Experimental
General
All bp’s and mp’s were uncorrected. The IR spectra were scanned with a Perkin-Elmer 783 spectrophotometer and only the pertinent values are expressed, in cm−1. The 1H-NMR spectra were recorded, either with a Varian EM 360L (60 MHz) or a Bruker AC-200 (200 MHz) spectrometer, using CDCl3 as the solvent. The chemical shift ( δ) and coupling constant (J) values are expressed in ppm and Hz only. The GLC analyses were carried out on a Shimadzu GC-7A chromatograph fitted with a flame ionization detector and glass packed column for routine analysis and a capillary column for the determination of isomeric compositions. The mass spectra (EI) were recorded at 70 eV with a Shimadzu GC-MS QP-1000A spectrometer. Unless otherwise mentioned, the organic extracts were dried over anhydrous Na2SO4.
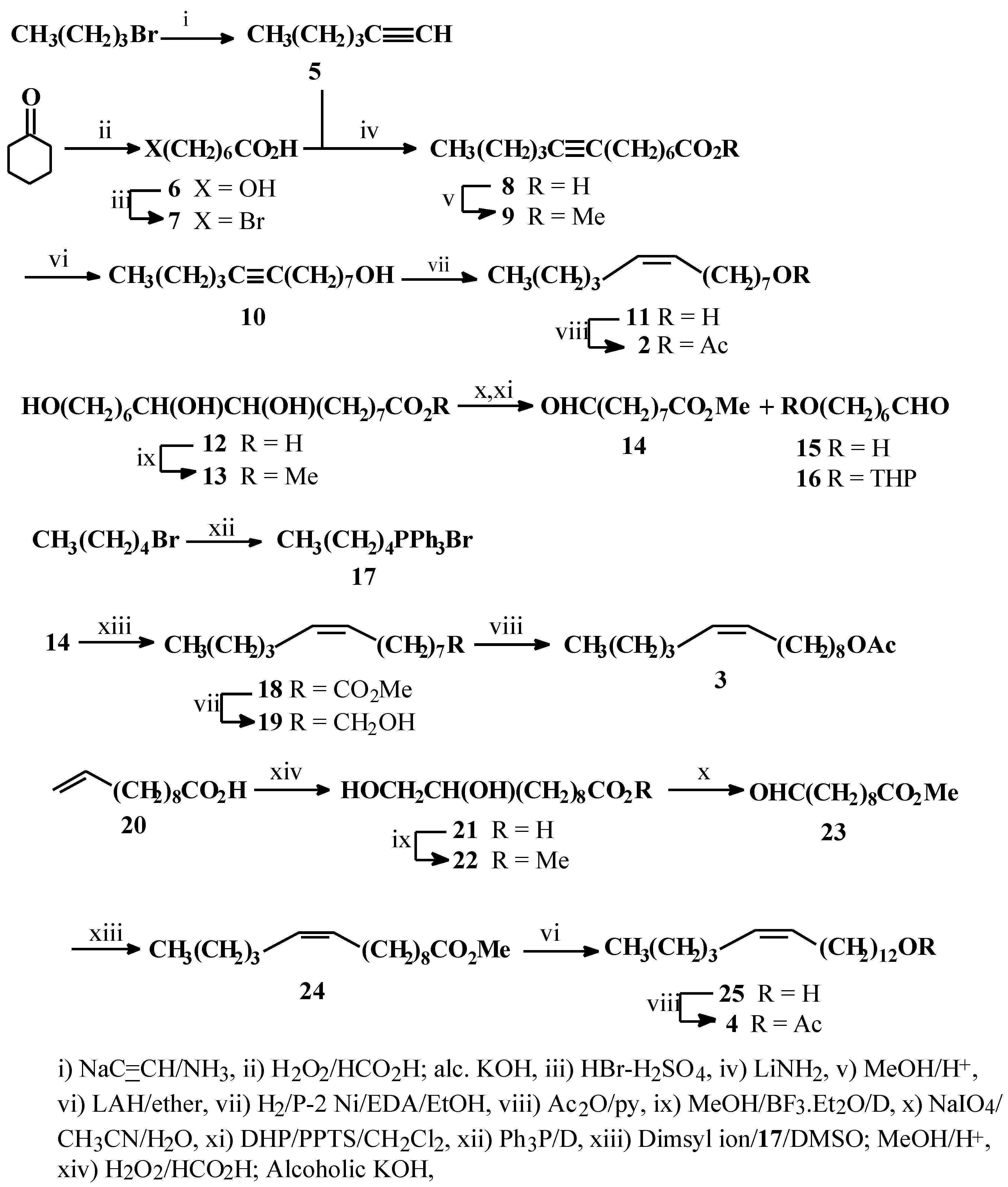
1-Hexyne (5)
Acetylene (gas) was bubbled through a stirred suspension of NaNH
2 [
20] [prepared from Na (21.0 g, 0.98 mol) using Fe(NO
3)
3 (0.5 g) as the catalyst] in liquid ammonia (750 mL) for 2 h. To the resultant grey solution of NaC≡CH, 1-bromobutane (96.0 g, 0.7 mol) was added over a period of 4 h. Stirring was continued for a further 4 h, the temperature of the cooling bath raised to -30 °C, kerosene (200 mL) added and the mixture left overnight. Next day, the reaction mixture was transferred into a separating funnel, washed with cold dil. HCl (2N), water, and dried over MgSO
4. The hexyne was distilled off (55 °C) from the extract under a slow stream of Ar. Redistillation of the fraction gave pure
