
Visible Light-Mediated Organoboron-Catalyzed Metal-Free Synthesis of Silanols from Silanes

 , and
, and
Abstract
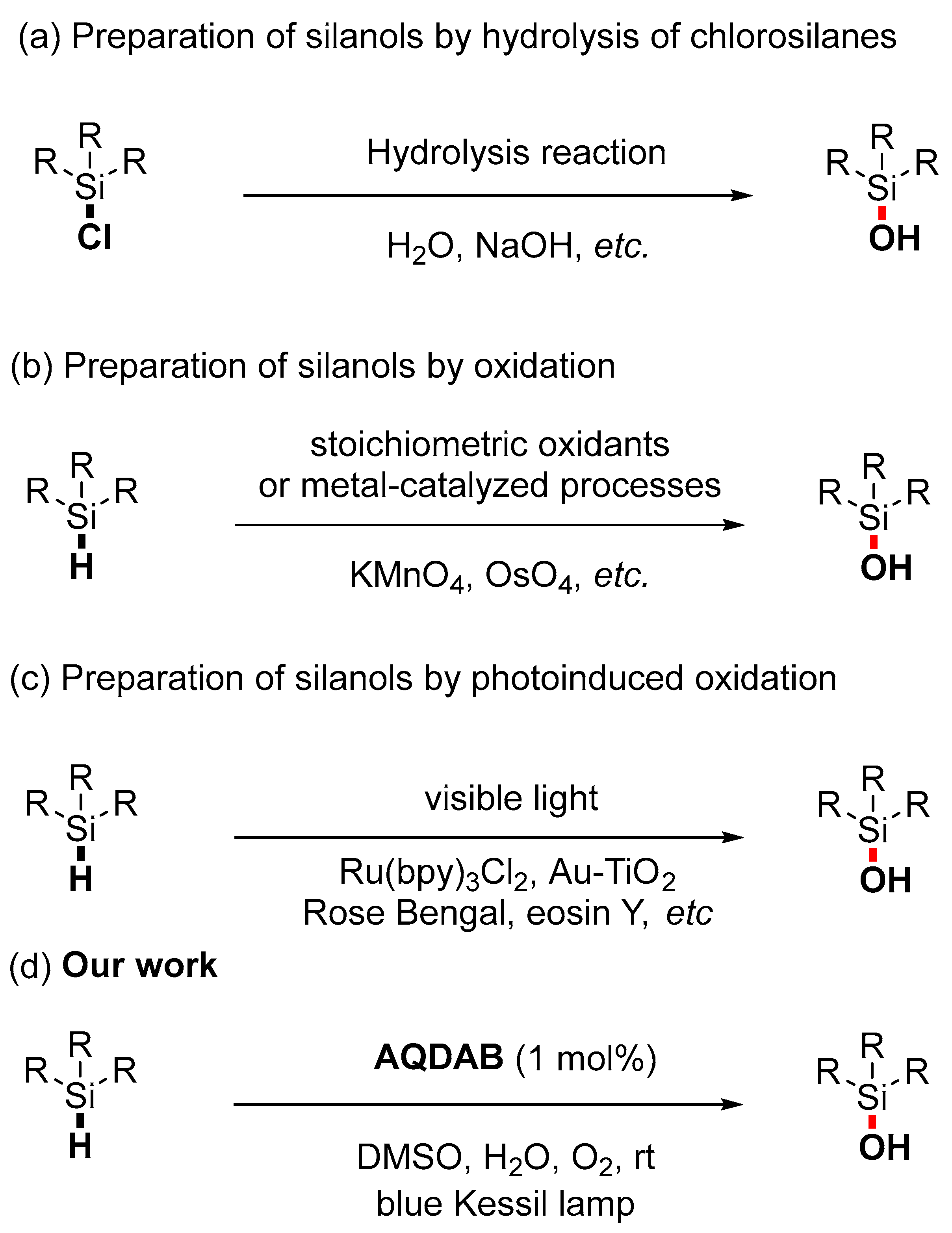
1. Introduction
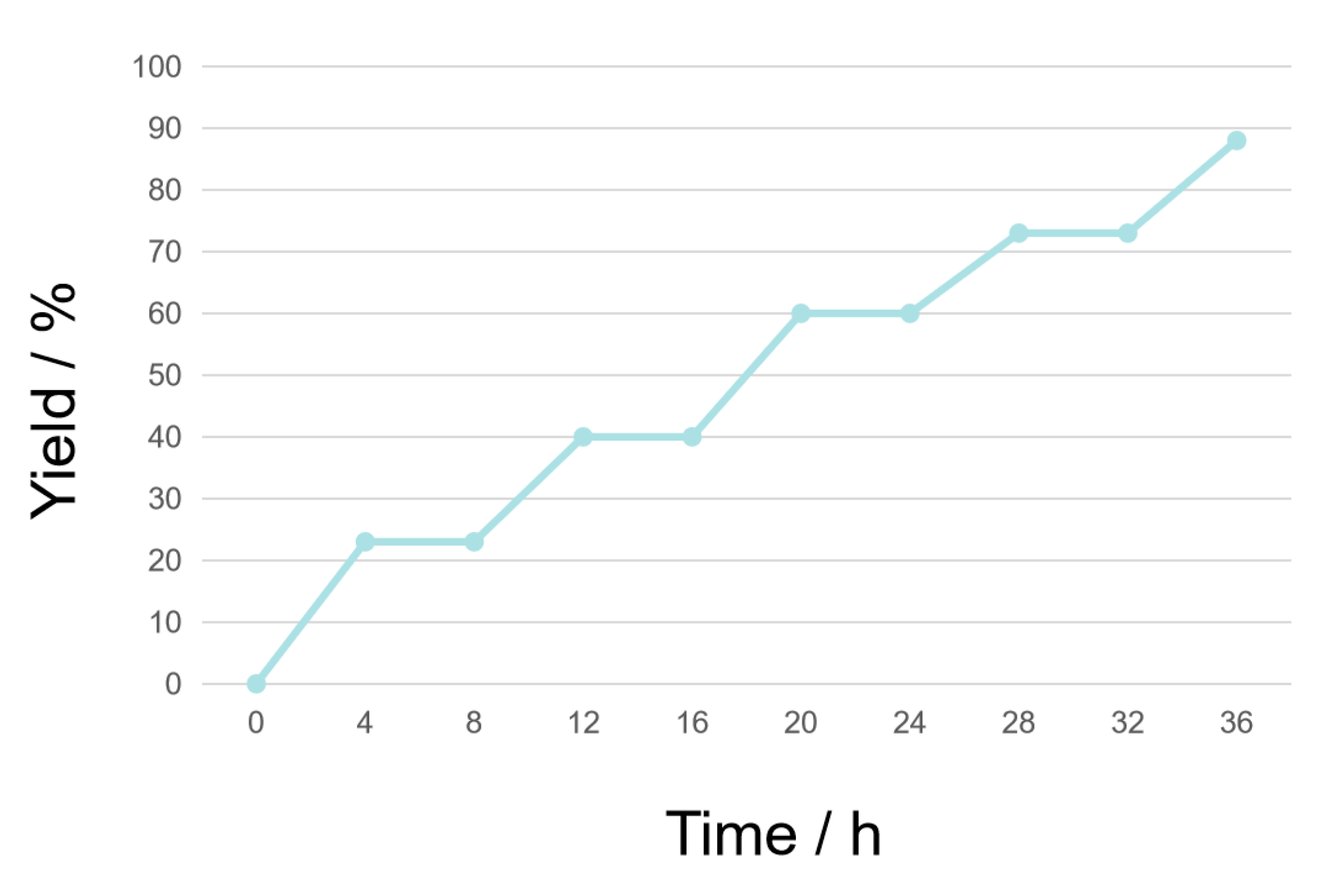
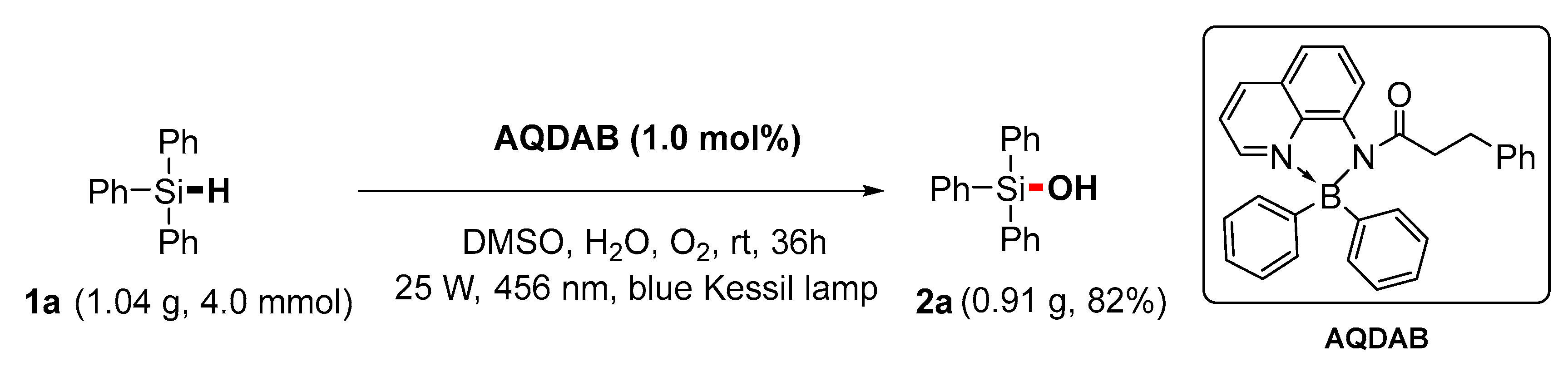
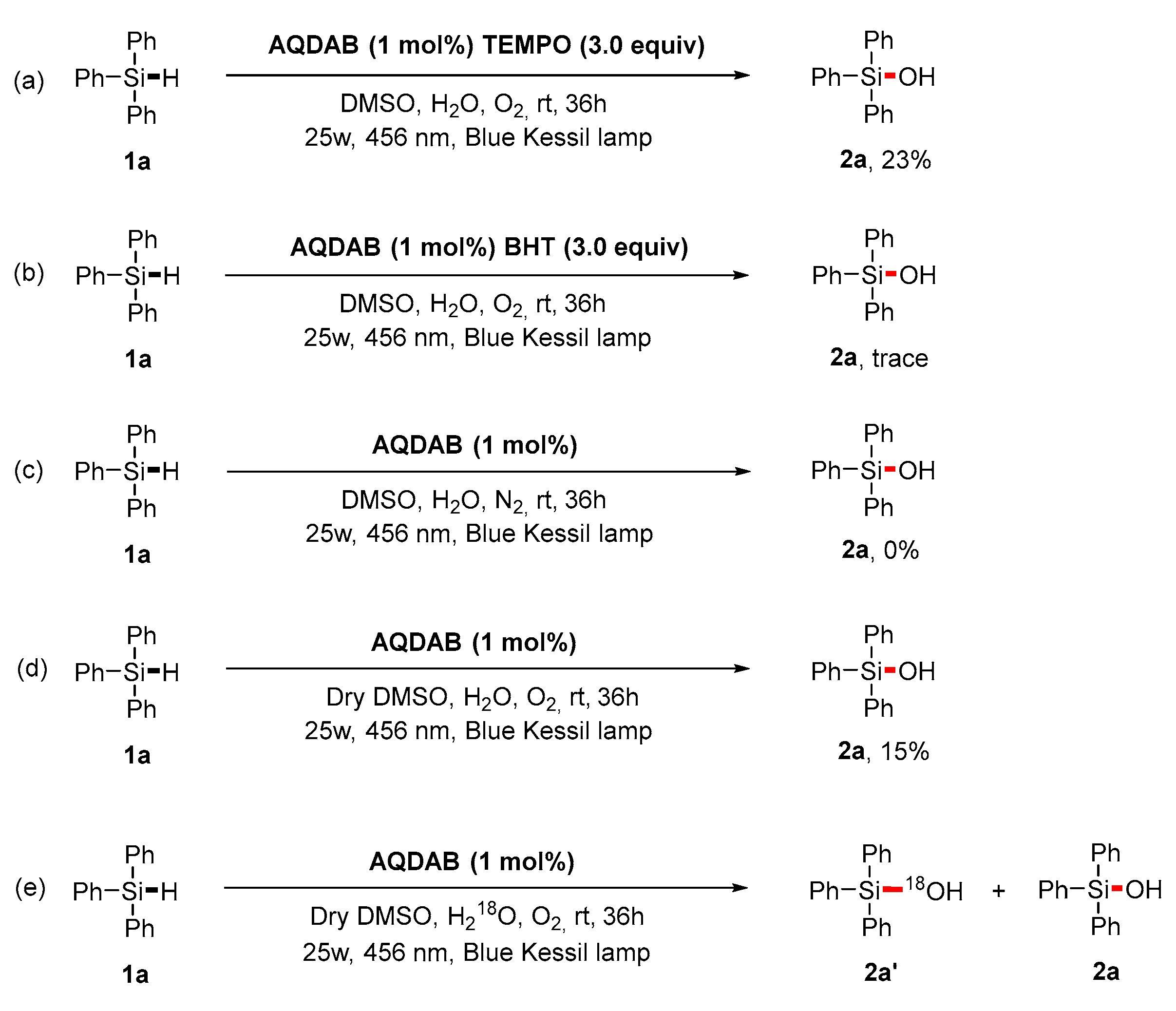
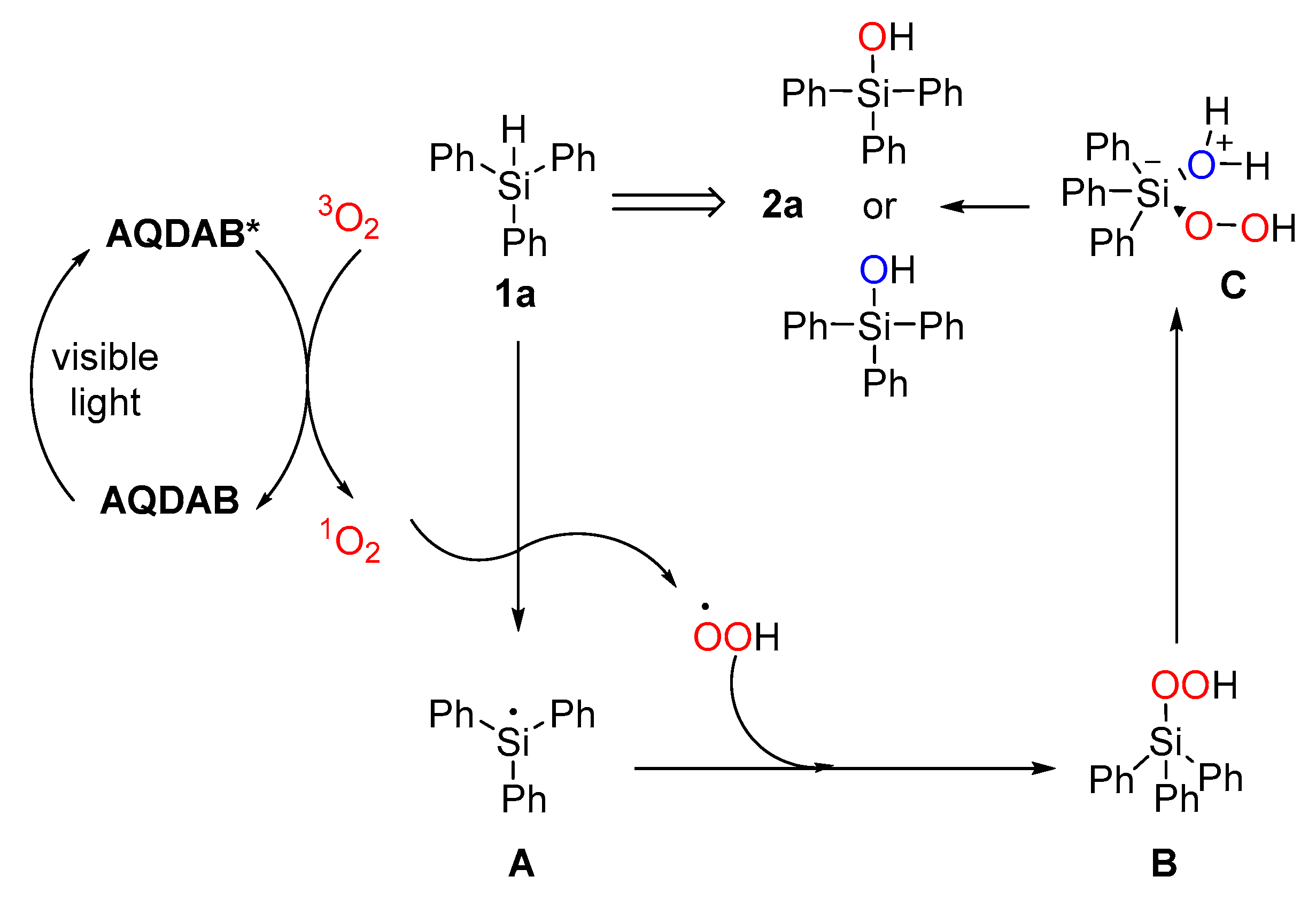
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Instruments
3.2. General Procedure for the Synthesis of Silanols
3.3. General Procedure for the Synthesis of AQDAB
3.3.1. Method A for the Synthesis of AQDAB
3.3.2. Method B for the Synthesis of AQDAB
3.3.3. Characterization Data of the AQDAB
3.4. General Procedure for the Synthesis of Starting Materials [45]
3.4.1. The Synthesis of the Starting Materials of 2a–2f, 2j, 2k, 2m–2r, 2v
3.4.2. The Synthesis of the Starting Materials of 2g–2i, 2l
3.5. Characterization Data of Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chandrasekhar, V.; Boomishankar, R.; Nagendran, S. Recent developments in the synthesis and structure of organosilanols. Chem. Rev. 2004, 104, 5847–5910. [Google Scholar] [CrossRef]
- Murugavel, R.; Voigt, A.; Walawalkar, M.G.; Roesky, H.W. Hetero-and metallasiloxanes derived from silanediols, disilanols, silanetriols, and trisilanols. Chem. Rev. 1996, 96, 2205–2236. [Google Scholar] [CrossRef]
- Denmark, S.E.; Regens, C.S. Palladium-catalyzed cross-coupling reactions of organosilanols and their salts: Practical alternatives to boron-and tin-based methods. Acc. Chem. Res. 2008, 41, 1486–1499. [Google Scholar] [CrossRef]
