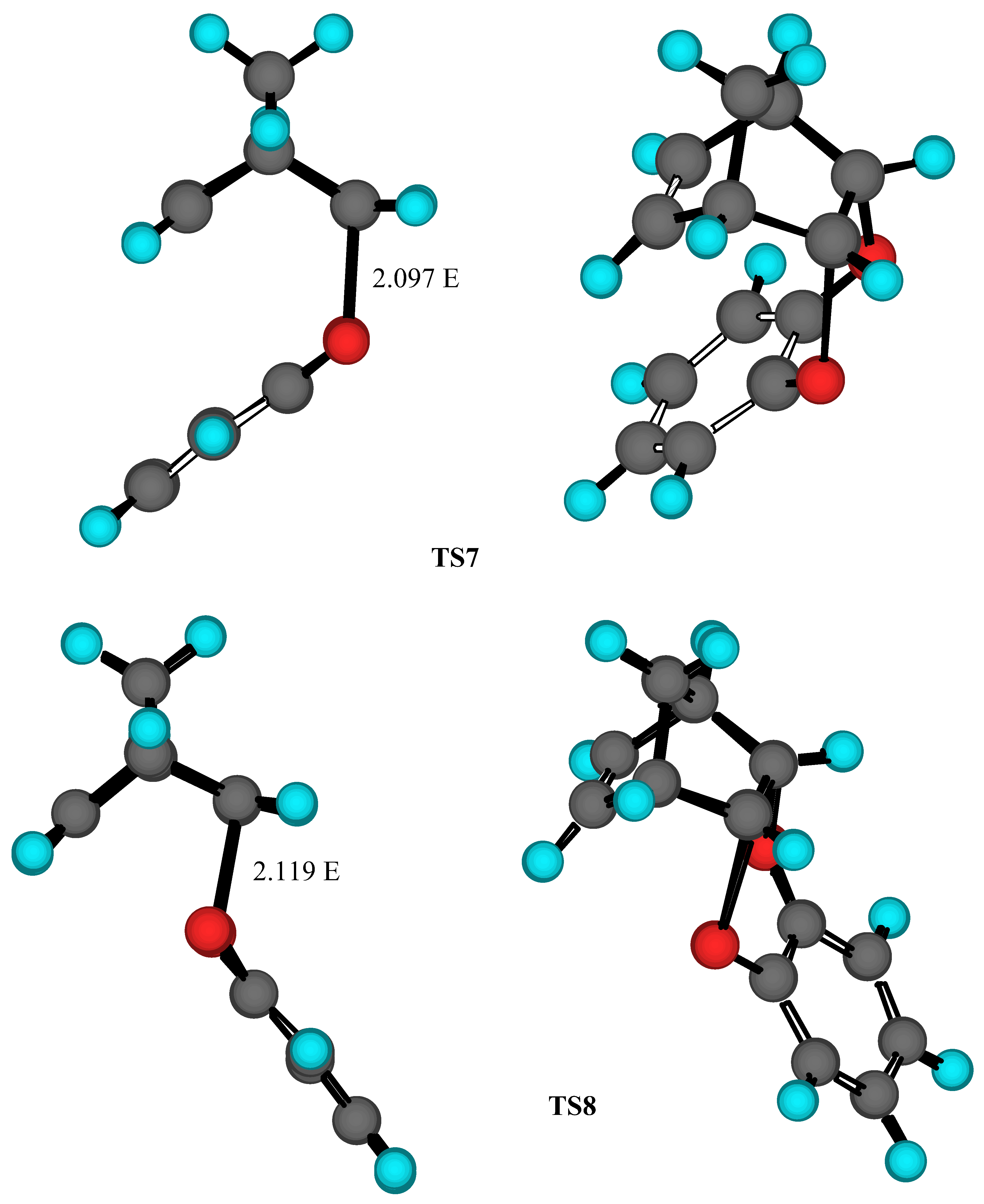
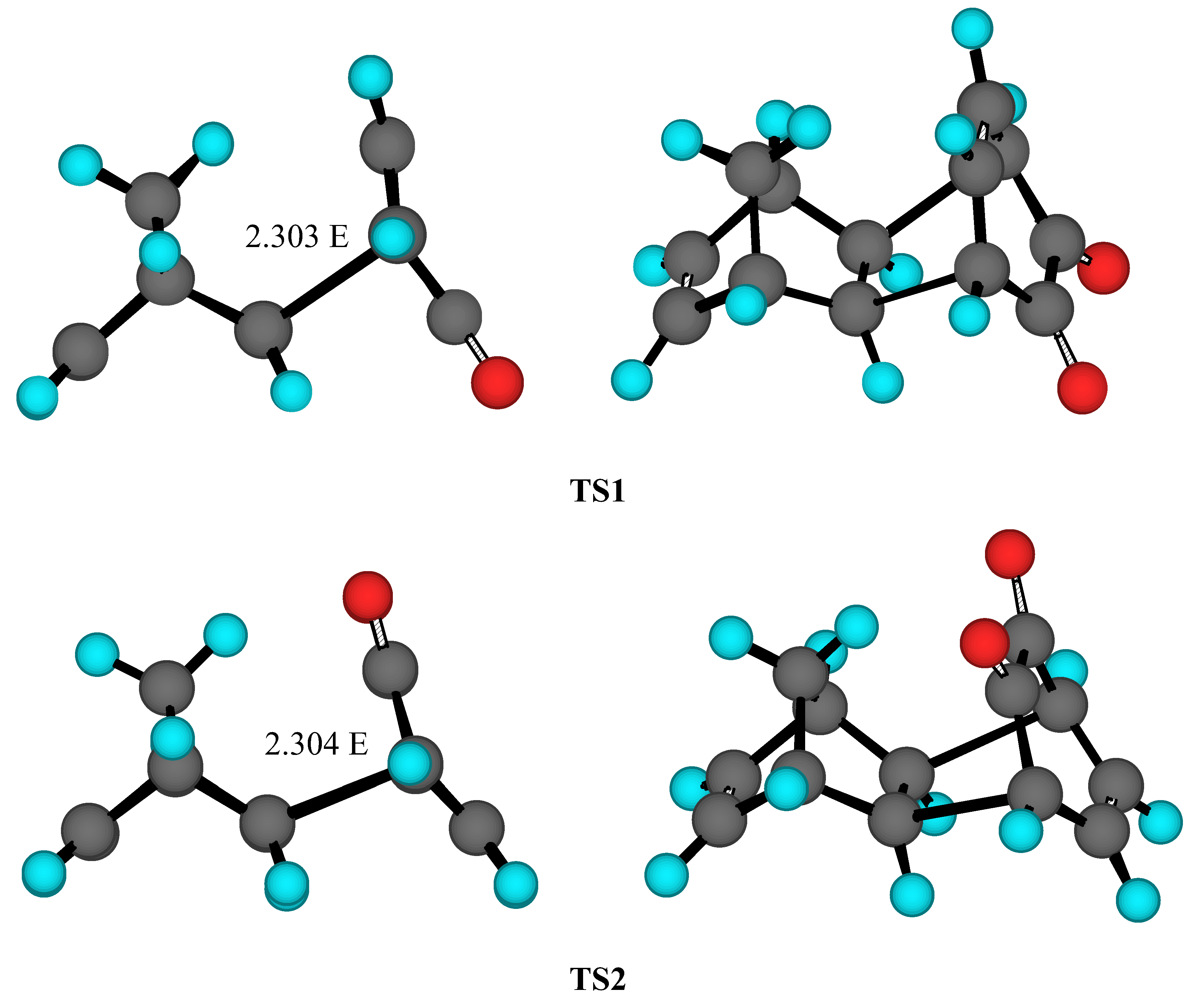
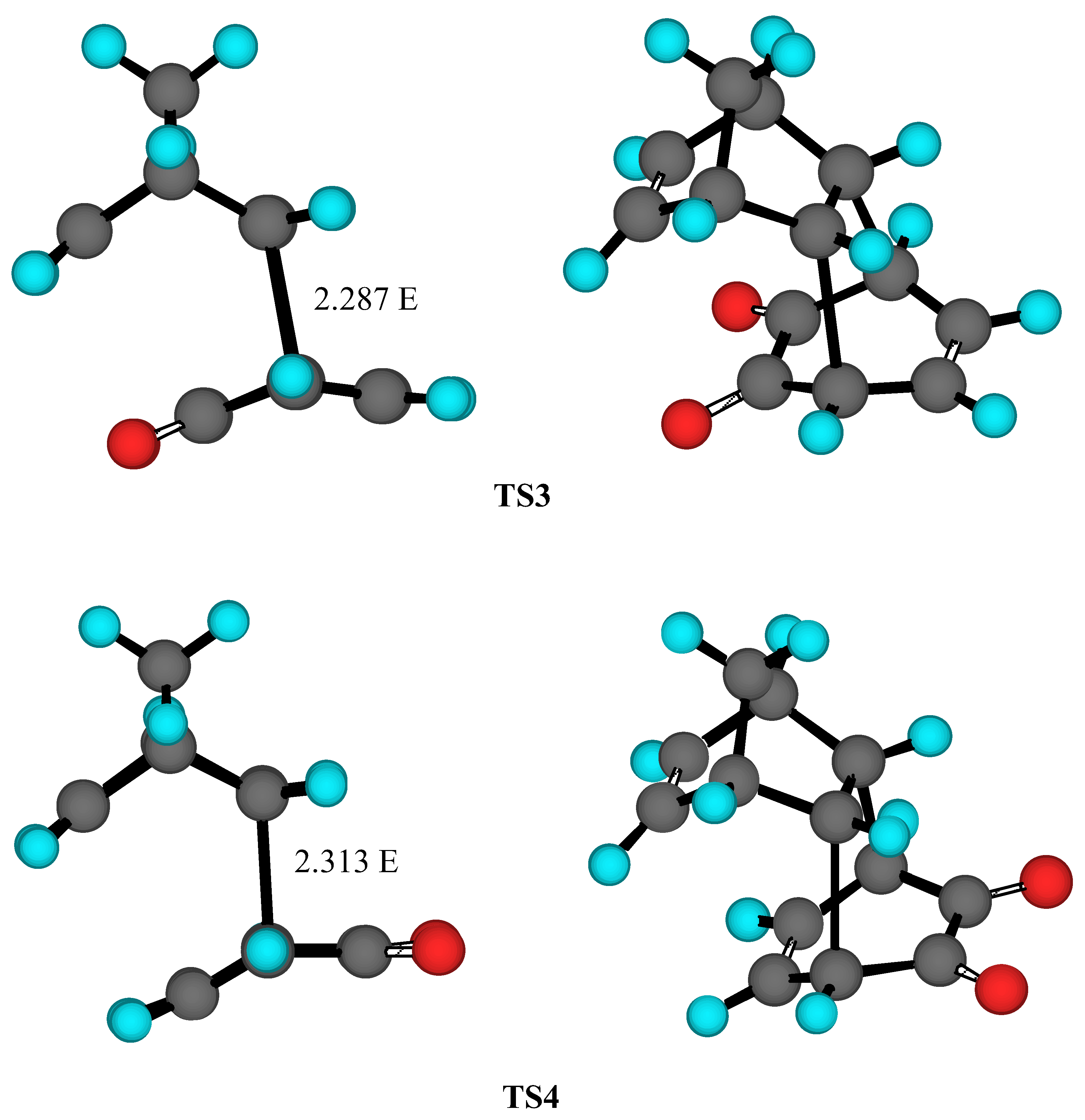
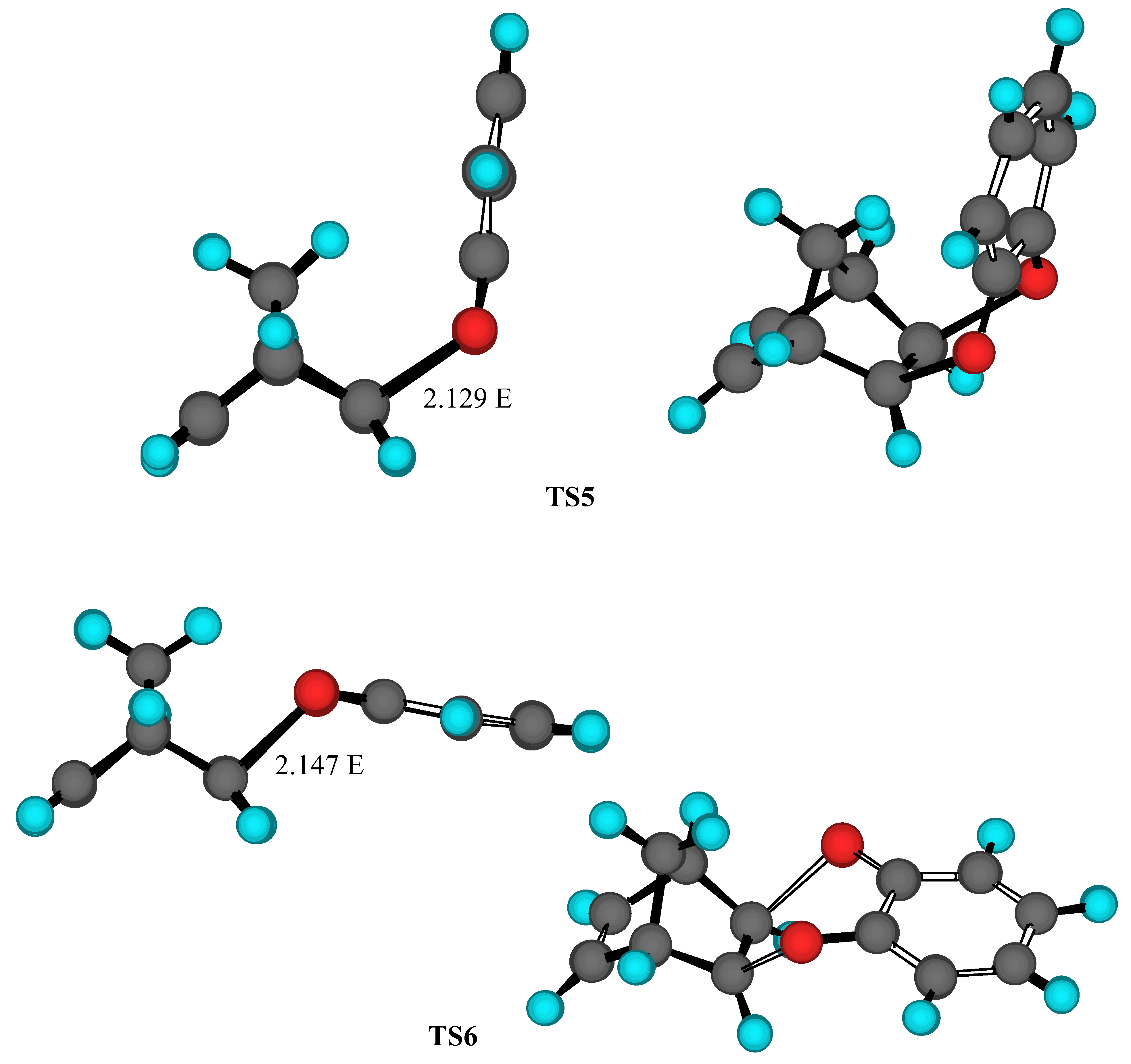
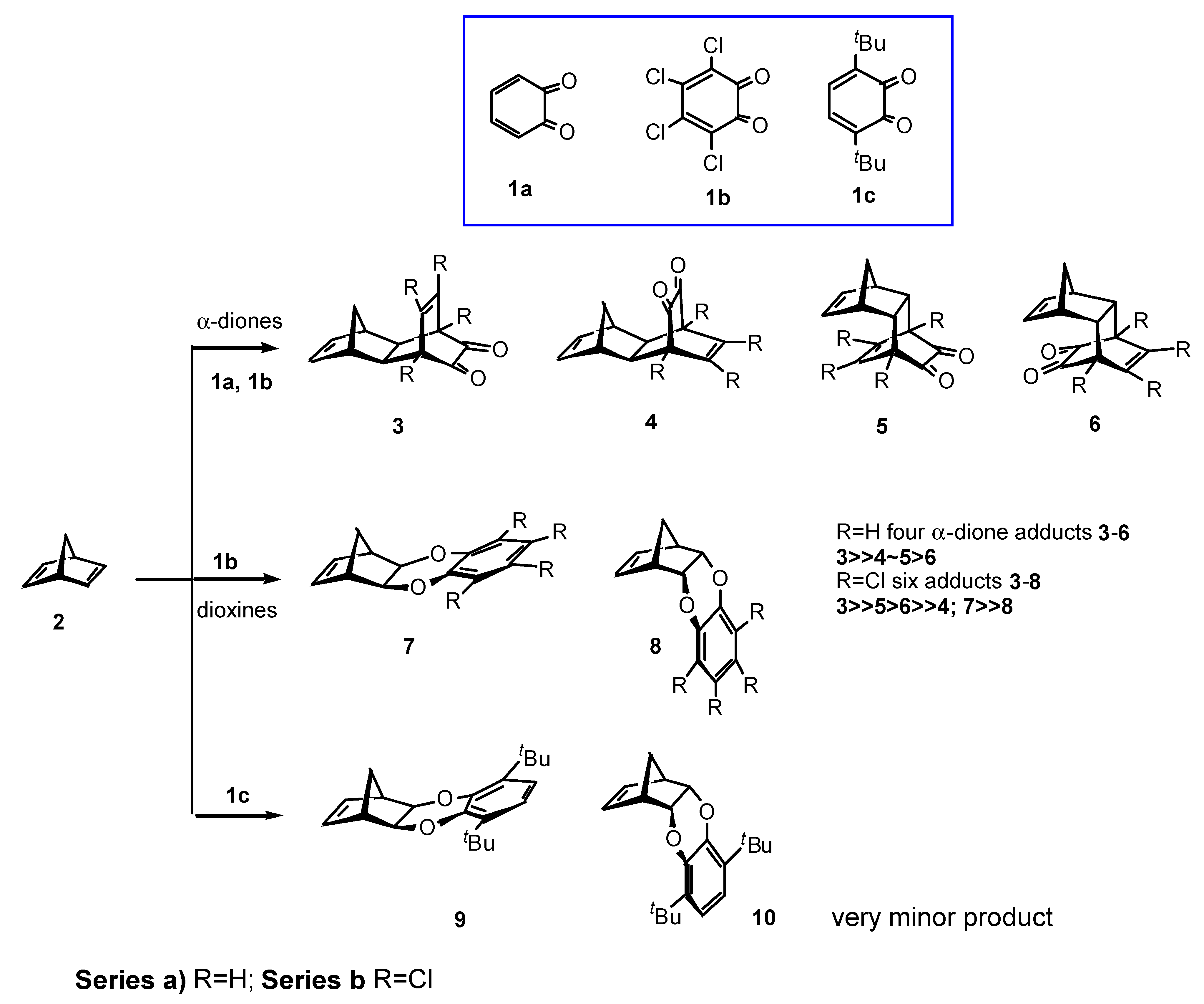
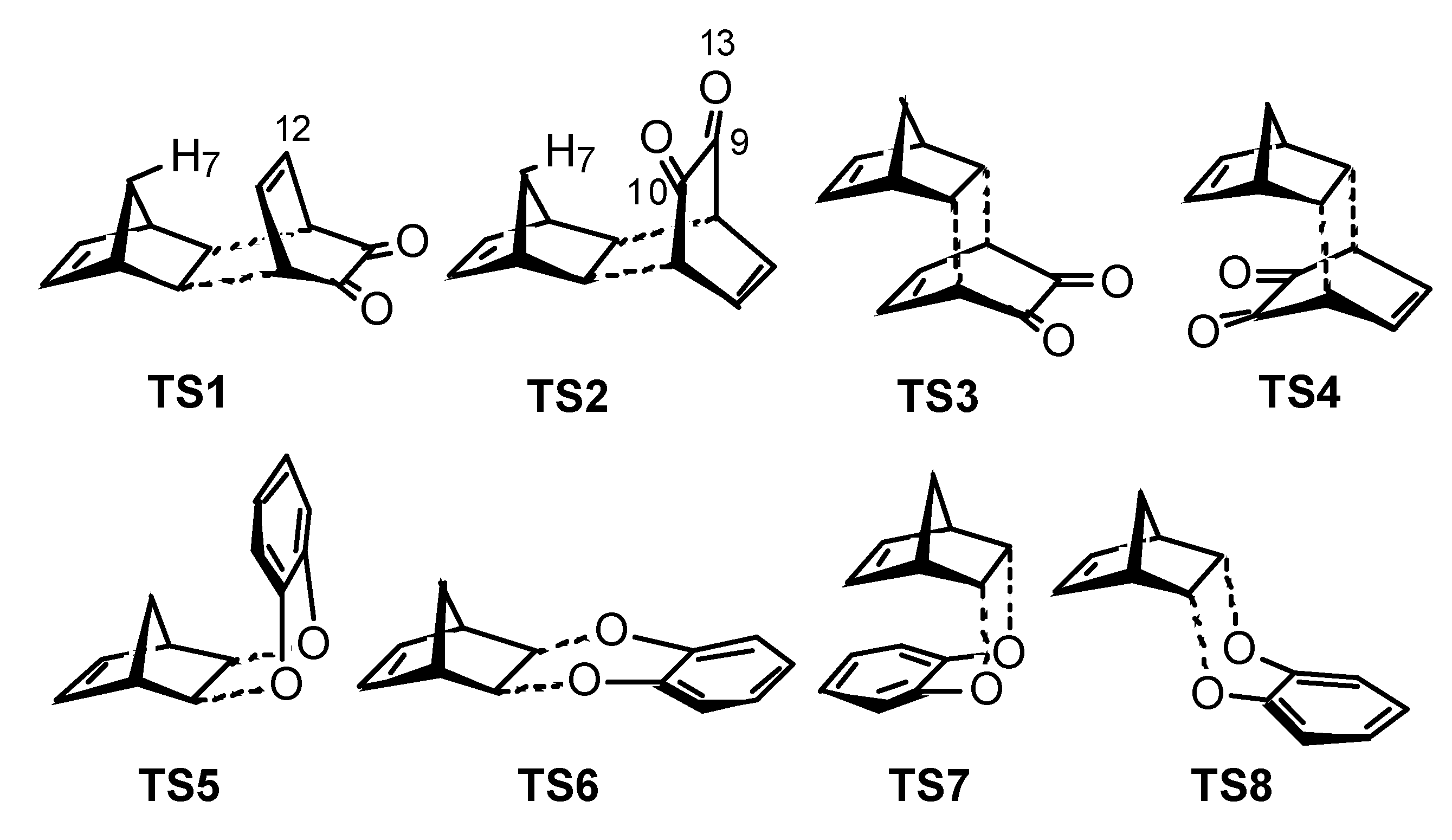
Results and Discussion
Conclusions
Acknowledgements
References
- Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett 1998, 366.
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon, Oxford, 1990. [Google Scholar] Wasserman, W. Diels - Alder Reactions. Elsevier: N. York, 1965. [Google Scholar] Fringuelli, F.; Taticchi, A. Dienes in the Diels - Alder Reaction; Wiley: N. York, 1990. [Google Scholar]
- Johnston, M. R.; Latter, M. J.; Warrener, R. N.; Golic, M.; McKavanagh, D.; Margetic, D. ECSOC-4 paper A0057.
- Warrener, M. R.; Johnston, M. R.; Schultz, A. C.; Golic, M.; Houghton, M.; Gunter, M. J. Synlett 1998, 590. Nunn, E. E.; Wilson, W. S.; Warrener, R. N. Tetrahedron Lett. 1972, 2, 175.
- Houk., K. N.; Li, Y.; Evansceck, J. D. Angew. Chem. Int. Ed. Engl. 1992, 31, 1–682.
- Goldstein, E.; Beno, B.; Houk, K. N. J. Am. Chem. Soc. 1996, 118, 6036. Houk, K. N.; Beno, B. R.; Nendel, M.; Black, K.; Yoo, H. Y.; Wilsey, S.; Lee, J. K. J. Mol. Struct. (Theochem) 1997, 398–399, 169. Domingo, L. R.; Arno, M.; Andres, J. J. Am. Chem. Soc. 1998, 120, 1617. Domingo, L. R.; Picher, M. T.; Andres, J. J. Org. Chem. 2000, 65, 3473. Domingo, L. R.; Picher, M. T.; Andres, J.; Oliva, M. J. Org. Chem. 1999, 64, 3026. Liu, J.; Niwayama, S.; You, Y.; Houk, K. N. J. Org. Chem. 1998, 63, 1064. Bareone, V.; Arnaud, R.; Chavant, P. Y.; Vallee, Y. J. Org. Chem. 1996, 61, 5121. Konecny, R.; Doren, D. J. J. Am. Chem. Soc. 1997, 119, 11098. Jones, G. A.; Shephard, M. J.; Paddon-Row, M. N.; Beno, B. R.; Houk, K. N.; Redmond, K.; Carpenter, B. K. J. Am. Chem. Soc. 1999, 121, 4334.