5. Yield: 41.0 g (71.1%); bp: 70 °C, [lit [
13]. 71 °C]; GLC (60 °C, neat sample): R
t = 0.95 min (98%); IR: 3320, 2140 cm
−1; PMR: δ 0.9 (dist. t, 3H), 1.3-1.8 (m, 6H), 2.1 (s, 1H).
6-Bromoheptanoic acid (7)
To a stirred solution of performic acid [prepared by addition of 30% H2O2 (150 mL) to HCO2H (300 mL) at 0 °C] was added cycloheptanone (47.1 mL, 0.4 mol) over a period of 1.5 h. After 24 h, most of the HCO3H was removed in vacuo, water (200 mL) added and the mixture extracted with CHCl3 by continuous liquid-liquid extraction. Most of the solvent and excess performic acid were removed at atmospheric pressure and at 0.1 mm to give 6-hydroxyheptanoic acid (6). Yield 40.7 g (69.7%); IR: 3400-3000, 2800-2500, 1720 cm−1; 1H-NMR: δ 1.45 (br. s, partially D2O exchangeable OH, 9H), 2.4 (t, J = 6 Hz, 2H), 3.60 (t, J = 6 Hz, 2H), 10.2 (br. s, D2O exchangeable, 1H).
The above acid (36.5 g, 0.25 mol) was introduced into an acid mixture [prepared by slow addition of conc. H2SO4 (15 mL) to 48% HBr (48.1 g)] at 0 °C]. After refluxing for 8 h, the mixture was brought to room temperature, diluted with water and extracted with ether. The extract was washed with water, brine, dried and concentrated. The crude product was purified by vacuum distillation to furnish pure 7. Yield: 26.0 g (62%); bp: 120-125°C/ 0.2 mm; IR: 3400-3000, 2800-2500, 1710 cm−1; 1H-NMR: δ 1.3-1.9 (br. s and m, 8H), 2.4 (t, J = 6 Hz, 2H), 3.43 (t, J = 6 Hz, 2H), 11.2 (s, D2O exchangeable, 1H).
