Synthesis and Antiproliferative Activity of Novel Heterocyclic Glycyrrhetinic Acid Derivatives



1
Laboratory of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Coimbra, 3000-548 Coimbra, Portugal
2
Centre for Neuroscience and Cell Biology, 3004-504 Coimbra, Portugal
3
Department of Biochemistry and Molecular Biomedicine, Faculty of Biology, University of Barcelona, Diagonal 643, 08028 Barcelona, Spain
4
Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), 28029 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(4), 766; https://doi.org/10.3390/molecules24040766
Submission received: 16 January 2019
/
Revised: 5 February 2019
/
Accepted: 15 February 2019
/
Published: 20 February 2019
(This article belongs to the Section Medicinal Chemistry)
Abstract
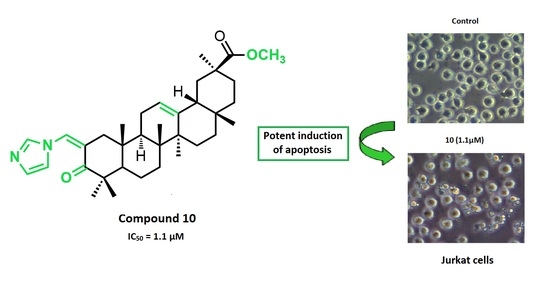
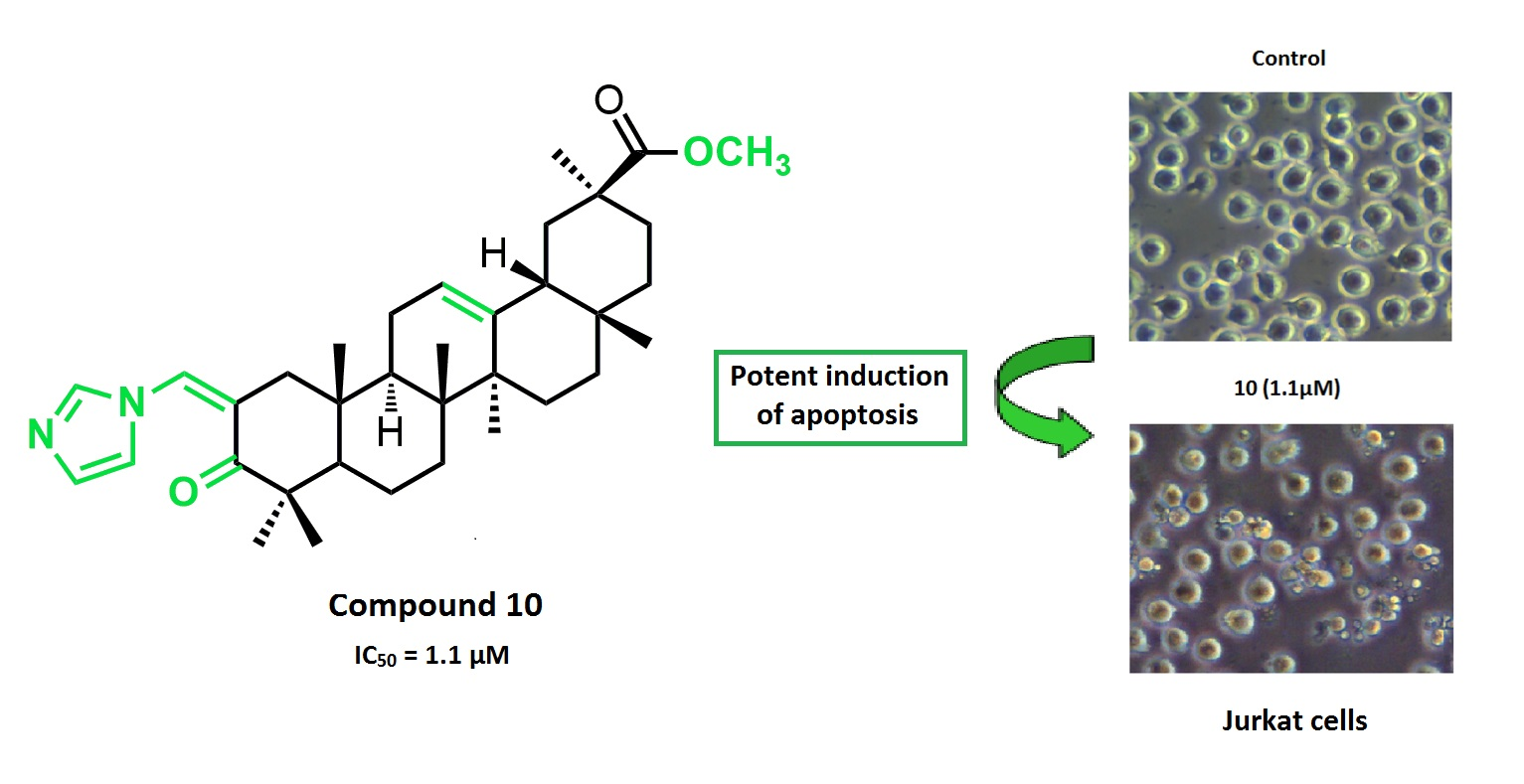
:A new series of glycyrrhetinic acid derivatives has been synthesized via the introduction of different heterocyclic rings conjugated with an α,β-unsaturated ketone in its ring A. These new compounds were screened for their antiproliferative activity in a panel of nine human cancer cell lines. Compound 10 was the most active derivative, with an IC50 of 1.1 µM on Jurkat cells, which is 96-fold more potent than that of glycyrrhetinic acid, and was 4-fold more selective toward that cancer cell line. Further biological studies performed in Jurkat cells showed that compound 10 is a potent inducer of apoptosis that activates both the intrinsic and extrinsic pathways.
1. Introduction
Triterpenoids are a large group of natural compounds that are widely found in nature and are particularly prevalent in plants. They display a wide spectrum of biological and pharmacological activities. Within this group, the pentacyclic triterpenoids are the most potent compounds and their anti-inflammatory, antiviral, and anticancer effects have been extensively studied. Moreover, there is an increasing interest in these triterpenoids as lead compounds for the development of new anticancer agents, which is reflected in the large number of scientific papers and patents emerging in this field [1,2,3,4,5,6,7,8,9,10,11].
Glycyrrhetinic acid (GA) 1 (Figure 1) is the aglycone of glycyrrhizin, a major pentacyclic triterpenoid saponin found in the roots of licorice (Glycyrrhiza species) [12]. This compound has a proven antiproliferative activity against several cancer cell lines and its anticancer effects were also observed in animal models [13,14,15,16]. Recent reports have shown that its cytotoxicity is mediated by the induction of apoptosis [16,17,18,19,20,21]. In addition, GA 1 can be extracted from the roots of licorice in high yields (up to 24%) [22]. These findings have increased its scientific interest as a scaffold for the development of new derivatives for potential cancer treatment [23,24,25,26,27].
In previous work performed by our group, introduction of imidazole, methyl-imidazole, and triazole heterocyclic rings was achieved in several positions of betulinic and ursolic acids, affording several derivatives with better antiproliferative activity compared with the parental compounds. The structure–activity relationship analysis revealed that the introduction of a heterocyclic ring in conjugation with an α,β-unsaturated ketone in ring A of the skeleton seems to provide the most potent derivatives [28,29].
Cycloaddition of azides and alkynes, commonly referred to as “click chemistry”, has acquired great interest in the development of novel heterocyclic compounds with varied biological activities [30,31,32,33]. This efficient and simple process has been successfully used in the synthesis of pentacyclic triterpenoid derivatives with improved anticancer activity [34,35,36,37].
The information provided above prompted us to synthesize new GA 1 derivatives via the introduction of different heterocyclic rings conjugated with an α,β-unsaturated ketone in its ring A. These novel semisynthetic derivatives were tested for their antiproliferative activity against a series of cancer cell lines. Further biological assays were performed for the most active compound 10 in the cancer cell line that yielded the best results (Jurkat cells), to elucidate its mechanism of action.
2. Results and Discussion
2.1. Chemistry
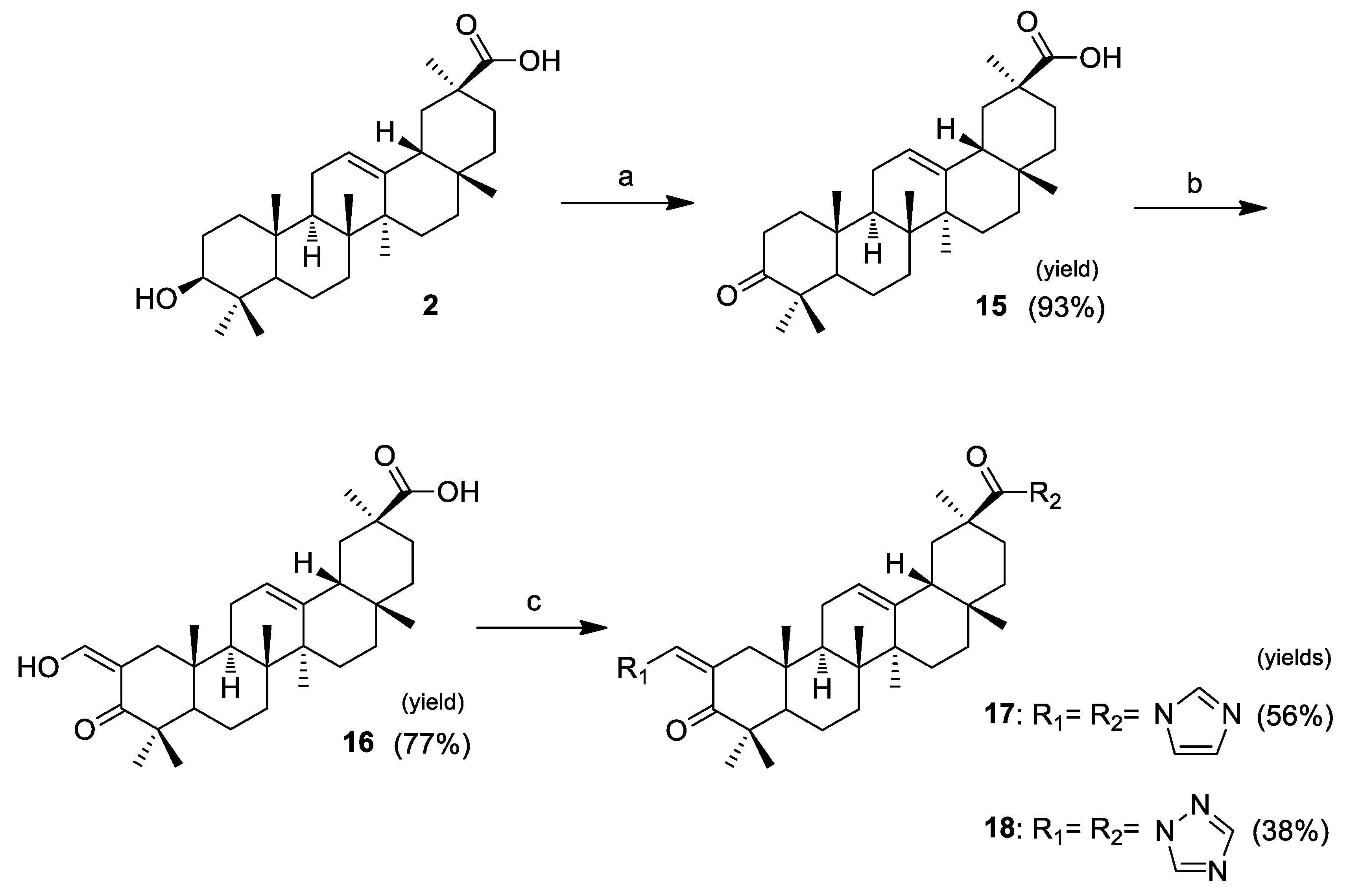
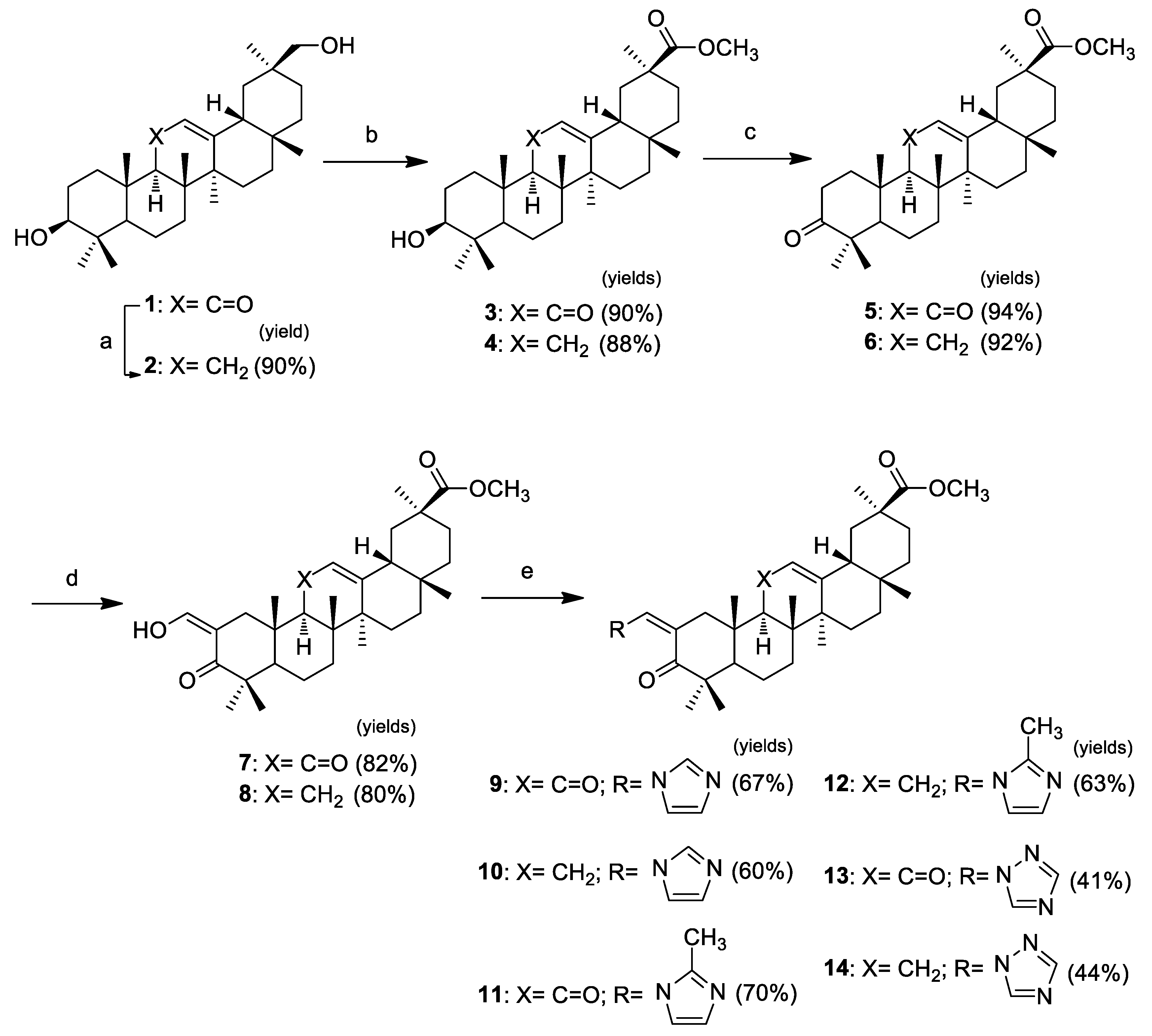
The synthesis of three pairs of novel GA 1 heterocyclic derivatives is summarized in Scheme 1. In addition to the intention to prepare heterocyclic derivatives with improved cytotoxicity, compounds 9–14 were synthesized to explore the effect of the keto group in position C-11 on the antiproliferative activity. Therefore, the first step performed in this sequence of reactions was the removal of the keto group via Clemmensen reduction with zinc dust and concentrated HCl in dioxane at room temperature, to afford 2 [38]. The following steps were executed in both original and reduced structures, providing successive pairs of derivatives. Methyl esters 3 and 4 were obtained from the reaction of compounds 1 and 2 with methyl iodide in the presence of potassium carbonate [39]. The 3β-hydroxyl group of these esters was oxidized using the Jones reagent [40], to give the 3-keto derivatives 5 and 6. The reaction of these derivatives with ethyl formate and sodium methoxide allowed the preparation of the 2-hydroxyvinyl-3-keto compounds 7 and 8 [40]. The heterocyclic derivatives 9–14 were prepared via the reaction of the vinyl alcohol at C-2 with the appropriate heterocyclic reagent, 1,1′-carbonyldiimidazole (CDI), 1,1′-carbonylbis(2′-methylimidazole) (CBMI), or 1,1′-carbonyl-di(1,2,4-triazole) (CDT) in reflux of THF under inert atmosphere [29], in yields ranging from 41% to 70%.