- Denmark, S.E. The interplay of invention, discovery, development, and application in organic synthetic methodology: A case study. J. Org. Chem. 2009, 74, 2915–2927. [Google Scholar] [CrossRef] [PubMed]
- Mewald, M.; Schiffner, J.A.; Oestreich, M. A New Direction in C–H Alkenylation: Silanol as a Helping Hand. Angew. Chem. Int. Ed. 2012, 51, 1763–1765. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, G.; Breit, B. Entfernbare dirigierende Gruppen in der organischen Synthese und Katalyse. Angew. Chem. 2011, 123, 2498–2543. [Google Scholar] [CrossRef]
- Tran, N.T.; Min, T.; Franz, A.K. Silanediol hydrogen bonding activation of carbonyl compounds. Chem. A Eur. J. 2011, 17, 9897–9900. [Google Scholar] [CrossRef]
- Schafer, A.G.; Wieting, J.M.; Mattson, A.E. Silanediols: A new class of hydrogen bond donor catalysts. Org. Lett. 2011, 13, 5228–5231. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon molecules with medicinal applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef]
- Tacke, R.; Schmid, T.; Hofmann, M.; Tolasch, T.; Francke, W. Sila-linalool as a pheromone analogue: A study on C/Si bioisosterism. Organometallics 2003, 22, 370–372. [Google Scholar] [CrossRef]
- Kim, J.K.; Sieburth, S.M. Synthesis and properties of a sterically unencumbered δ-silanediol amino acid. J. Org. Chem. 2012, 77, 2901–2906. [Google Scholar] [CrossRef]
- Cella, J.A.; Carpenter, J.C. Procedures for the preparation of silanols. J. Organomet. Chem. 1994, 480, 23–26. [Google Scholar] [CrossRef]