Methyl tridec-8-ynoate (9)
To a stirred solution of LiNH2 [prepared from Li (3.2 g, 0.45 mol) using Fe(NO3)3 (0.1 g) as the catalyst] in liquid ammonia (300 mL) was added the alkyne 5 (24.6 g, 0.3 mol) and the mixture stirred for 1 h. A solution of the acid 7 (20.9 g, 0.1 mol) in THF (150 mL) was then added dropwise over the course of 1 h. The reaction mixture was stirred for 4 h at the same temperature and left overnight. The residue was treated with dil. HCl (10:1) and extracted with ether. The ether layer was extracted thoroughly with 5% aqueous NaOH, then the ether layer separated and the aqueous layer acidified with dil. HCl. This was then re-extracted with ether, the new organic extract was washed with water, brine and dried. Concentration of the extract gave the crude acid 8 which was used for the next step without any purification .
Thus, a solution of the above acid in MeOH (100 mL) was refluxed in the presence of PTS (0.1 g) to furnish the ester 9 after the usual isolation and purification by column chromatography (silica gel, 5% EtOAc/hexane). Yield: 18.1 g (81%); IR: 1750, 1060, 720 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.32 (br. s, 12H), 2.1-2.2 (m, 4H), 2.32 (t, J = 6 Hz, 2H), 3.5 (s, 3H).
Tridec-8-yn-1-ol (10)
Reduction of the ester 9 (9.0 g, 0.04 mol) with LAH (1.15 g, 0.03 mol) in ether (100 mL) gave pure 10. Yield: 7.1 g (89.6%); bp: 110 °C (bath)/ 0.3 mm; IR: 3440, 3005, 1660 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.1-1.5 (br. s & m, 14H), 1.8-2.1 (m, 4H), 2.8 (s, D2O exchangeable, 1H), 3.3-3.5 (m, 3H), 5.3-5.5 (m, 2H).
(Z)-Tridec-8-en-1-ol (11)
To a stirred suspension of nickel acetate (1.24 g, 5.0 mmol) in EtOH (50 mL), a solution of NaBH4 (295.0 mg, 75.0 mmol) was added. A black colloidal suspension of P-2 Ni appeared immediately with the evolution of hydrogen. When the evolution of gas ceased, a mixture of the alkynol 10 (11.2 g, 0. 05 mol) and ethylenediamine (0.9 g) was added and the mixture stirred under a slight positive pressure of hydrogen. After the required hydrogen uptake, the mixture was diluted with hexane and passed through a pad of celite to remove the catalyst. The clear solution was concentrated and the product purified by distillation to afford 11. Yield: 9.6 g (85%); bp: 90 °C/0.3 mm; IR: 3350, 3010, 1650 cm−1; 1H-NMR: δ 0.95 (dist. t, 3H), 1.2-1.7 (m, 14H), 1.9 (s, D2O exchangeable OH, 1H), 2.0-2.3 (m, 4H), 3.65 (t, J = 6 Hz, 2H), 5.3-5.5 (m, 2H).
(Z)-8-Tridecenyl acetate (2)
A mixture of 11 (5.94 g, 0.03 mol), Ac2O (10.0 mL, 0.01 mol) and pyridine (15 mL) was stirred for 12 h at room temperature. Ice-cold water was added to the mixture and stirring continued for 1 h. The mixture was extracted with ether, the ether layer washed successively with water, aqueous NaHCO3, water, aqueous HCl, water and brine and finally dried. Removal of the solvent followed by distillation furnished pure 2. Yield: 6.0 g (83%); bp: 130° (bath) C/0.3 mm; GLC (3% OV-17, 40 mL N2/min, temp. 180 °C): Rt = 5.04 min (98%); IR: 3010, 1740, 1240 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.3 (br. s, 14H), 2.0-2.3 (m containing a s at δ 2.1, 7H), 4.3 (t, J = 6 Hz, 2H), 5.2-5.5 (m, 2H).