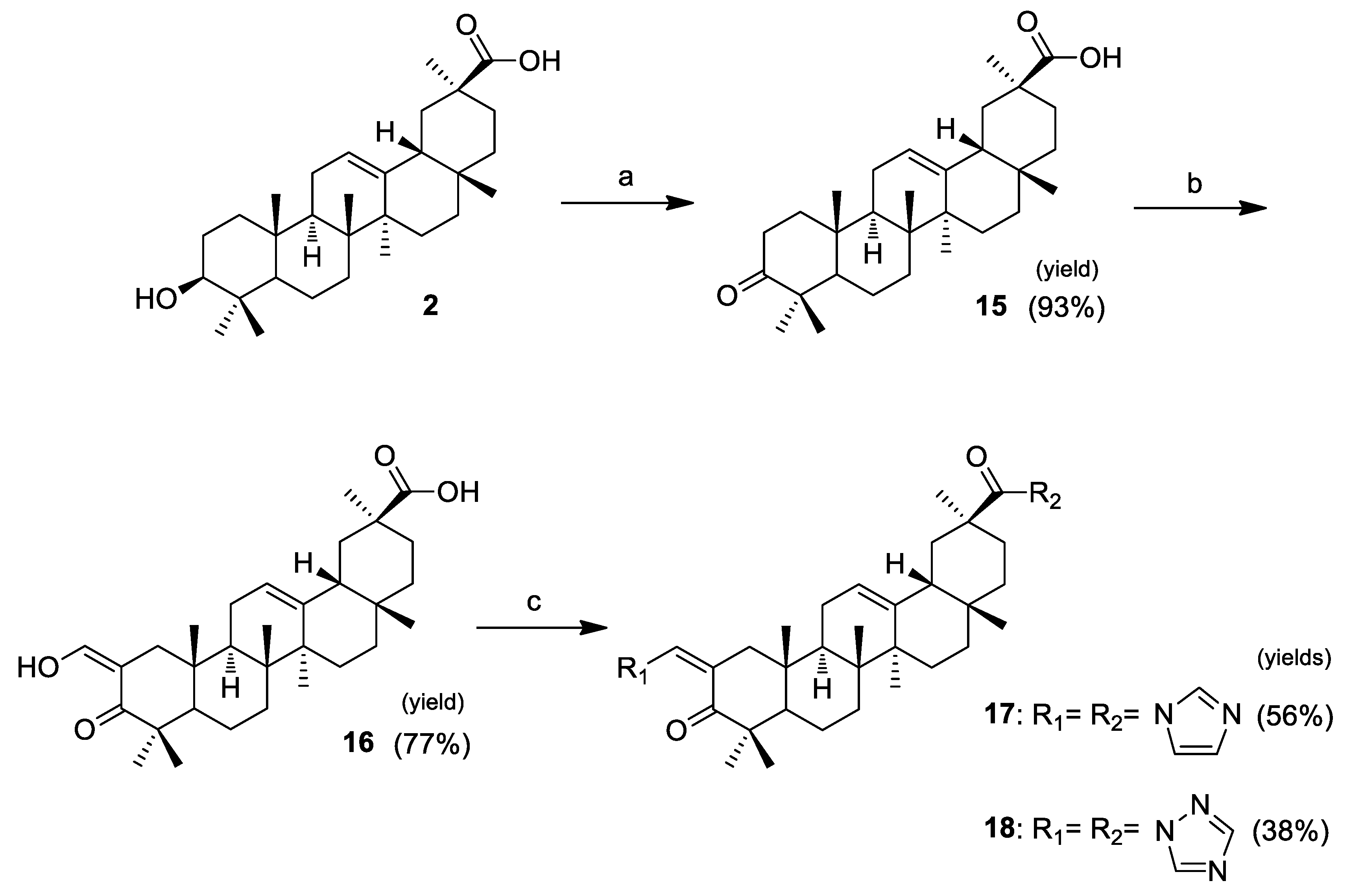
Compounds 17 and 18 were synthesized as depicted in Scheme 2. These derivatives were prepared to evaluate if the presence of another heterocyclic ring at C-30 would benefit the antiproliferative activity. The steps of this sequence of reactions were performed using the same methods of oxidation, reaction with ethyl formate, and introduction of heterocyclic rings described in Scheme 1. These novel heterocyclic derivatives, 17 and 18, were obtained in yields of 56% and 38%, respectively.
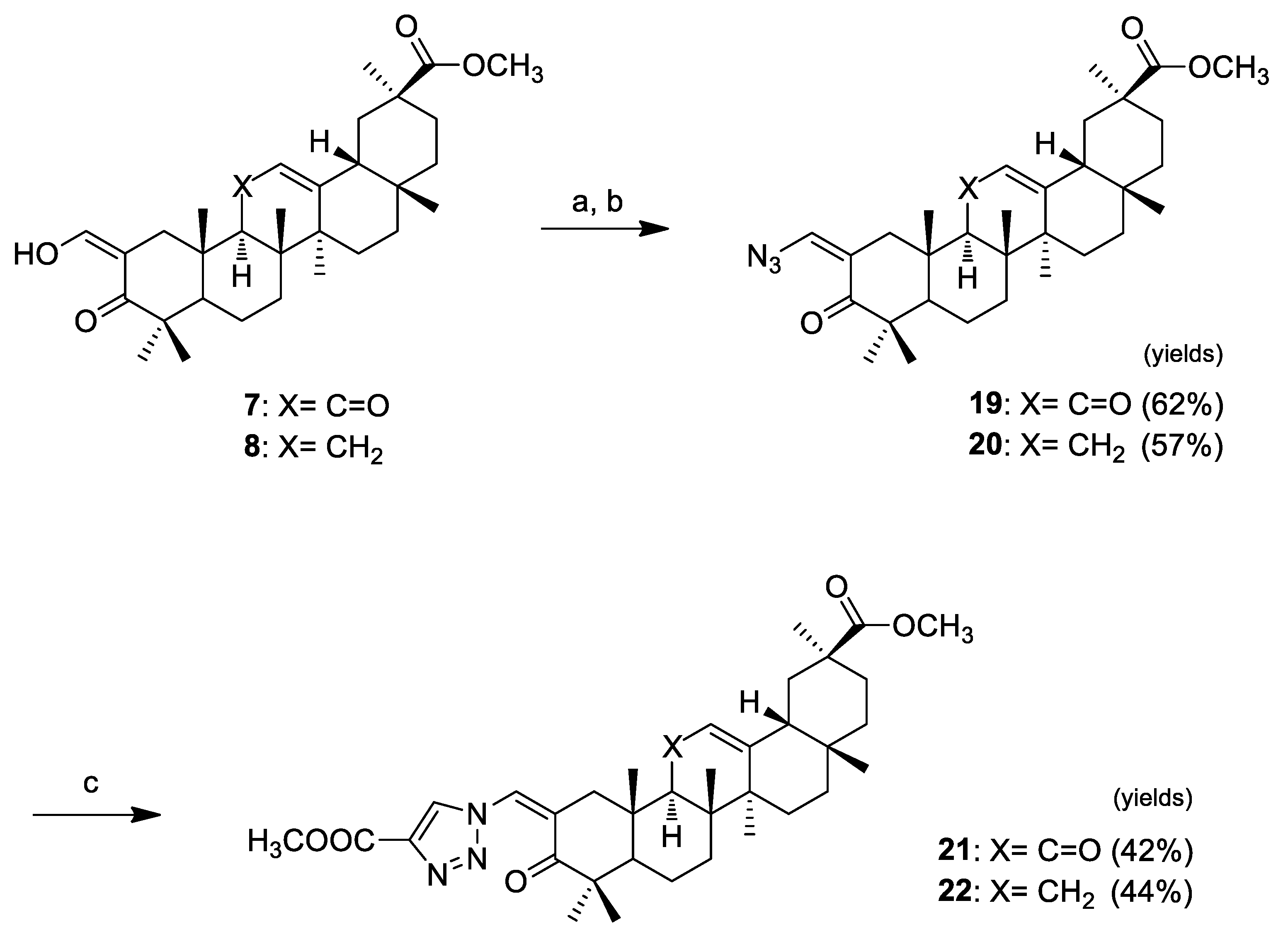
Click chemistry was handled as shown in Scheme 3. We decided to synthesize 1,4-substituted-triazolyl derivatives in conjugation with an α,β-unsaturated ketone in ring A of pentacyclic triterpenoids via Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAc) with terminal alkynes. We prepared the azido derivatives 19 (62%) and 20 (57%) by tosylation of the vinyl alcohol at C-2 and subsequent replacement by azide. The triazolyl derivatives 21 and 22 were obtained via the “click reaction” of those compounds with methyl propiolate, catalyzed by CuI, in THF, at 65 °C (which were the best reaction conditions observed here), in yields of 42% and 44%, respectively.
Full structural elucidation of the new glycyrrhetinic acid derivatives was achieved using infrared spectroscopy (IR), nuclear magnetic resonance (NMR), and mass spectrometry (MS). The data obtained from these analytical techniques, for the known compounds 2–8, were in agreement with those reported in the literature [38,39,40].
The removal of the keto group at position C-11, via Clemmensen reduction, was confirmed by the absence of a band around 1664 cm−1 on the IR spectrum, which is present in glycyrrhetinic acid, and corresponds to the carbonyl group in ring C. On the 13C NMR spectrum, a δ signal around 200 ppm, which corresponds to the presence of that carbonyl group, was absent, and the δ signals for carbons C-12 and C-13 were present at higher field than that observed in compounds with the carbonyl group.
The preparation of compounds 7, 8, and 16 was confirmed by the presence of a δ signal at ~8.6 ppm in the 1H NMR spectra, corresponding to the proton in the exocyclic double bond at C-2. On the 13C NMR spectrum, the presence of this moiety was confirmed by the observation of a δ signal at ~188 ppm, corresponding to the exocyclic carbon, and a δ signal at ~106 ppm, corresponding to the C-2 carbon.
The successful preparation of the heterocyclic derivatives 9–14, 17 and 18, and 21 and 22, was confirmed by the presence of a δ signal at ~7.7–8.0 ppm, corresponding to the proton of the exocyclic double bond at C-2 in the 1H NMR spectrum. On the 13C NMR spectrum the presence of this moiety was confirmed by the presence of a δ signal at ~127–131 ppm, corresponding to the exocyclic carbon.
The presence of heterocyclic ring(s) can be detected by the presence of extra δ signals in 1H and 13C NMR spectra. On the 1H NMR spectra, the imidazole group had three specific protons that appeared at values higher than 7 ppm, the methyl-imidazole group had two protons ranging from 6.9 to 7.7 ppm, the triazole group had two proton signals with a δ higher than 8 ppm, and the triazole group on compounds 21 and 22 had only one proton at ~8.3–8.4 ppm. In the 13C NMR spectra, the δ signals of the carbons of heterocyclic rings were present at values ranging from 118 to 153 ppm, varying in accordance with the different heterocyclic rings.