- Cho, H.M.; Jeon, S.H.; Lee, H.K.; Kim, J.H.; Park, S.; Choi, M.-G.; Lee, M.E. Facile syntheses, structural characterizations, and isomerization of disiloxane-1, 3-diols. J. Organomet. Chem. 2004, 689, 471–477. [Google Scholar] [CrossRef]
- McN, S. The study of methods to synthesize silylated calixarenes in the 1, 3-alternate conformation. J. Org. Chem 1993, 58, 7584–7586. [Google Scholar]
- Spialter, L.; Austin, J.D. Ozone: A New Cleavage Reagent for Organosilanes. J. Am. Chem. Soc. 1965, 87, 4406. [Google Scholar] [CrossRef]
- Sommer, L.; Ulland, L.A.; Parker, G. Stereochemistry of asymmetric silicon. XX. Hydroxylation and carbene insertion reactions of R3SiH. J. Am. Chem. Soc. 1972, 94, 3469–3471. [Google Scholar] [CrossRef]
- Adam, W.; Mello, R.; Curci, R. O-Atom Insertion into Si–H Bonds by Dioxiranes: A Stereospecific and Direct Conversion of Silanes into Silanols. Angew. Chem. Int. Ed. Engl. 1990, 29, 890–891. [Google Scholar] [CrossRef]
- Valliant-Saunders, K.; Gunn, E.; Shelton, G.R.; Hrovat, D.A.; Borden, W.T.; Mayer, J.M. Oxidation of tertiary silanes by osmium tetroxide. Inorg. Chem. 2007, 46, 5212–5219. [Google Scholar] [CrossRef]
- Lickiss, P.D.; Lucas, R. Oxidation of sterically hindered organosilicon hydrides using potassium permanganate. J. Organomet. Chem. 1996, 521, 229–234. [Google Scholar] [CrossRef]