Methyl aleuritate (13)
A solution of aleuritic acid (
12) (76.0 g, 0.25 mol) in MeOH (300 mL) containing BF
3.Et
2O (15 mL) was refluxed for 4 h. Most of the solvent was removed in vacuo, the residue taken up in ether and the ether layer washed with water, brine, dried and concentrated. The product was purified by chromatography (silica gel, 0-10% EtOAc/hexane) to furnish
13. Yield: 62.8 g (79.5%); mp: 75-78°C, (lit. [
16] mp: 73 °C); IR: 3440, 1750 and 1480 cm
−1;
1H-NMR: δ 1.32 (br. s, 22H), 2.21 (t,
J = 7 Hz), 3.68 (t,
J = 7 Hz, 2H), 3.7 (s, 3H), 3.8-4.0 (m, 2H).
Pentyltriphenylphosphonium bromide (17)
A solution of triphenylphosphine (262.0 g, 1.0 mol) and 1-bromopentane (151.0 g, 1.0 mol) in CH
3CN (500 mL) was refluxed for 30 h. After removing most of the solvent, benzene (300 mL) was added and the mixture refluxed for 3 h. After cooling, the upper layer was decanted to remove the unreacted reactants, the separated white crystalline solid was collected, washed with benzene and dried under vacuum (0.1 mm) to afford
17. Yield: 367.0 g (88.8%), mp 165 °C, (lit. [
17] mp: 167-168°C).
Methyl (Z)-9-tetradecenoate (18)
The aldehyde 14 (11.2 g, 0.06 mol) was reacted with the phosphorane generated from the phosphonium salt 17 (27.2 g, 0.066 mol) using a dimsyl ion as the base (0.072 mol) in DMSO (100 mL). Subsequent work-up followed by column chromatography of the residue (silica gel, 0-10% EtOAc/hexane) gave 18. Yield: 7.5 g (52%); bp: 140°C (bath)/ 0.5 mm; IR: 3010, 1745, 1650 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.3 (br. s, 14H), 2.0-2.5 (m, 6H), 3.65 (s, 3H), 5.35 (t, J = 7 Hz, 2H).
(Z)-9-Tetradecen-1-ol (19)
To a stirred suspension of LiAlH4 (1.14 g, 0.03 mol) in anhydrous ether (100 mL) compound 18 (7.2 g, 0.03 mol) in ether (50 mL) was slowly added. The mixture was refluxed untill the completion of the reaction (cf. TLC, 8 h) and quenched by dropwise addition of aqueous saturated Na2SO4 solution (7-8 mL). The organic layer was separated from the white granular solid which was thoroughly washed with ether. After drying, the organic layer was concentrated and the resulting alcohol 19 purified by evaporative distillation. Yield: 5.5 g (86.5%); bp: 125 °C/0.3 mm; IR: 3330, 3010, 1640, 1080 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.3 (br. s, 16H), 1.5 (s, D2O exchangeable, 1H), 1.8-2.2 (m, 4H), 3.45 (t, J = 7 Hz, 2H), 5.5 (t, J = 7 Hz, 2H).
(Z)-9-Tetradecenyl acetate (3)
A mixture of 19 (4.24 g, 0.02 mol) and Ac2O (15 mL) in pyridine (10 mL) was stirred for 12 h at room temperature. Water was added to the mixture which was stirred for another 1 h and extracted with ether. The ether layer was washed with 5% aqueous HCl, water, 10% aqueous NaHCO3, water and brine. After drying, the extract was concentrated in vacuo and the residue distilled to obtain pure 3. Yield: 4.2 g (83%); bp: 130 °C/0.1 mm; GLC (3% OV-17, 40 mL N2/min, temp. 180 °C): Rt = 7.4 min (96%); GLC (capillary, OV-17, 50 M x 0.25 mm, split (1:100), 2 mL N2/min, temp. 210 °C): Rt = 13.2 min; IR: 3020, 1740, 1650, 1240 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.3 (br. s, 16H), 1.9-2.3 (m containing a s at δ 2.1, 7H), 4.05 (t, J = 7 Hz, 2H), 5.2-5.5 (m, 2H). Anal. Calcd. for C16H30O2: C 75.6, H 11.8; Found: C 75.5, H 11.9.