2.2. Biological Activity
2.2.1. Antiproliferative Activity
Several cancer lines were cultured and used in experiments aimed at evaluating the potential cytotoxicity of the synthesized compounds against human cancers. This evaluation was based on the determination of IC50 values using 3-(4,5-dimethylthiozol-2-yl)-3,5-dipheryl tetrazolium bromide (MTT) or triphenyl tetrazolium chloride (XTT) assays after 72 h of treatment with the compounds.
All compounds were screened for their antiproliferative activity on the HT-29 (colon adenocarcinoma) cell line. As shown in Table 1, most of the novel heterocyclic derivatives (9–14, 17, 18, 21, and 22) showed improved cytotoxicity compared with the parental compound GA 1 and the intermediate compounds. The methylation of compounds 1 and 2 provided derivatives (3 and 4) that exhibited an important increment in antiproliferative activity, but lacked selectivity towards tumor cells [39]. The intermediates 7, 8, 16, 19, and 20, which preceded the final heterocyclic derivatives, were also tested on the A549 (lung adenocarcinoma) cell line (Table 1). Analysis of the IC50 values of the heterocyclic derivatives with respect to their substrates in the two cancer cell lines showed that the introduction of a heterocyclic ring provided more potent compounds, with the exception of compound 13.
The novel compounds 9–14, 17, 18, 21, and 22, and the parental compound GA 1 were further tested in seven additional human cancer cell lines: MIAPaCa 2 (pancreas adenocarcinoma), HeLa (cervix adenocarcinoma), A375 (melanoma), MCF7 (breast adenocarcinoma), HepG2 (hepatocellular carcinoma), SH-SY5Y (neuroblastoma), and Jurkat (acute T cell leukemia) cells (Table 2). The synthesis of compounds 17 and 18 was performed to investigate the effect of the combined introduction of heterocyclic rings at the vinyl alcohol at the C-2 and at the C-30 positions. This combination resulted in a loss of cytotoxicity in all tested lines compared with compounds 10 and 14, respectively. The preparation of the four pairs of derivatives 9–14, 21, and 22 allowed us to evaluate the effect of the keto group at position C-11 on the antiproliferative activity. The results of the assays in all tested cell lines were consistent and revealed that the compounds from which the keto group was removed from ring C were more potent. The type of heterocyclic ring conjugated with the α,β-unsaturated ketone in ring A also influenced the antiproliferative activity. Within the group of heterocyclic derivatives with a reduced ring C, compound 10, which bears an imidazole ring, was the most active derivative in all tested cell lines. This compound was 31- to 96-fold more potent than GA 1, depending on the cancer cell line. Moreover, with the exception of SH-SY5Y cells, compound 10 showed a similar or slightly improved antiproliferative activity against all cancer cell lines compared to the chemotherapy agent cisplatin [41,42,43,44,45,46] (Table 1 and Table 2).
The selectivity towards cancer cells was studied for GA 1 and compound 10 by incubating them with a human nontumoral cell line (BJ) (Table 2). Compound 10 and GA 1 showed IC50 values that were 6.3 and 1.6 times lower on Jurkat cells than on the nontumoral BJ cells, respectively. Therefore, the heterocyclic derivative 10 was more selective towards malignant cells than its parental compound 1.
2.2.2. Cell Viability over Time
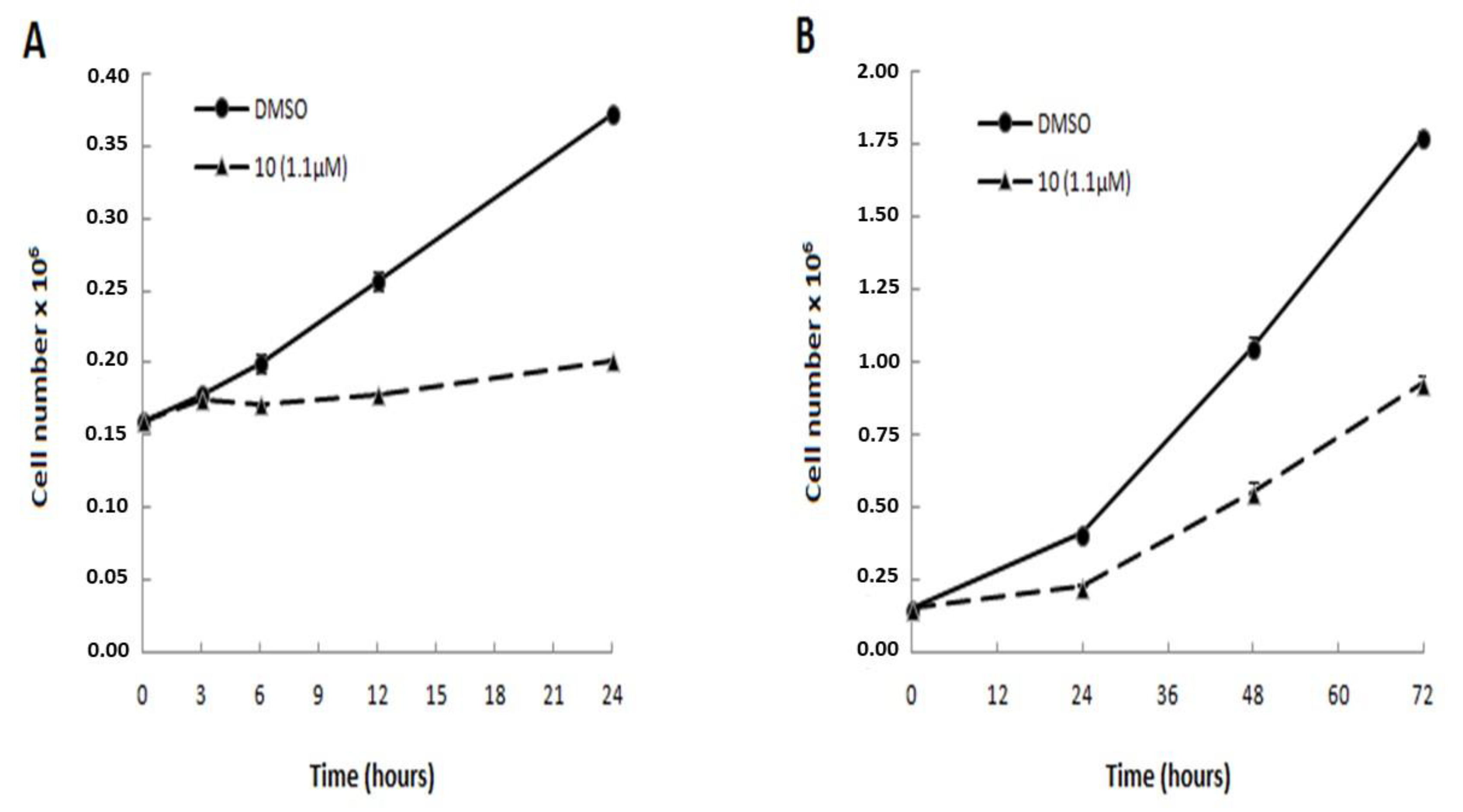
The Jurkat cell line, which exhibited the best results regarding antiproliferative activity among the tested cancer cell lines, was used to study the mechanism of action of the most potent compound synthesized here, the heterocyclic derivative 10. Trypan blue cell-counting assays were conducted to assess the antiproliferative profile of this compound over time. The counting of viable cells was performed after incubation of Jurkat cells with compound 10, at a concentration corresponding to its IC50 value at 72 h of treatment, or with only the vehicle (control with DMSO). As shown in Figure 2, compound 10 affected the cell growth of Jurkat cells as early as at 6h of treatment. At this incubation time, a reduction of 18% of cell viability was observed in cells treated with compound 10 compared with cells that were not treated with the same compound. This effect on cell viability increased during next hours, until cell viability was 45% lower than it was in control cells at 24 h, and reached 50% at 72 h of treatment. Based on the antiproliferative profile obtained in these Trypan Blue counting experiments, we established the incubation times for the subsequent biological studies that were performed in this study.
2.2.3. Analysis of Cell Cycle Distribution and Apoptosis
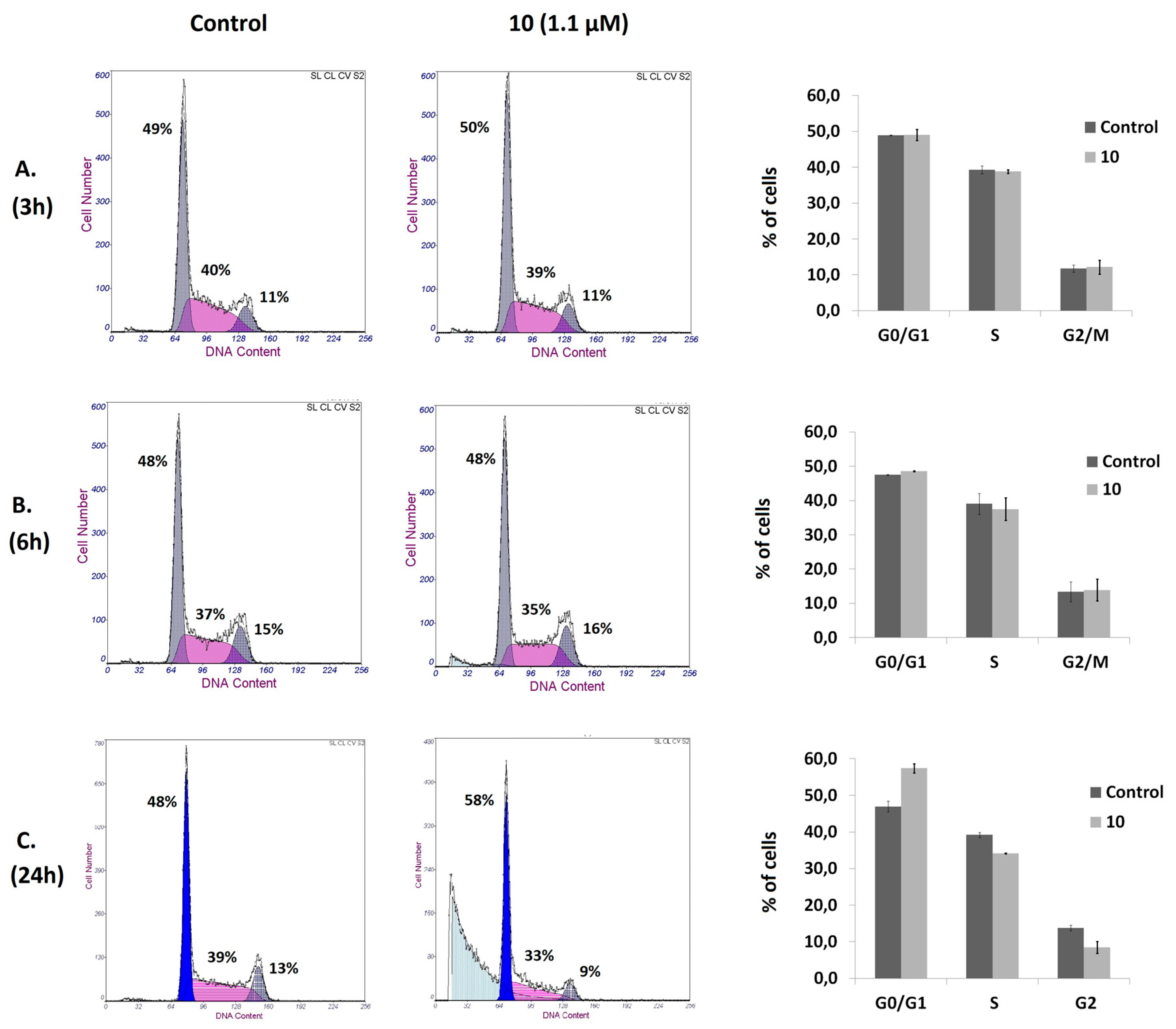
Cell cycle and apoptosis assays were performed to elucidate the underlying antiproliferative mechanisms of compound 10. To examine the effects on the cell cycle pattern, Jurkat cells were treated with compound 10, at a concentration corresponding to its IC50 value at 72 h of treatment, for 3, 6, and 24 h. The study was performed using flow cytometry, and the calculation of the fraction of cells in phases G0/G1, S, and G2/M was executed using the fraction of live cells. No significant changes on the cell cycle distribution were detected after 3 and 6 h of exposure; treatment for 24 h increased the population at the G0/G1 phase, with a concomitant decrease in the populations of cells at the S and the G2/M phases with respect to the untreated cells (Figure 3). Hypodiploid sub-G0 peaks that correspond to cells with DNA fragmentation were observed after 6 and 24 h of incubation. These results suggest that compound 10 has the ability to induce cell death in Jurkat cells.