- Cavicchioli, M.; Montanari, V.; Resnati, G. Oxyfunctionalization reactions by perfluoro cis-2, 3-dialkyloxaziridines. Enantioselective conversion of silanes into silanols. Tetrahedron Lett. 1994, 35, 6329–6330. [Google Scholar] [CrossRef]
- Spialter, L.; Pazdernik, L.; Bernstein, S.; Swansiger, W.A.; Buell, G.R.; Freeburger, M.E. Mechanism of the reaction of ozone with the silicon-hydrogen bond. J. Am. Chem. Soc. 1971, 93, 5682–5686. [Google Scholar] [CrossRef]
- Schubert, U.; Lorenz, C. Conversion of Hydrosilanes to Silanols and Silyl Esters Catalyzed by [Ph3PCuH]6. Inorg. Chem. 1997, 36, 1258–1259. [Google Scholar] [CrossRef]
- Ison, E.A.; Corbin, R.A.; Abu-Omar, M.M. Hydrogen production from hydrolytic oxidation of organosilanes using a cationic oxorhenium catalyst. J. Am. Chem. Soc. 2005, 127, 11938–11939. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Ko, S.; Chang, S. Highly selective and practical hydrolytic oxidation of organosilanes to silanols catalyzed by a ruthenium complex. J. Am. Chem. Soc. 2000, 122, 12011–12012. [Google Scholar] [CrossRef]
- Lee, Y.; Seomoon, D.; Kim, S.; Han, H.; Chang, S.; Lee, P.H. Highly efficient iridium-catalyzed oxidation of organosilanes to silanols. J. Org. Chem. 2004, 69, 1741–1743. [Google Scholar] [CrossRef]
- Mitsudome, T.; Noujima, A.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Supported gold nanoparticle catalyst for the selective oxidation of silanes to silanols in water. Chem. Commun. 2009, 35, 5302–5304. [Google Scholar] [CrossRef]