- Warrener, R. N.; Russell, R. A.; Margetic, D. Synlett 1997, 1, 38. Margetic, D.; Warrener, R. N.; Malpass, J. R. Internet Journal of Computational Chemistry. Bachrach, S., Ed.; 2 - 30 November 1998. http://hackberry.chem.niu.edu/. Sun, G.; Butler, D. N.; Warrener, R. N.; Margetic, D.; Malpass, J. R. Article 062; Electronic Conference on Heterocyclic Chemistry '98; Rzepa, H. S., Kappe, O., Eds.; Imperial College Press, 1998; ISBN ISBN-981-02-3549-1. http://www.ch.ic.ac.uk /ectoc/echet98. Margetic, D.; Warrener, R. N.; Tiekink, E. R. T. Article 064; Electronic Conference on Heterocyclic Chemistry '98; Rzepa, H. S., Kappe, O., Eds.; Imperial College Press, 1998; ISBN ISBN-981-02-3549-1. http://www.ch.ic.ac.uk/ectoc/echet98. Margetic, D.; Warrener, R. N.; Malpass, J. R. Fifth Electronic Conference on Computational Chemistry (ECCC5); Bachrach, S., Ed.; November 2 - 30 1998; http://hackberry.chem.niu.edu/. Golic, M.; Butler, D. N.; Warrener, R. N.; Margetic, D. Third International Electronic Conference on Synthetic Organic Chemistry (ECSOC-3); http://www.reprints.net/ecsoc-3.htmSeptember 1-30 1999.
- Spartan v. 5.0, Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370, Irvine, CA, 92612, 1997.
- Gaussian 98, Revision A.5, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
- Roothan, C. C. J. Rev. Mod. Phys. 1951, 23, 69.
- Hehre, W. J.; Radom, L.; Schleyer, P.v.R.; Pople, J. Ab Initio Molecular Orbital Theory; Wiley: N. York, 1986. [Google Scholar]
- McIver, J. W.; Komornicki, A. J. Am. Chem. Soc. 1972, 94, 2625–2629.
- Becke, A. D. J. Chem. Phys. 1993, 98, 1372.
- Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
- Marchand, A. P.; Ganguly, B.; Shukla, R. Tetrahedron 1998, 54, 4477. Sustmann, R.; Sicking, W. J. Am. Chem. Soc. 1996, 118, 12562.
- For origins and consequences of norbornene π - facial stereoselectivity see: Borden, W. T. Chem. Rev. 1989, 89, 1095. and references cited therein.
- Bach, R. D.; McDouall, J. J. W.; Schlegel, H. B. J. Org. Chem. 1989, 54, 2931.
- Hehre, W. R. Practical Strategies for Electronic Structure Calculations; Wavefunction, Inc.; 18401 Von Karman Suite 370, Irvine, California 92715; 1995. [Google Scholar]
- Loncharich, R. J.; Brown, F. K.; Houk, K. N. J. Org. Chem. 1989, 54, 1129–1134. Birney, D. M.; Houk, K. N. J. Am. Chem. Soc. 1990, 112, 4127–4133.
- Nair, V.; Kumar, S. Tetrahedron 1996, 52, 4029. Nair, V.; Kumar, S. J. Chem. Soc. Chem. Commun. 1994, 1341. Nair, V.; Kumar, S. Synlett 1996, 1143.
- Sample Availability: No samples available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



© 2000 MDPI (http://www.mdpi.org). All rights reserved.
Share and Cite
Margetic, D.; Johnston, M.R.; Warrener, R.N. High-level Computational Study of the Site-, Facial- and Stereoselectivities for the Diels-Alder Reaction Between o-Benzoquinone and Norbornadiene. Molecules 2000, 5, 1417-1428. https://doi.org/10.3390/51201417
Margetic D, Johnston MR, Warrener RN. High-level Computational Study of the Site-, Facial- and Stereoselectivities for the Diels-Alder Reaction Between o-Benzoquinone and Norbornadiene. Molecules. 2000; 5(12):1417-1428. https://doi.org/10.3390/51201417
Chicago/Turabian StyleMargetic, Davor, Martin R. Johnston, and Ronald N. Warrener. 2000. "High-level Computational Study of the Site-, Facial- and Stereoselectivities for the Diels-Alder Reaction Between o-Benzoquinone and Norbornadiene" Molecules 5, no. 12: 1417-1428. https://doi.org/10.3390/51201417
APA StyleMargetic, D., Johnston, M. R., & Warrener, R. N. (2000). High-level Computational Study of the Site-, Facial- and Stereoselectivities for the Diels-Alder Reaction Between o-Benzoquinone and Norbornadiene. Molecules, 5(12), 1417-1428. https://doi.org/10.3390/51201417