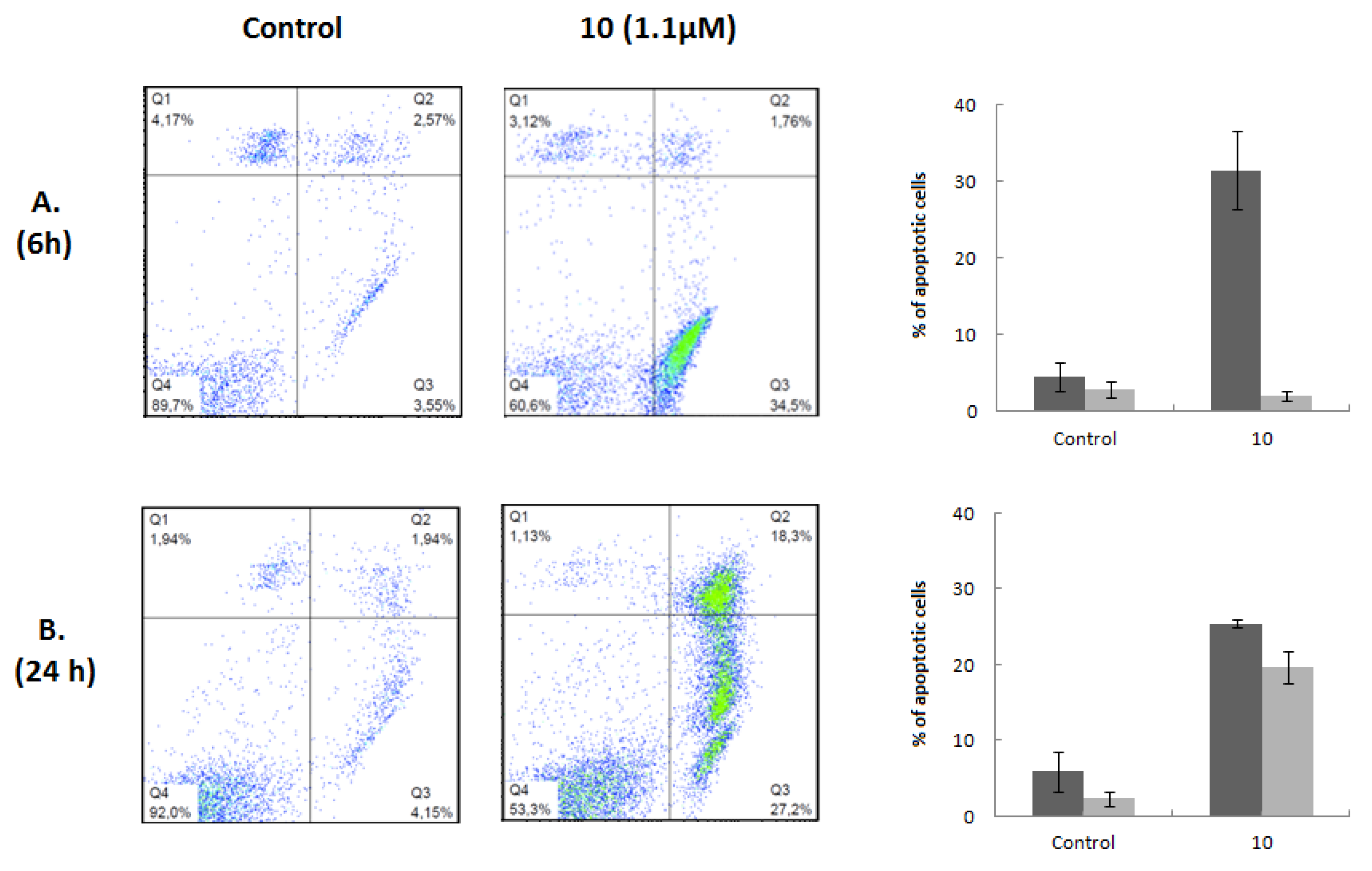
Apoptosis assays were then conducted to elucidate the mechanism underlying the cytotoxic effect of this heterocyclic derivative. The Annexin V-FITC/PI flow cytometry assay employs the ability of Annexin V to bind to phosphatidylserine (PS) and the ability of propidium iodide (PI) to enter cells with damaged cell membranes and to bind to DNA. Early apoptosis is characterized by the loss of membrane asymmetry, with translocation of PS from the inner to the outer membrane, prior to the loss of membrane integrity. Therefore, this assay allows the discrimination of live cells (Annexin-V−/PI−) from early apoptotic (Annexin-V+/PI−), late apoptotic (Annexin-V+/PI+), or necrotic (Annexin-V−/PI+). Considering the results obtained in the cell viability and cell cycle experiments, we performed this assay on Jurkat cells after 6 and 24 h of treatment with compound 10 at a concentration corresponding to its IC50 value at 72 h of treatment. Flow cytometry dot plots representing Annexin V and PI staining are shown in Figure 4. Compound 10 induced 32% of cells to enter the early stage of apoptosis after 6 h of treatment. No significant changes were observed in the late apoptotic and necrotic populations. Exposure to the compound for 24 h increased the early apoptotic population by 25% and the late apoptotic population by 20%. The percentage of necrotic cells did not change significantly. We also performed this assay after 48 and 72 h of treatment; the Annexin V/PI profiles observed for these incubation times were similar to those obtained at 24 h (data not shown). The results of the Annexin V-FITC/PI assays are in good agreement with the hypodiploid sub-G0 peaks and their respective magnitudes, observed in the cell cycle experiments. Moreover, the fact that the appearance of G0/G1 arrest occurred after the beginning of the apoptotic events indicates that cell death was caused by a primary effect of compound 10 and not by the activation of apoptosis as a consequence of the inability of cells to overcome cell growth arrest and proceed through the cell cycle.
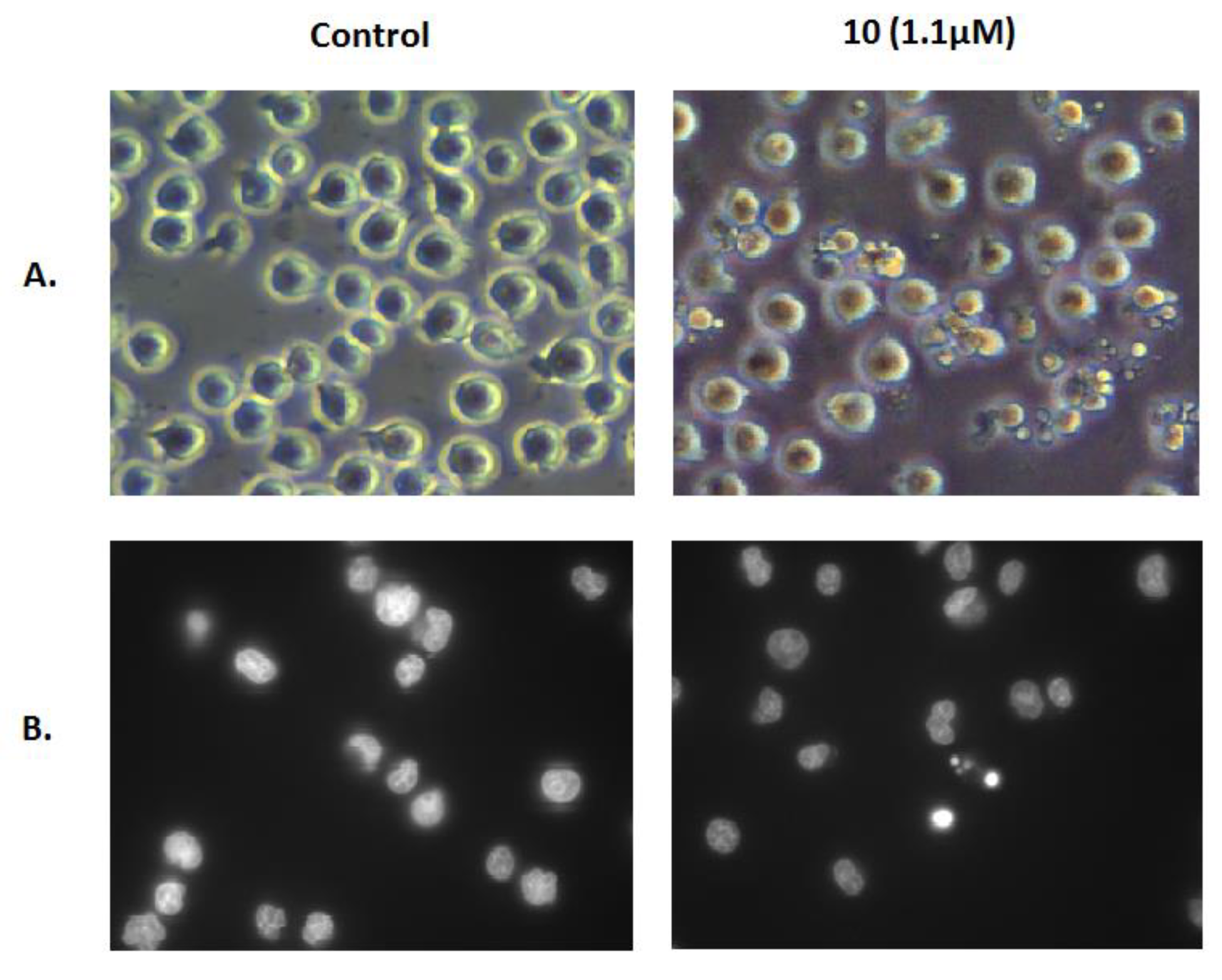
We further confirmed the induction of apoptosis by examining its characteristic morphological changes in Jurkat cells treated with the same concentration of compound 10 for 6 h. As shown in Figure 5, apoptotic bodies were observed in the treated cells using a phase-contrast microscope, and Hoechst 33342 staining showed that some of those cells exhibited a highly condensed nuclear morphology. In contrast, untreated cells presented intact plasma membranes and normal nuclear morphology.
Taken together, the results described above reveal that compound 10 is a potent inducer of apoptosis.
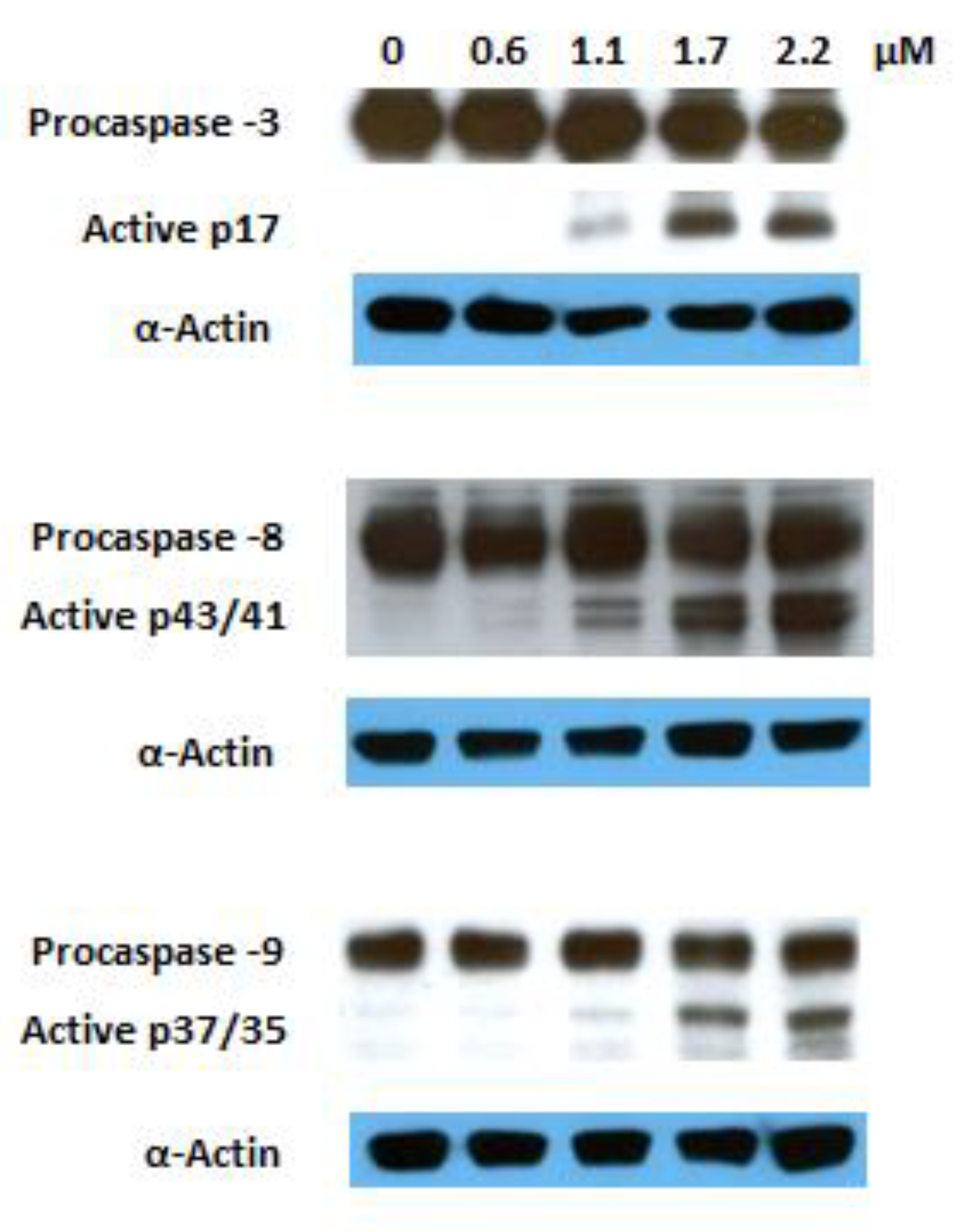
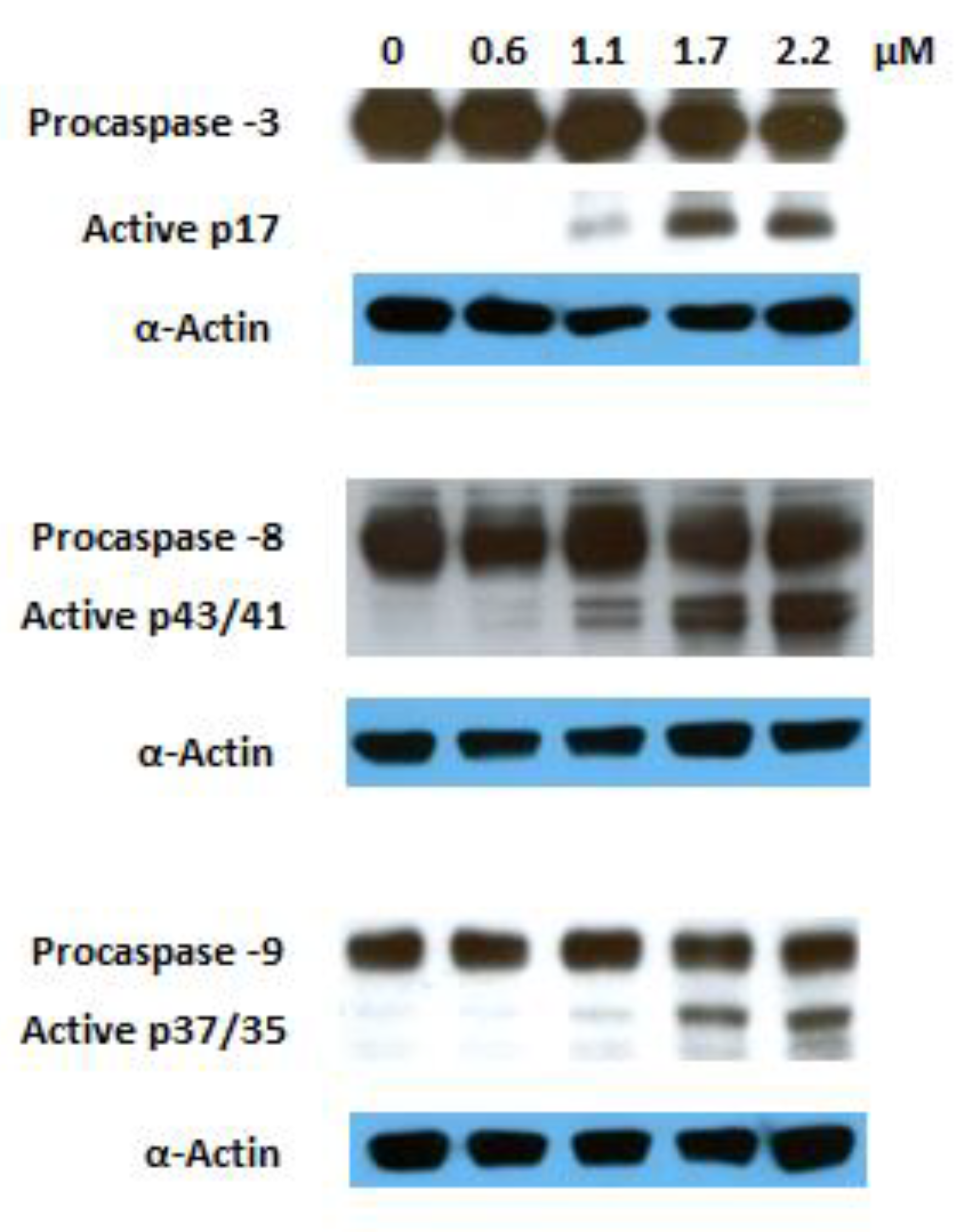
The activation of several caspases is involved in the execution phase of apoptosis. Caspase 8 is the upstream caspase for the extrinsic (death receptor) pathway, whereas caspase 9 is that of the intrinsic (mitochondrial) pathway. These pathways converge to caspase 3, the action of which is required for the morphological changes that are associated with apoptosis [47]. Previous reports have shown that GA 1 induces apoptosis involving both the extrinsic and intrinsic pathways [17,18,48,49,50,51]. To explore the mechanism via which the GA heterocyclic derivative 10 triggers this programmed cell death, we investigated its effects on the activation of caspases 3, 8, and 9 via Western blotting. The proteolytic cleavage of all mentioned caspases was observed after 6 h of treatment at a dose corresponding to the IC50 recorded at 72 h (Figure 6). These results indicate that the induced apoptosis is mediated by the activation of the both extrinsic and intrinsic pathways.