- John, J.; Gravel, E.; Hagège, A.; Li, H.; Gacoin, T.; Doris, E. Catalytic oxidation of silanes by carbon nanotube–gold nanohybrids. Angew. Chem. 2011, 33, 7675–7678. [Google Scholar] [CrossRef]
- Kikukawa, Y.; Kuroda, Y.; Yamaguchi, K.; Mizuno, N. Diamond-Shaped [Ag4]4+ Cluster Encapsulated by Silicotungstate Ligands: Synthesis and Catalysis of Hydrolytic Oxidation of Silanes. Angew. Chem. Int. Ed. 2012, 51, 2434–2437. [Google Scholar] [CrossRef]
- Adam, W.; Corma, A.; García, H.; Weichold, O. Titanium-Catalyzed Heterogeneous Oxidations of Silanes, Chiral Allylic Alcohols, 3-Alkylcyclohexanes, and Thianthrene 5-Oxide: A Comparison of the Reactivities and Selectivities for the Large-Pore Zeolite Ti-β, the Mesoporous Ti-MCM-41, and the Layered Alumosilicate Ti-ITQ-2. J. Catal. 2000, 196, 339–344. [Google Scholar]
- Adam, W.; Mitchell, C.M.; Saha-Möller, C.R.; Weichold, O. Host−Guest Chemistry in a Urea Matrix: Catalytic and Selective Oxidation of Triorganosilanes to the Corresponding Silanols by Methyltrioxorhenium and the Urea/Hydrogen Peroxide Adduct. J. Am. Chem. Soc. 1999, 121, 2097–2103. [Google Scholar] [CrossRef]
- Adam, W.; Saha-Möller, C.R.; Weichold, O. NaY zeolite as host for the selective heterogeneous oxidation of silanes and olefins with hydrogen peroxide catalyzed by methyltrioxorhenium. J. Org. Chem. 2000, 65, 2897–2899. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, R.; Kamata, K.; Mizuno, N. Highly selective oxidation of organosilanes to silanols with hydrogen peroxide catalyzed by a lacunary polyoxotungstate. Angew. Chem. 2009, 121, 9062–9066. [Google Scholar] [CrossRef]