Methyl 10,11-dihydroxyundecanoate (22)
A solution of HCO3H [prepared from 30% H2O2 (25 mL) and HCO2H (15 mL) at 0 °C] was slowly added to 10-undecenoic acid (20) (18.4 g, 0.1 mol). The reaction mixture was stirred for 8 h at 40 °C and subsequently at room temperature overnight. The mixture was distilled in vacuo (10 mm), the residue diluted with water and extracted with ether. The ether layer was washed with water, dried (MgSO4) and concentrated.
The residue was treated with 5% aqueous KOH (100 mL) and heated on a steam bath for 1 h, cooled and poured into excess cold dil. HCl (1N) under vigorous stirring (maintaining the temperature ~20 °C). The mixture was extracted with ether which was washed with water, dried and concentrated to give the crude diol acid
21 which was recrystallized from EtOH/H
2O. Yield: 13.2 g (55%); mp: 86-87 °C, (lit. [
19] mp: 84-85 °C); IR: 3500-3300, 1710, 1460 cm
−1;
1H-NMR: δ 1.3 (br. s, 14H), 1.9-2.4 (m partially D
2O exchangeable, 4H), 3.5-3.8 (m, 3H), 9.8 (s, D
2O exchangeable, 1H).
Esterification of 21 (13.0 g, 0.06 mol) with MeOH (100 mL) in the presence of BF3.Et2O (5 mL) gave the diolester 22. A part of this was distilled to prepare an analytical sample. Yield: 6.0 g (~ quant.); bp: 145° (bath) C/0.2 mm; IR: 3450, 1750 cm−1; 1H-NMR: δ 1.33 (m, 14H), 2.2-2.5 (m, 2H), 3.2-3.7 (m, partially D2O exchangeable, 5H), 3.8 (s, 3H). Anal. Calcd. For C12H24O4: C 62.03, H 10.41; Found: C 62.12, H 10.31.
Methyl (10Z)-pentadecenoate (24)
As described earlier, the Wittig reaction between the aldehyde 23 (9.0 g, 0.045 mol) and the phosphorane generated from 17 (20.7 g, 0.05 mol) using a dimsyl ion as the base (0.054 mol) in DMSO (50 mL) and subsequent workup gave 24. The ester was purified by column chromatography over silica gel (10% EtOAc/hexane). Yield: 5.8 g (51%); bp: 135-140 °C (bath)/1 mm; GLC (3% OV-17, 180 °C): Rt = 7.2 min (98%); IR: 3010, 2950, 2870, 1745 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.3 (br. s, 16H), 2.0-2.4 (m, 4H), 2.3 (t, J = 6 Hz, 2H), 3.59 (s, 3H), 5.2-5.3 (m, 2H).
(Z)-Pentadec-10-en-1-ol (25)
Reduction of 24 (5.0 g, 0.02 mol) with LAH (0.5 g, 0.026 mol) in ether (100 mL) gave the alcohol 25. Yield: 3.6 g (80%); bp: 110-120 °C/0.3 mm; IR: 3350, 3010, 1660, 1070 cm−1; 1H-NMR: δ 0.9 (dist. t, 3H), 1.33 (br. s, 18H), 1.62 (s, D2O exchangeable, 1H), 1.8-2.2 (m, 4H), 3.68 (t, J = 6 Hz, 2H), 5.33 (t, J = 5 Hz, 2H).
(Z)-Pentadec-10-en-1-yl acetate (4)
Acetylation of 25 (3.4 g, 0.015 mol) with Ac2O (8.0 mL) and pyridine (12.0 mL) gave the pheromone 4. yield: 3.3 g (82%); bp: 110 °C (bath)/0.5 mm; GLC (180 °C): Rt = 9.6 min (96%); IR: 3000, 1740, 1240 cm−1; PMR: δ 0.85 (dist. t, 3H), 1.32 (br. s, 18H), 1.95 (s, 3H), 2.0-2.2 (m, 4H), 3.8-4.1 (m, 2H), 5.2-5.3 (m, 2H).

{kind=link}