Apoptosis is a highly regulated process that occurs in physiological conditions and plays a major role in the pathogenesis of many diseases. In fact, acquired resistance towards apoptosis is a hallmark of most types of cancer. Despite its involvement in the carcinogenesis, apoptosis is a widespread target of many treatment strategies, including the development of novel chemotherapeutic molecules [47,52]. The p53 tumor suppressor protein is a critical component of the apoptotic signaling circuitry in both pathways. The loss of its function is the most common strategy employed by cancer cells to evade death. In fact, approximately 50% of human cancers bear P53 gene mutations and, in the majority of the remaining cases, its function is compromised by different mechanisms [52,53,54]. The fact that Jurkat cells are a cancer p53-deficient cell line [55] indicates that compound 10 can activate the apoptotic machinery in a p53-independent manner.
Taken together, our results suggest that the induction of apoptosis involving both the intrinsic and extrinsic pathways is the main mechanism responsible for the antiproliferative activity of the GA heterocyclic derivative 10. Efforts are currently underway to elucidate further its mechanism of action.
3. Materials and Methods
3.1. Chemistry
Glycyrrhetinic acid and all reagents were purchased from Sigma-Aldrich Co (St. Louis, MO, USA). The solvents used in the reactions were obtained from Merck Co (kenilworth, NJ, USA). and were purified and dried according to the literature procedures. The solvents used in workups were purchased from VWR Portugal (Radnor, PA, USA). Thin-layer chromatography (TLC) analysis was performed in Kieselgel 60HF254/Kieselgel 60G. Purification of compounds by flash column chromatography (FCC) was carried out using Kiesegel 60 (230–400 mesh, Merck) (kenilworth, NJ, USA). Melting points were determined using a BUCHI melting point B-540 apparatus and were uncorrected. IR spectra were obtained on a Fourier transform spectrometer. 1H and 13C NMR spectra (see Supplementary Materials) were recorded on a Bruker Avance-400 Digital NMR spectrometer, in CDCl3,, with Me4Si as the internal standard. Chemical shifts values (δ) are given in parts per million (ppm) and coupling constants (J) are presented in hertz (Hz). Massa spectra were obtained using a Quadrupole/Ion Trap Mass Spectrometer (QIT-MS) (LCQ Advantage MAX, THERMO FINNINGAN). Elemental analysis was performed in an Analyzer Elemental Carlo Erba 1108 by chromatographic combustion.
3β-hydroxy-olean-12-en-30-oic acid (2): Compound 2 was prepared according to the literature [38], from 1 to give a white solid (90%). m.p.: 315–317 °C.
Methyl 3β-hydroxy-11-oxo-olean-12-en-30-oate (3): Compound 3 was prepared according to the literature [39], from 1 to give a colorless solid (90%). m.p.: 254–256 °C
Methyl 3β-hydroxy-olean-12-en-30-oate (4): Compound 4 was prepared from 2, using the same method as for the preparation of 3, with the obtention of a white solid (88%). m.p.: 239–242 °C.
Methyl 3,11-dioxo-olean-12-en-30-oate (5): Compound 5 was prepared according to the literature [40], from 3 to give a white solid (94%). m.p.: 248–250 °C.
Methyl 3-oxo-olean-12-en-30-oate (6): Compound 6 was prepared from 4, using the same method as for the preparation of 5, with the obtention of a white solid. (92%). m.p.: 185–188 °C
Methyl 2-hydroxymethylene-3,11-dioxo-olean-12-en-30-oate (7): Compound 7 was prepared according to the literature [40], from 5 to give a colorless solid (82%). m.p.: 231–234 °C.
Methyl 2-hydroxymethylene-3-oxo-olean-12-en-30-oate (8): Compound 8 was prepared from 6, using the same method as for the preparation of 7, with the obtention of a white solid (80%). m.p.: 136–139 °C.
Methyl 2-(1H-imidazol-1-yl)-methylene-3,11-dioxo-olean-12-en-30-oate (9): To a solution of compound 7 (300 mg, 0.59 mmol) in anhydrous THF (5 mL), CDI (191 mg, 1.18 mmol) was added. After 4 h under magnetic stirring at reflux temperature and N2 atmosphere, the reaction was completed. Water (50 mL) and ethyl acetate (50 mL) were added to the reaction mixture. The aqueous phase was further extracted with ethyl acetate (2 × 50 mL). The combined organic extract was then washed with water (2 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered and evaporated to dryness. The resulting solid was subjected to FCC [petroleum ether/ ethyl acetate from (1:1) to (1:2)] to afford 9 as a white solid. (67%). m.p.: 249–251 °C. IR νmax/cm−1 (KBr): 3113, 2953, 1728, 1685, 1649, 1601, 1518, 1485, 1458, 1385, 1306, 1028. 1H NMR (400MHz, CDCl3): δ 7.76 (1H,s), 7.66 (1H, s), 7.33 (1H,s), 7.10 (1H, s), 5.75 (1H, s), 4.19 (1H, d, J = 16.5), 3.67 (3H, s), 2.52 (1H, s), 1.39 (3H, s), 1.18 (3H, s), 1.16 (3H, s), 1.13 (6H, s), 1.12 (3H, s), 0.81 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.2, 199.2, 176.8, 170.6, 139.1, 130.7, 130.3, 128.4, 122.1, 119.0, 59.2, 52.8, 51.7, 48.4, 45.3, 44.8, 44.0, 43.3, 43.2, 41.2, 37.6, 35.9, 31.8, 31.3, 31.0, 29.7, 28.5, 28.2, 26.5, 26.3, 23.1, 22.3, 19.5, 17.9, 15.5. ESI-MS m/z: 561.71 ([M + H]+, 100%). Found C 74.45, H 8.80, N 5.05, calcd for C35H48N2O4.0: C 74.96, H 8.63, N 5.00%.
Methyl 2-(1H-imidazol-1-yl)-methylene-3-oxo-olean-12-en-30-oate (10): The method followed that described for compound 9 but using compound 8 (300 mg, 0.60 mmol) and CDI (195 mg, 1.20 mmol) in anhydrous THF (5 mL) at reflux for 5 h. The resulting solid was purified by FCC with petroleum ether/ethyl acetate (1:1) and afforded compound 10 as a white solid (60%). m.p.: 151–154 °C. IR νmax/cm−1 (KBr): 3118, 2951, 1730, 1687, 1610, 1518, 1487, 1458, 1383, 1306, 1028. 1H NMR (400MHz, CDCl3): δ 7.78 (1H,s), 7.70 (1H,s), 7.22 (1H,s), 7.15 (1H,s), 5.34 (1H, t, J = 3.4), 3.68 (3H, s), 2.90 (1H, d, J = 16.0), 1.18 (6H, s), 1.13 (6H, s), 1.02 (3H, s), 0.91 (3H, s), 0.78 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.7, 177.6, 144.6, 138.7, 130.7, 130.5, 122.9, 122.0, 119.2, 52.6, 51.6, 48.4, 45.4, 45.2, 44.3, 43.2, 42.8, 41.9, 39.7, 38.3, 36.0, 32.0, 31.7, 31.3, 29.8, 28.5, 28.2, 27.0, 26.1, 25.7, 23.8, 22.5, 20.3, 16.4, 15.6. ESI-MS m/z: 547.73 ([M + H]+, 100%). Found C 76.72, H 9.43, N 4.98, calcd for C35H50N2O3: C 76.88, H 9.22, N 5.12%.
Methyl 2-(2′-methyl-1H-imidazol-1-yl)-methylene-3,11-dioxo-olean-12-en-30-oate (11): To a solution of compound 7 (300 mg, 0.59 mmol) in anhydrous THF (5 mL), CBMI (231 mg, 1.18 mmol) was added. After 2 h under magnetic stirring at reflux temperature and N2 atmosphere, the reaction was completed. The workup was performed according to the same method as for 9. The resulting solid was submitted to FCC [petroleum ether/ ethyl acetate from (1:1) to (1:4)] to afford 11 as a white solid (70%). m.p.: 146–149 °C. IR νmax/cm−1 (KBr): 3116, 2953, 1730, 1686, 1657, 1608, 1541, 1502, 1458, 1412, 1385. 1H NMR (400MHz, CDCl3): δ 7.64 (1H,s), 7.29 (1H,s), 6.94 (1H,s), 5.75 (1H,s), 4.17 (1H, d, J = 16.3), 3.67 (3H, s), 2.52 (1H, s), 2.47 (3H, s), 1.39 (3H, s), 1.19 (3H, s), 1.17 (3H, s), 1.14 (9H, s), 0.82 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.4, 199.3, 176.8, 170.5, 147.3, 129.9, 128.7, 128.5, 122.3, 118.1, 59.2, 53.0, 51.8, 48.5, 45.4, 44.9, 44.0, 43.4, 42.6, 41.2, 37.7, 36.0, 31.8, 31.3, 31.1, 29.7, 28.6, 28.3, 26.5, 26.3, 23.2, 22.4, 19.5, 18.0, 15.4, 13.7. ESI-MS m/z: 575.86 ([M + H]+, 100%). Found C 74.91, H 9.01, N 4.78, calcd for C36H50N2O4: C 75.22, H 8.77, N 4.87%.
Methyl 2-(2′-methyl-1H-imidazol-1-yl)-methylene-3-oxo-olean-12-en-30-oate (12): The method followed that described for compound 11 but using compound 8 (300 mg, 0.60 mmol) and CBMI (235 mg, 1.20 mmol) in anhydrous THF (5 mL) at reflux for 4 h. The resulting solid was subjected to FCC (petroleum ether/ ethyl acetate from (1:1) to (1:4)) to afford 12 as a white solid (63%). m.p.: 134–137 °C. IR νmax/cm−1 (KBr): 3118, 2951, 1730, 1687, 1606, 1541, 1502, 1456, 1412, 1383. 1H NMR (400MHz, CDCl3): δ 7.66 (1H,s), 7.14 (1H,s), 7.00 (1H,s), 5.33 (1H, t, J = 3.4), 3.67 (3H, s), 2.87 (1H, d, J = 15.9), 2.48 (3H, s), 1.18 (6H, s), 1.15 (3H, s), 1.12 (3H, s), 1.01 (3H, s), 0.90 (3H, s), 0.78 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.7, 177.6, 147.3, 144.6, 130.2, 128.3, 123.8, 122.0, 118.2, 52.7, 51.6, 48.4, 45.4, 45.3, 44.3, 42.8, 42.5, 41.9, 39.7, 38.3, 36.1, 32.0, 31.7, 31.3, 29.7, 28.5, 28.2, 26.9, 26.1, 25.7, 23.7, 22.6, 20.3, 16.4, 15.5, 13.6. ESI-MS m/z: 561.77 ([M + H]+, 100%). Found C 76.85, H 9.67, N 4.64, calcd for C36H52N2O3: C 77.10, H 9.35, N 5.00%.