- Limnios, D.; Kokotos, C.G. Organocatalytic oxidation of organosilanes to silanols. ACS Catal. 2013, 3, 2239–2243. [Google Scholar] [CrossRef]
- He, P.; Zhang, F.; Si, X.; Jiang, W.; Shen, Q.; Li, Z.; Zhu, Z.; Tang, S.; Gui, Q.-W. Visible-Light-Induced Aerobic Oxidation of Tertiary Silanes to Silanols using Molecular Oxygen as an Oxidant. Synthesis 2022, 55, 765–772. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Duan, P.; Zhang, W. Highly Active and Selective Photocatalytic Oxidation of Organosilanes to Silanols. ACS Sustain. Chem. Eng. 2022, 10, 4642–4649. [Google Scholar] [CrossRef]
- Cao, J.; Yang, X.; Ma, L.; Lu, K.; Zhou, R. Metal-free hydrogen evolution cross-coupling enabled by synergistic photoredox and polarity reversal catalysis. Green Chem. 2021, 23, 8988–8994. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.; Liu, L.-C.; Jiang, C.; He, T.; He, W. Metal-free visible-light-mediated aerobic oxidation of silanes to silanols. Sci. China Chem. 2018, 61, 1594–1599. [Google Scholar] [CrossRef]
- Lv, H.; Laishram, R.D.; Chen, J.; Khan, R.; Zhu, Y.; Wu, S.; Zhang, J.; Liu, X.; Fan, B. Metal-Free Visible-Light-Induced Atom-Transfer Radical Addition Reaction of Alkenes/Alkynes with ICH2CN. Chem. Commun. 2021, 57, 3660–3663. [Google Scholar] [CrossRef] [PubMed]