Methyl 2-(1H-triazol-1-yl)-methylene-3,11-dioxo-olean-12-en-30-oate (13): To a solution of compound 7 (300 mg, 0.59 mmol) in anhydrous THF (6 mL), CDT (581 mg, 3.54 mmol) was added. The workup was performed after 24 h of reaction, under magnetic stirring at reflux temperature and N2 atmosphere, using the same procedure as for compound 9. The resulting solid was purified by FCC with petroleum ether/ ethyl acetate (1:2) and afforded 13 as a white solid (41%). m.p.: 148–151 °C. IR νmax/cm−1 (KBr): 3122, 2953, 1730, 1691, 1657, 1618, 1508, 1460, 1385. 1H NMR (400MHz, CDCl3): δ 8.40 (1H,s), 8.06 (1H,s), 7.80 (1H,s), 5.76 (1H,s), 4.42 (1H, d, J = 18.6), 3.69 (3H, s), 2.57, 1.40 (3H, s), 1.22 (3H, s), 1.17 (3H, s), 1.15 (3H, s), 1.14 (6H, s), 0.82 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.4, 199.1, 176.9, 170.1, 152.7, 145.3, 128.7, 128.6, 125.1, 59.1, 53.0, 51.8, 48.5, 45.4, 44.9, 44.0, 43.7, 43.4, 41.2, 37.7, 35.7, 31.9, 31.4, 31.1, 29.8, 28.6, 28.3, 26.6, 26.4, 23.1, 22.4, 19.6, 18.0, 15.5. ESI-MS m/z: 274.48 (87%), 302.51 (15), 318.49 (100), 346.54 (22), 362.37 (37), 374.60 (11), 562.68 ([M + H]+, 93), 563.71 ([M + 2H] 2+, 26), 575.80 (24), 622.48 (15), 624.80 (21).
Methyl 2-(1H-triazol-1-yl)-methylene-3-oxo-olean-12-en-30-oate (14): The method followed that described for compound 13 but using compound 8 (300 mg, 0.60 mmol) and CDT (591 mg, 3.60 mmol) in anhydrous THF (6 mL) at reflux for 26 h. The resulting solid was purified by FCC with petroleum ether/ ethyl acetate (3:1) and afforded 14 as a white solid (44%). m.p.: 137–140 °C. IR νmax/cm−1 (KBr): 3122, 2951, 1732, 1689, 1622, 1512, 1456, 1383. 1H NMR (400MHz, CDCl3): δ 8.34 (1H,s), 8.07 (1H,s), 7.78 (1H,s), 5.36 (1H, t, J = 3.2), 3.69 (3H, s), 3.41 (1H, d, J = 17.6), 1.20 (3H, s), 1.19 (3H, s), 1.14 (3H, s), 1.13 (3H, s), 1.03 (3H, s), 0.92 (3H, s), 0.79 (3H, s). 13C NMR (100MHz, CDCl3): δ 207.4, 177.8, 153.1, 146.0, 144.5, 128.4, 125.6, 122.5, 52.9, 51.7, 48.6, 45.5, 45.3, 44.4, 43.5, 43.0, 42.0, 39.8, 38.5, 35.9, 32.2, 31.8, 31.4, 29.9, 28.7, 28.4, 27.1, 26.2, 25.8, 23.9, 22.6, 20.5, 16.5, 15.9. ESI-MS m/z: 274.53 (89%), 302.57 (23), 318.52 (100), 330.56 (14), 346.56 (27), 362.53 (25), 374.60 (16), 548.68 ([M + H]+, 93), 549.70 ([M + 2H] 2+, 28), 561.81 (23), 578.36 (20).
3-Oxo-olean-12-en-30-oic acid (15): Compound 15 was prepared from 2, using the same method as for the preparation of 5, with the obtention of a white solid (93%). m.p.: 245-248 °C. IR νmax/cm−1 (KBr): 3396, 2949, 1732, 1703, 1460, 1385. 1H NMR (400MHz, CDCl3): δ 5.31 (1H,t, J = 3.2), 2.33-2.59 (2H, m), 1.20 (3H, s), 1.14 (3H, s), 1.09 (3H, s), 1.06 (3H, s), 1.05 (3H, s), 1.01 (3H, s), 0.82 (3H, s). 13C NMR (100MHz, CDCl3): δ 217.9, 183.4, 144.2, 122.5, 55.3, 48.0, 47.4, 46.8, 44.0, 42.4, 41.6, 39.7, 39.2, 38.2, 36.6, 34.1, 32.1, 31.9, 31.0, 28.6, 28.1, 26.9, 26.4, 26.0, 25.8, 23.5, 21.4, 19.6, 16.6, 15.1. ESI-MS2 (25%) m/z: 274.98 ([M + H − C12H20O]+, 16%), 408.61 ([M + H − H2O − C2H4]+ or [M + H − HCOOH]+, 39); 437 ([M + H − H2O]+, 7), 455.42 ([M+H]+, 100).
2-Hydroxymethylene-3-oxo-olean-12-en-30-oic acid (16): The method followed that described for compound 7 but using compound 15 (850 mg, 1.87 mmol), ethyl formate (1.1 mL, 13.09 mmol) and sodium methoxide (1010 mg, 18.70 mmol), with the obtention of a white solid (77%). m.p.: 154–158 °C. IR νmax/cm−1 (KBr): 3442, 2953, 1731, 1699, 1643, 1587, 1456, 1383. 1H NMR (400MHz, CDCl3): δ 14.91 (1H, d, J = 3.0), 8.59 (1H, d, J = 2.7), 5.35 (1H, t, J = 3.4), 2.30 (1H, d, J = 14.4), 1.21 (3H, s), 1.20 (3H, s), 1.16 (3H, s), 1.13 (3H, s), 1.02 (3H, s), 0.93 (3H, s), 0.83 (3H, s). 13C NMR (100MHz, CDCl3): δ 190.8, 188.4, 183.4, 144.3, 122.6, 105.8, 52.1, 48.1, 45.6, 44.1, 42.6, 41.7, 40.2, 39.6, 39.4, 38.3, 36.2, 32.0, 31.9, 31.1, 28.7, 28.5, 28.2, 27.0, 26.1, 25.8, 23.5, 20.9, 19.6, 16.6, 14.7. ESI-MS2 (25%) m/z: 437.35 ([M + H − H2O − C2H4]+ or [M + H − HCOOH]+, 44%), 465.36 ([M + H − H2O]+, 100), 483.32 ([M + H]+, 56).
30-(1H-Imidazol-1-yl)-3,30-dioxo-olean-12-en-2-(1H-imidazol-1-yl)-methylene (17): Compound 17 was prepared using the same method as for the preparation of 9, but using compound 16 (300 mg, 0.62 mmol) and CDI (302 mg, 1.86 mmol) in anhydrous THF (6 mL) at reflux for 6 h. The resulting solid was submitted to FCC (petroleum ether/ ethyl acetate from (1:2) to (1:8)) to afford 17 as a white solid (56%). m.p.: 162–165 °C. IR νmax/cm−1 (KBr): 3124, 2937, 1713, 1687, 1610, 1522, 1489, 1456, 1383, 1308, 1227, 1173, 1030. 1H NMR (400MHz, CDCl3): δ 8.30 (1H, s), 7.78 (1H, s), 7.70 (1H, s), 7.58 (1H, s), 7.24 (1H, s), 7.17 (1H, s), 7.08 (1H, s), 5.19 (1H, t, J = 3.4), 2.90 (1H, d, J = 16.0), 1.44 (3H, s), 1.19 (3H, s), 1.18 (3H, s), 1.13 (3H, s), 0.99 (3H, s), 0.90 (3H, s), 0.79 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.6, 174.0, 143.8, 138.7, 137.2, 130.7, 130.6, 129.8, 122.8, 122.7, 119.2, 117.4, 52.5, 48.1, 46.6, 45.4, 45.2, 44.4, 43.2, 41.9, 39.6, 37.5, 35.9, 32.9, 32.2, 31.7, 29.8, 27.9, 27.9, 27.2, 25.9, 25.5, 23.7, 22.4, 20.3, 16.4, 15.6. ESI-MS2 (25%) m/z: 487.54 ([M + H − C3H3N2 − C2H4]+ or [M + H − C4H3N2O]+, 100%), 555.31 ([M + H − C2H4]+, 2), 583.44 ([M + H]+, 16).
30-(1H-Triazol-1-yl)-3,30-dioxo-olean-12-en-2-(1H-triazol-1-yl)-methylene (18): The method followed that described for compound 17 but using compound 16 (300 mg, 0.62 mmol) and CDT (1018 mg, 6.20 mmol) in anhydrous THF (6 mL) at reflux for 38 h. The resulting solid was submitted to FCC (petroleum ether/ ethyl acetate from (2:1) to (1:1)) to afford 18 as a white solid (38%). m.p.: 142–145 °C. IR νmax/cm−1 (KBr): 3126, 2954, 1713, 1695, 1618, 1512, 1456, 1383, 1279, 1161, 1132. 1H NMR (400MHz, CDCl3): δ 8.39 (1H, s), 8.25 (2H, s), 8.09 (1H, s), 7.80 (1H, s), 5.39 (1H, t, J = 3.4), 3.39 (1H, d, J = 17.5), 1.21 (3H, s), 1.20 (6H, s), 1.14 (3H, s), 1.03 (3H, s), 0.92 (3H, s), 0.82 (3H, s). 13C NMR (100MHz, CDCl3): δ 207.6, 182.0, 152.8, 146.9 (2), 146.0, 144.5, 128.4, 125.8, 122.6, 52.9, 48.4, 45.5, 45.3, 44.2, 43.5, 42.8, 42.0, 39.8, 38.4, 35.9, 32.2, 31.8, 31.3, 29.9, 28.9, 28.4, 27.1, 26.2, 25.8, 23.9, 22.6, 20.5, 16.5, 15.9. ESI-MS2 (35%) m/z: 488.42 ([M + H − C2H3N3 − C2H4]+ or [M + H − C3H3N3O]+, 100%), 514.64 ([M + H − C4H6O]+, 5), 584.67 (M+, 4), 585.77 ([M + H]+, 3).
Methyl 2-azidomethylene-3,11-dioxo-olean-12-en-30-oate (19): To a solution of compound 7 (700 mg, 1.37 mmol) in dichloromethane (14 mL), triethylamine (Et3N) (573 µL, 4.11 mmol) and tosyl chloride (TsCl) (654 mg, 3.43 mmol) were added. After 3.5 h, under magnetic stirring at room temperature, the reaction was completed. The solvent of the reaction mixture was removed under reduced pressure and the residue was suspended in acetone (14 mL); sodium azide (NaN3) (134 mg, 2.06 mmol) was added. After 5 h under magnetic stirring at room temperature, the reaction was completed. The acetone was removed under reduced pressure and water (60 mL) and ethyl acetate (60 mL) were added to the residue. The aqueous phase was further extracted with ethyl acetate (2 × 60 mL). The combined organic extract was then washed with water (3 × 50 mL), dried over Na2SO4, filtered and evaporated to the dryness, to afford a solid. The solid was subjected to flash FCC with petroleum ether/-ethyl acetate (15:1) to afford 19 as a white solid (62%). m.p.: 167–170 °C. IR νmax/cm−1 (KBr): 2951, 2127, 1730, 1659, 1460, 1387. 1H NMR (400MHz, CDCl3): δ 7.37 (1H, d, J = 1.5), 5.72 (1H, s), 3.76 (1H, d, J = 16.5), 3.69 (3H,s), 2.46 (1H,s), 1.37 (3H,s), 1.15 (3H,s), 1.14 (3H,s), 1.11 (3H,s), 1.09 (6H,s), 0.81 (3H,s). 13C NMR (100MHz, CDCl3): δ 205.0, 199.1, 176.9, 169.7, 137.2, 128.6, 123.0, 59.1, 53.4, 51.8, 48.3, 45.2, 44.9, 44.0, 43.3, 41.2, 40.6, 37.7, 35.7, 31.8, 31.6, 31.1, 29.3, 28.6, 28.3, 26.5, 26.4, 23.2, 22.5, 19.5, 18.0, 15.2. ESI-MS2 (35%) m/z: 462.38 ([M + H − C3H6O2]+, 100%), 508.46 ([M + H − CO]+ or [M + H − C2H4]+, 12), 518.37 ([M + H − H2O]+, 43), 536.61 ([M + H]+, 7).
Methyl 2-azidomethylene-3-oxo-olean-12-en-30-oate (20): Compound 20 was prepared using the same method as for the preparation of 19, but using compound 8 (700 mg, 1.41 mmol) in dichloromethane (14 mL), Et3N (492 µL, 3.53 mmol), TsCl (538 mg, 2.82 mmol), and then NaN3 (138 mg, 2.12 mmol) in acetone (14 mL). The resulting solid was subjected to flash FCC with petroleum ether/ ethyl acetate (20:1) to afford 20 as a white solid (57%). m.p.: 186–189 °C. IR νmax/cm−1 (KBr): 2943, 2116, 1730, 1672, 1589, 1452, 1383. 1H NMR (400MHz, CDCl3): δ 7.36 (1H, d, J = 1.6), 5.33 (1H, t, J = 3.4), 3.68 (3H,s), 2.66 (1H, d, J = 16.6), 1.15 (3H,s), 1.13 (3H,s), 1.09 (6H,s), 1.01 (3H,s), 0.88 (3H,s), 0.79 (3H,s). 13C NMR (100MHz, CDCl3): δ 205.7, 177.6, 144.3, 137.1, 123.2, 122.4, 53.1, 51.6, 48.3, 45.2, 45.1, 44.3, 42.8, 41.8, 40.2, 39.6, 38.3, 35.7, 32.0, 31.8, 31.3, 29.2, 28.5, 28.2, 27.0, 26.0, 25.7, 23.6, 22.6, 20.3, 16.4, 15.4. ESI-MS2 (35%) m/z: 462.48 ([M + H − H3COH − C2H4]+ or [M + H − H3CCOOH]+, 32%), 504.31 ([M + H − H2O]+, 100), 522.38 ([M + H]+, 7).