- Zu, W.; Day, C.; Wei, L.; Jia, X.; Xu, L. Dual aminoquinolate diarylboron and nickel catalysed metallaphotoredox platform for carbon–oxygen bond construction. Chem. Commun. 2020, 56, 8273–8276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zu, W.; Tian, Q.; Cao, Z.; Wei, Y.; Xu, L. A nickel/organoboron catalyzed metallaphotoredox platform for C (sp 2)–P and C (sp 2)–S bond construction. Org. Chem. Front. 2022, 9, 1070–1076. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, J.; Xu, L. 3D porous copper skeleton supported zinc anode toward high capacity and long cycle life zinc ion batteries. ACS Sustain. Chem. Eng. 2020, 8, 13894–13899. [Google Scholar] [CrossRef]
- Wei, L.; Wei, Y.; Zhang, J.; Xu, L. Visible-light-mediated organoboron-catalysed metal-free dehydrogenation of N-heterocycles using molecular oxygen. Green Chem. 2021, 23, 4446–4450. [Google Scholar] [CrossRef]
- Cui, H.; Wei, W.; Yang, D.; Zhang, Y.; Zhao, H.; Wang, L.; Wang, H. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent DSS-induced colitis in mice. Green Chem. 2017, 19, 3520–3524. [Google Scholar] [CrossRef]
- Rahaman, R.; Das, S.; Barman, P. Visible-light-induced regioselective sulfenylation of imidazopyridines with thiols under transition metal-free conditions. Green Chem. 2018, 20, 141–147. [Google Scholar] [CrossRef]
- Liang, H.; Wang, L.J.; Ji, Y.X.; Wang, H.; Zhang, B. Selective electrochemical hydrolysis of hydrosilanes to silanols via anodically generated silyl cations. Angew. Chem. Int. Ed. 2021, 60, 1839–1844. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Variations from the Standard Conditions | Yield (b) |