Compound 21: To a solution of compound 19 (350 mg, 0.65 mmol) in THF (7 mL), methyl propiolate (64.4 µL, 0.72 mmol), and CuI (12.4 mg, 0.065 mmol) were added. After 4 h under magnetic stirring at 65 °C, the reaction was completed. The reaction mixture was filtered and evaporated to dryness. The resulting solid was purified by FCC (petroleum ether/ ethyl acetate from (8:1) to (2:1)) to afford 21 as a white solid (42%). m.p.: 184–186 °C. IR νmax/cm−1 (KBr): 3136, 2951, 1747, 1726, 1691, 1651, 1620, 1554, 1458, 1385, 1209, 1034. 1H NMR (400MHz, CDCl3): δ 8.42 (1H, s), 7.99 (1H, s), 5.74 (1H,s), 4.17 (1H, d, J = 17.5), 3.96 (3H, s), 3.69 (3H,s), 2.53 (1H,s), 1.41 (3H, s), 1.22 (3H, s), 1.16 (3H,s), 1.14 (9H,s), 0.81 (3H,s). 13C NMR (100MHz, CDCl3): δ 205.8, 198.8, 176.9, 170.3, 160.7, 140.1, 128.8, 128.5, 128.2, 127.7, 59.0, 53.0, 52.4, 51.8, 48.5, 45.6, 44.9, 44.0, 43.6, 43.4, 41.2, 37.7, 35.9, 31.9, 31.3, 31.1, 29.7, 28.6, 28.3, 26.5, 26.4, 23.1, 22.3, 19.5, 17.9, 15.6. ESI-MS m/z: 620.59 ([M + H]+, 100%). Found C 69.39, H 8.21, N 6.66, calcd for C36H49N3O6: C 69.76, H 7.97, N 6.78 %.
Compound 22: Compound 22 was prepared using the same method as for the preparation of 21, but using compound 20 (350 mg, 0.67 mmol), methyl propiolate (66.0 µL, 0.74 mmol), and CuI (12.8 mg, 0.067 mmol). The resulting solid was purified by FCC [petroleum ether / ethyl acetate from (8:1) to (2:1)] to afford 22 as a white solid (44%). m.p.: 125–127 °C. IR νmax/cm−1 (KBr): 3143, 2953, 1730, 1699, 1620, 1556, 1456, 1385, 1221, 1034. 1H NMR (400MHz, CDCl3): δ 8.30 (1H, s), 7.90 (1H, s), 5.32 (1H, t, J = 3.4), 3.98 (3H, s), 3.68 (3H,s), 3.18 (1H, d, J = 18.5), 1.21 (3H, s), 1.18 (3H,s), 1.15 (3H, s), 1.13 (3H, s), 1.01 (3H,s), 0.92 (3H,s), 0.78 (3H, s). 13C NMR (100MHz, CDCl3): δ 206.6, 177.6, 160.7, 144.3, 139.8, 129.2, 128.5, 128.3, 122.2, 52.7, 52.5, 51.6, 48.4, 45.5, 45.2, 44.2, 43.6, 42.7, 41.8, 39.6, 38.3, 35.9, 32.0, 31.6, 31.3, 29.6, 28.5, 28.2, 26.9, 26.0, 25.6, 23.7, 22.4, 20.3, 16.3, 15.8. ESI-MS m/z: 606.33 ([M + H]+, 100%) Found C 71.58, H 8.58, N 6.88, calcd for C36H51N3O5: C 71.37, H 8.49, N 6.94 %.
3.2. Biology
HT-29, A549, MIA Paca 2, HeLa, A375, MCF7, HepG2, SH-SY5Y, Jurkat, and BJ cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD). Dulbecco’s Modified Eagle’s Medium (DMEM), Roswell Park Memorial Institute (RPMI)-1640 medium, Phosphate Buffered Saline (PBS), glucose 45%, human insulin 10 mg/mL, dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) powder, Trypan Blue (TB) 0.4%, propidium iodide (PI), Hoescht 33342, and protease inhibitor cocktail were obtained from Sigma-Aldrich Co. (St. Luis, MO, USA). Minimum Essential Medium (MEM), penicillin/streptomycin (P/S) and L-glutamine were purchased from Gibco-BRL (Eggenstein, Germany). Sodium pyruvate, trypsin/EDTA (0.05%/0.02%), MEM-Eagle nonessential amino acids 100x and ECL Western Blotting Detection Kit Reagent were obtained from Biological Industries (Kibbutz Beit Haemek, Israel). Fetal Bovine Serum (FBS) was obtained from PAA Laboratories (Pasching, Austria), the Cell Proliferation Kit II (XTT kit) was purchased from Roche (Roche Molecular Biochemicals, Indianapolis, IN, USA), and annexin V-FITC was obtained from Bender MedSystems (Vienna, Austria). Primary antibodies against caspases 3 (#9662), 8 (#9746) and 9 (#9502) were purchased from Cell Signaling Technology (Beverly, MA, USA) and primary antibody against α-actin (#69100) was obtained from MP Biomedicals (Santa Ana, CA, USA). Secondary antibodies HRP-conjugated goat anti-rabbit (NA934) and HRP-conjugated rabbit anti-mouse (P02060) were purchased from Amersham Biosciences (Uppsala, Sweden) and DAKO (Copenhagen, Denmark), respectively.
Stock solutions of 20 mM in DMSO of the synthesized compounds were prepared and stored at −20 °C. Working solutions were prepared in culture medium and appropriate amounts of DMSO were included in controls; all solutions had a final concentration of 0.5% DMSO.
3.2.1. Cell Culture
HT-29, A549, MIA Paca 2, HeLa, A375, and SH-SY5Y cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% P/S. HepG2 and BJ cells were grown in DMEM supplemented with 10% heat-inactivated FBS, 1% P/S and 1 mM sodium pyruvate. Jurkat cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 1% P/S, and 2 mM L-glutamine. MCF7 cells were maintained in MEM supplemented with 10% heat-inactivated FBS, 0.1% P/S, 1 mM sodium pyruvate, 2 mM L-glutamine, 1x MEM-Eagle nonessential amino acids, 0.01mg/mL insulin human and 10 mM glucose.
All cell cultures were incubated in a 5% CO2 humidified atmosphere at 37 °C.
3.2.2. Antiproliferative Activity Assays
The antiproliferative activity of the synthesized compounds on HT-29, A549, MIA Paca 2, HeLa, A375, MCF7, HepG2, and BJ cells was determined by the MTT assay. Exponentially growing cells were plated in 96-well plates at a density of 1–8 × 103 cells/ well. After 24 h of incubation, the growth medium was replaced with fresh medium containing either the compounds dissolved in DMSO at different concentrations or only with DMSO, in triplicate, and the cells were continued to culture for 72 h. After incubation with the compounds, the medium was removed and 100 µL of MTT solution (0.5 mg/mL) were added to each well and the plates were incubated for 1 h. MTT was removed and 100 µL of DMSO was added to dissolve the formazan crystals. The absorbance was immediately read at 550 nm on an ELISA plate reader (Tecan Sunrise MR20-301, TECAN, Austria). For SH-SY5Y and Jurkat cells, the antiproliferative activity was determined by XTT assay. SH-SY5Y cells were plated with 1.6 × 104 cells/well in 96-well plates in 100 µL medium. The compounds at different concentrations or only the vehicle (medium with 0.5% DMSO), in 100 μL, were added 24 h after seeding, in triplicate, and incubated for 72 h. For Jurkat cells, 5.5 × 103 cells/well were plated simultaneously with the addition of the different concentrations of compounds or vehicle, in triplicate, and were allowed to incubate for 72 h. In both cases, after that incubation period, 100 μL of the XTT labeling mixture was added to each well and the plates were incubated again for 4 h. Then, the absorbance was read at 450 nm on the ELISA plate reader.
Concentrations that inhibit cell proliferation by 50% (IC50) represent an average of a minimum of three independent experiments and were expressed as means ± standard deviation (SD).
3.2.3. Cell Viability over Time Assays
The viability over time of Jurkat cells, treated with compound 10, was assessed by TB cell counting experiments. For these assays, 1.6 × 105 cells were plated per well in 6-well plates simultaneously with the addition of compound 10, in the concentration of its IC50 value at 72 h, or only vehicle, in a total volume of 2 mL of medium. After each incubation time (3, 6, 12, 24, 48, and 72h), the suspension of cells of each well was collected. Equal amounts of TB solution and suspension were mixed and the mixture was placed in a Neubauer chamber in order to perform the counting. The number of cells determined for each condition, in each incubation time, represents an average of three independent experiments, with two replicates per experiment.
3.2.4. Cell Cycle Analysis
Cell cycle was assessed by flow cytometry using a fluorescence-activated cell sorter (FACS). Jurkat cells were plated in 6-well plates at a density of 1.6 × 105 cells, simultaneously with the addition of compound 10, at a concentration corresponding to its IC50 value at 72 h of treatment, or with only the vehicle, in a total volume of 2 mL of medium. The cells were allowed to incubate for 3, 6, and 24 h. After incubation, cells were collected and centrifuged. The supernatant was removed and the pellet was resuspended in 1 mL of TBS containing 1mg/mL PI, 10mg/mL RNase free of DNase, and 0.1% Igepal CA-630, for 1 h, at 4 °C. FACS analysis was performed at 488 nm in an Epics XL flow cytometer (Coulter Corporation, Hialeah, FL, USA). Data were collected and analyzed using the Multicycle software (Phoenix Flow Systems, San Diego, CA, USA). Three independent experiments were performed, with two replicates per experiment.
3.2.5. Annexin V-FITC/PI Flow Cytometry Assay
Apoptosis was assessed by flow cytometry using a FACS. The same number of Jurkat cells taken for the cell cycle assay was treated as described above. The cells were allowed to incubate for 6 and 24 h. After incubation, cells were collected and centrifuged. The supernatant was removed and the pellet was resuspended in 95 µL of binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Annexin V-FITC conjugate (3 µL) was added and cells were incubated for 30 min, at room temperature, in darkness. After incubation, 0.8 mL of binding buffer was added. Just before the FACS analysis, cells were stained with 20 µL of 1mg/mL PI solution.
Three independent experiments were performed, with two replicates per experiment.
3.2.6. Observation of Morphological Changes
The morphological changes were observed by fluorescence microscopy using Hoechst staining. Jurkat cells were plated in 6-well plates at a density of 1.6 × 105 cells, simultaneously with the addition of compound 10, at a concentration corresponding to its IC50 value at 72 h of treatment, or with only the vehicle, in a total volume of 2 mL of medium (six wells per condition). The cells were incubated for 6 h. Before collecting, morphological changes were observed under a phase-contrast microscope. Cells were collected by centrifugation, washed twice with PBS and stained with 500 µL of Hoechst 33342 solution (2 µg/mL in PBS) for 15 min, at room temperature, in darkness. Finally, cells were washed and resuspended in 10 µL PBS. The samples were mounted on a slide and observed with a fluorescence microscope (DMRB, Leica Microssystems, Wetzlar, Germany) with a DAPI filter. Three independent experiments were conducted.
3.2.7. Western Blot Analysis
Jurkat cells (1.6 × 106 cells) were cultured in 25 cm2 flasks and treated for 6 h with compound 10 at different concentrations. After incubation, the cells were washed twice with ice-cold PBS and resuspended in lysis buffer containing 20 mM Tris/ acetate, pH 7.5, 270 mM sucrose, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, and 1% protease inhibitor cocktail. The samples were sonicated, incubated on ice for 20 min, and centrifuged at 13,200 rpm for 12 min at 4 °C, and the pellets were discarded. The supernatants were assayed for protein concentration using a BCA kit (Thermo fisher Scientific, Waltham, MA, USA). Protein extracts (50 μg) were separated on 15% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to a polyvinylnitrocellulose transfer membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked by incubation in TBS buffer (20 mM Tris, pH = 7.5 and 132 mM NaCl) containing 0.1% of Tween and 5% of BSA or nonfat milk, for 1 h, at room temperature. Then, membranes were blotted with the primary antibodies anti-caspase 3, 8 and 9 (1:1000) overnight at 4 °C. The blots were washed three times with TBS-0.1% Tween and incubated with the appropriate secondary antibodies, for 1 h, at room temperature. Before protein detection, membranes were washed again five times with TBS-0.1% Tween. Immunocomplexes were visualized using the Immobilon ECL Western Blotting Detection Kit Reagent. Three independent experiments were performed.
4. Conclusions
In this study, we synthesized a series of novel GA derivatives via the introduction of different heterocyclic rings conjugated with an α,β-unsaturated ketone in its ring A. Screening for antiproliferative activity in a panel of nine human cancer cell lines showed that the most active compound 10 was 31- to 96-fold more potent than GA. This derivative was also more selective towards tumor cells. Further biological studies performed in Jurkat cells suggest that potent apoptosis induction involving both the intrinsic and extrinsic pathways is the main mechanism underlying the antiproliferative activity of compound 10. The enhanced potency and selectivity, and the biological activity of this new GA heterocyclic derivative warrant further preclinical evaluation.
Supplementary Materials
1H-NMR and 13C-NMR spectra of selected compounds are available online.
Author Contributions
Conceptualization, J.A.R.S. and S.M.; Synthesis and Structural Characterization, D.P.S.A. and J.A.R.S.; Biological Experiments, D.P.S.A. and S.M.; Writing—Original Draft Preparation, D.P.S.A.; Writing—Review and Editing, J.A.R.S., M.C., and S.M.; Supervision, J.A.R.S. and S.M; Project Administration, J.A.R.S. and M.C.; Funding Acquisition, J.A.R.S. and M.C.