| 1 | None | 88% |
| 2 | without AQDAB | N.R. |
| 3 | without O2 (under N2) | N.R. |
| 4 | without Kessil lamp | N.R. |
| 5 | air instead of O2 | 17% |
| 6 | DMA instead of DMSO | 20% |
| 7 | DMF instead of DMSO | 28% |
| 8 | MeCN instead of DMSO | trace |
| 9 | DCM instead of DMSO | trace |
| 10 | reaction time: 24 h | 68% |
| 11 | white Kessil lamp | N.R. |
| 12 | AQDAB (0.5 mol%) | 85% |
| 13 | AQDAB (2 mol%) | 87% |
 | ||
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Cao, X.; Wei, L.; Zhang, J.; Zhang, J.; Liu, P.; Xu, L.; Li, P. Visible Light-Mediated Organoboron-Catalyzed Metal-Free Synthesis of Silanols from Silanes. Molecules 2023, 28, 4082. https://doi.org/10.3390/molecules28104082
Yang J, Cao X, Wei L, Zhang J, Zhang J, Liu P, Xu L, Li P. Visible Light-Mediated Organoboron-Catalyzed Metal-Free Synthesis of Silanols from Silanes. Molecules. 2023; 28(10):4082. https://doi.org/10.3390/molecules28104082
Chicago/Turabian StyleYang, Jinbo, Xiangxue Cao, Lanfeng Wei, Jianshu Zhang, Jinli Zhang, Ping Liu, Liang Xu, and Pengfei Li. 2023. "Visible Light-Mediated Organoboron-Catalyzed Metal-Free Synthesis of Silanols from Silanes" Molecules 28, no. 10: 4082. https://doi.org/10.3390/molecules28104082
APA StyleYang, J., Cao, X., Wei, L., Zhang, J., Zhang, J., Liu, P., Xu, L., & Li, P. (2023). Visible Light-Mediated Organoboron-Catalyzed Metal-Free Synthesis of Silanols from Silanes. Molecules, 28(10), 4082. https://doi.org/10.3390/molecules28104082