Funding
Jorge A. R. Salvador thanks PT2020 (Programa Operacional do Centro 2020), project nº 3269, drugs2CAD, and the financial support by FEDER (European Regional Development Fund) through the COMPETE 2020 Programme (Operational Programme for Competitiveness and Internationalisation). Jorge A. R. Salvador also wishes to thank Universidade de Coimbra for financial support. Daniela P. S. Alho thanks FEDER (Programa Operacional Factores de Competitividade—COMPETE 2020) and Fundação para a Ciência e Tecnologia (FCT) through Projecto Estratégico: UID/NEU/04539/2013 and the financial support for the PhD grant SFRH/BD/66020/2009. Marta Cascante and Silvia Marin thank MINECO-European Commission FEDER—Una manera de hacer Europa (SAF2017-89673-R) and Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR)—Generalitat de Catalunya (2017SGR1033). Marta Cascante also acknowledges the support received through the prize “ICREA Academia” for excellence in research, funded by ICREA Foundation—Generalitat de Catalunya. The authors acknowledge UC-NMR facility, which is supported by FEDER and FCT funds through the grants REEQ/481/QUI/2006, RECI/QEQ-FI/0168/2012, and CENTRO-07-CT62-FEDER-002012, and Rede Nacional de Ressonância Magnética Nuclear (RNRMN), for NMR data.
Acknowledgments
The authors are grateful to Laboratório de Espectometria de Massa (LEM)-CEF/UC integrated in the Rede Nacional de Espectrometria de Massa (RNEM) of Portugal for the MS analysis, and Centro de Apoio Científico e Tecnolóxico á Investigación (CACTI), Universidade de Vigo, for elemental analysis. The authors acknowledge Centres Científics I Tecnològics de la UB (CCiTUB) for flow cytometry analysis support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dzubak, P.; Hajduch, M.; Vydra, D.; Hustova, A.; Kvasnica, M.; Biedermann, D.; Markova, L.; Urban, M.; Sarek, J. Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat. Prod. Rep. 2006, 23, 394–411. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Moreira, V.M.; Goncalves, B.M.F.; Leal, A.S.; Jing, Y.K. Ursane-type pentacyclic triterpenoids as useful platforms to discover anticancer drugs. Nat. Prod. Rep. 2012, 29, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Leal, A.S.; Alho, D.P.S.; Goncalves, B.M.F.; Valdeira, A.S.; Mendes, V.I.S.; Jing, Y.K. Highlights of pentacyclic triterpenoids in the cancer settings. In Studies in Natural Products Chemistry; AttaUrRahman, F.R.S., Ed.; Elsevier Science Bv: Amsterdam, The Netherland, 2014; Volume 41, pp. 33–73. [Google Scholar]
- Chudzik, M.; Korzonek-Szlacheta, I.; Krol, W. Triterpenes as potentially cytotoxic compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.R.; Leal, A.S.; Valdeira, A.S.; Goncalves, B.M.F.; Alho, D.P.S.; Figueiredo, S.A.C.; Silvestre, S.M.; Mendes, V.I.S. Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur. J. Med. Chem. 2017, 142, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Schwarz, S.; Per, V.; Kowitsch, A.; Siewert, B.; Csuk, R. Incorporation of a michael acceptor enhances the antitumor activity of triterpenoic acids. Eur. J. Med. Chem. 2015, 101, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, B.M.F.; Salvador, J.A.R.; Marin, S.; Cascante, M. Synthesis and anticancer activity of novel fluorinated asiatic acid derivatives. Eur. J. Med. Chem. 2016, 114, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Mendes, V.I.S.; Bartholomeusz, G.A.; Ayres, M.; Gandhi, V.; Salvador, J.A.R. Synthesis and cytotoxic activity of novel a-ring cleaved ursolic acid derivatives in human non-small cell lung cancer cells. Eur. J. Med. Chem. 2016, 123, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Urea derivates of ursolic, oleanolic and maslinic acid induce apoptosis and are selective cytotoxic for several human tumor cell lines. Eur. J. Med. Chem. 2016, 119, 1–16. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine b conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef]
- Spivak, A.; Khalitova, R.; Nedopekina, D.; Dzhemileva, L.; Yunusbaeva, M.; Odinokov, V.; D’Yakonov, V.; Dzhemilev, U. Synthesis and evaluation of anticancer activities of novel c-28 guanidine-functionalized triterpene acid derivatives. Molecules 2018, 23, 22. [Google Scholar] [CrossRef]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of glycyrrhiza sp and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.H.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, P.H.; Zhao, W.X.; Su, W.X.; Zhang, Z.Q.; Zhang, L.; Liu, J.M.; Ren, G.L.; Yin, Z.Y.; Wang, X.M. 18 beta-glycyrrhetinic acid inhibits hepatocellular carcinoma development by reversing hepatic stellate cell-mediated immunosuppression in mice. Int. J. Cancer 2013, 132, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.K.; Khan, R.; Ali, N.; Khan, A.Q.; Rehman, M.U.; Tahir, M.; Lateef, A.; Nafees, S.; Mehdi, S.J.; Rashid, S.; et al. 18-glycyrrhetinic acid alleviates 2-acetylaminofluorene-induced hepatotoxicity in wistar rats: Role in hyperproliferation, inflammation and oxidative stress. Hum. Exp. Toxicol. 2015, 34, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Shen, Y.; Qiu, R.F.; Chen, Z.L.; Chen, Z.H.; Chen, W.B. 18 beta-glycyrrhetinic acid exhibits potent antitumor effects against colorectal cancer via inhibition of cell proliferation and migration. Int. J. Oncol. 2017, 51, 615–624. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Lee, M.S.; Han, E.S.; Lee, S.J. 18 beta-glycyrrhetinic acid induces apoptotic cell death in siha cells and exhibits a synergistic effect against antibiotic anti-cancer drug toxicity. Life Sci. 2008, 83, 481–489. [Google Scholar] [CrossRef]
- Sharma, G.; Kar, S.; Palit, S.; Das, P.K. 18 beta-glycyrrhetinic acid (concur) induces apoptosis through modulation of akt/foxo3a/bim pathway in human breast cancer mcf-7 cells. J. Cell. Physiol. 2012, 227, 1923–1931. [Google Scholar] [CrossRef]
- Wang, D.; Wong, H.K.; Feng, Y.B.; Zhang, Z.J. 18beta-glycyrrhetinic acid induces apoptosis in pituitary adenoma cells via ros/mapks-mediated pathway. J. Neuro-Oncol. 2014, 116, 221–230. [Google Scholar] [CrossRef]
- Hasan, S.K.; Siddiqi, A.; Nafees, S.; Ali, N.; Rashid, S.; Ali, R.; Shahid, A.; Sultana, S. Chemopreventive effect of 18 beta-glycyrrhetinic acid via modulation of inflammatory markers and induction of apoptosis in human hepatoma cell line (hepg2). Mol. Cell. Biochem. 2016, 416, 169–177. [Google Scholar] [CrossRef]
- Cai, Y.; Zhao, B.X.; Liang, Q.Y.; Zhang, Y.Q.; Cai, J.Y.; Li, G.F. The selective effect of glycyrrhizin and glycyrrhetinic acid on topoisomerase ii alpha and apoptosis in combination with etoposide on triple negative breast cancer mda-mb-231 cells. Eur. J. Pharmacol. 2017, 809, 87–97. [Google Scholar] [CrossRef]
- Baltina, L.A. Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr. Med. Chem. 2003, 10, 155–171. [Google Scholar] [CrossRef]
- Guo, W.B.; Yan, M.M.; Xu, B.; Chu, F.H.; Wang, W.; Zhang, C.Z.; Jia, X.H.; Han, Y.T.; Xiang, H.J.; Zhang, Y.Z.; et al. Design, synthesis, and biological evaluation of the novel glycyrrhetinic acid-cinnamoyl hybrids as anti-tumor agents. Chem. Cent. J. 2016, 10, 11. [Google Scholar] [CrossRef]
- Li, Y.; Feng, L.; Song, Z.F.; Li, H.B.; Huai, Q.Y. Synthesis and anticancer activities of glycyrrhetinic acid derivatives. Molecules 2016, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.L.; Bai, L.F.; Zhu, H.L.; Zhang, W.M.; Lv, P.C. Synthesis and biological evaluation of glycyrrhetic acid derivatives as potential vegfr2 inhibitors. ChemMedChem 2017, 12, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wu, G.R.; Zhang, X.Y.; Yan, M.M.; Zhao, R.; Xue, N.N.; Fang, K.; Wang, H.; Chen, M.; Guo, W.B.; et al. An overview of structurally modified glycyrrhetinic acid derivatives as antitumor agents. Molecules 2017, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Huai, X.D.; Zheng, Q.X.; Wang, W.; Li, H.J.; Huai, Q.Y. Design and preparation of derivatives of oleanolic and glycyrrhetinic acids with cytotoxic properties. Drug Des. Dev. Ther. 2018, 12, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.C.; Salvador, J.A.R.; Marin, S.; Cascante, M.; Moreira, J.N.; Dinis, T.C.P. Synthesis and structure-activity relationship study of novel cytotoxic carbamate and n-acylheterocyclic bearing derivatives of betulin and betulinic acid. Bioorg. Med. Chem. 2010, 18, 4385–4396. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Wang, R.; Salvador, J.A.R.; Jing, Y.K. Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21(waf1) and noxa in pancreatic cancer cells. Bioorg. Med. Chem. 2012, 20, 5774–5786. [Google Scholar] [CrossRef]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef]
- Musumeci, F.; Schenone, S.; Desogus, A.; Nieddu, E.; Deodato, D.; Botta, L. Click chemistry, a potent tool in medicinal sciences. Curr. Med. Chem. 2015, 22, 2022–2050. [Google Scholar] [CrossRef]
- Ma, N.; Wang, Y.; Zhao, B.X.; Ye, W.C.; Jiang, S. The application of click chemistry in the synthesis of agents with anticancer activity. Drug Des. Dev. Ther. 2015, 9, 1585–1599. [Google Scholar] [CrossRef]
- Meghani, N.M.; Amin, H.H.; Leel, B.J. Mechanistic applications of click chemistry for pharmaceutical drug discovery and drug delivery. Drug Discov. Today 2017, 22, 1604–1619. [Google Scholar] [CrossRef]
- Pertino, M.W.; Lopez, C.; Theoduloz, C.; Schmeda-Hirschmann, G. 1,2,3-triazole-substituted oleanolic acid derivatives: Synthesis and antiproliferative activity. Molecules 2013, 18, 7661–7674. [Google Scholar] [CrossRef] [PubMed]
- Dangroo, N.A.; Singh, J.; Rath, S.K.; Gupta, N.; Qayum, A.; Singh, S.; Sangwan, P.L. A convergent synthesis of novel alkyne-azide cycloaddition congeners of betulinic acid as potent cytotoxic agent. Steroids 2017, 123, 1–12. [Google Scholar] [CrossRef]
- Pokorny, J.; Borkova, L.; Urban, M. Click reactions in chemistry of triterpenes - advances towards development of potential therapeutics. Curr. Med. Chem. 2018, 25, 636–658. [Google Scholar] [CrossRef] [PubMed]
- Pertino, M.W.; Petrera, E.; Alche, L.E.; Schmeda-Hirschmann, G. Synthesis, antiviral and cytotoxic activity of novel terpenyl hybrid molecules prepared by click chemistry. Molecules 2018, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Wang, Y.T.; Gao, Y.; Li, L.; Guo, X.; Liu, D.; Jing, Y.K.; Zhao, L.X. Synthesis of methyl 2-cyano-3,12-dioxo-18 beta-olean-1,9(11)-dien-30-oate analogues to determine the active groups for inhibiting cell growth and inducing apoptosis in leukemia cells. Org. Biomol. Chem. 2014, 12, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Csuk, R. Synthesis and antitumour activity of glycyrrhetinic acid derivatives. Bioorg. Med. Chem. 2010, 18, 7458–7474. [Google Scholar] [CrossRef]
- Rao, G.; Kondaiah, P.; Singh, S.K.; Ravanan, P.; Sporn, M.B. Chemical modifications of natural triterpenes-glycyrrhetinic and boswellic acids: Evaluation of their biological activity. Tetrahedron 2008, 64, 11541–11548. [Google Scholar] [CrossRef]
- Chu, F.H.; Xu, X.; Li, G.L.; Gu, S.; Xu, K.; Gong, Y.; Xu, B.; Wang, M.N.; Zhang, H.Z.; Zhang, Y.Z.; et al. Amino acid derivatives of ligustrazine-oleanolic acid as new cytotoxic agents. Molecules 2014, 19, 18215–18231. [Google Scholar] [CrossRef] [PubMed]
- Porchia, M.; Dolmella, A.; Gandin, V.; Marzano, C.; Pellei, M.; Peruzzo, V.; Refosco, F.; Santini, C.; Tisato, F. Neutral and charged phosphine/scorpionate copper(i) complexes: Effects of ligand assembly on their antiproliferative activity. Eur. J. Med. Chem. 2013, 59, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, B.M.F.; Salvador, J.A.R.; Marin, S.; Cascante, M. Synthesis and biological evaluation of novel asiatic acid derivatives with anticancer activity. RSC Advances 2016, 6, 3967–3985. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Y.; Halliday, G.C.; Berry, P.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Del Bello, F.; Piergentili, A.; Newell, D.R.; et al. Structurally diverse mdm2-p53 antagonists act as modulators of mdr-1 function in neuroblastoma. Br. J. Cancer 2014, 111, 716–725. [Google Scholar] [CrossRef]
- Antunovic, M.; Kriznik, B.; Ulukaya, E.; Yilmaz, V.T.; Mihalic, K.C.; Madunic, J.; Marijanovic, I. Cytotoxic activity of novel palladium-based compounds on leukemia cell lines. Anti-Cancer Drugs 2015, 26, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Rajic, Z.; Zorc, B.; Raic-Malic, S.; Ester, K.; Kralj, M.; Pavelic, K.; Balzarini, J.; De Clercq, E.; Mintas, M. Hydantoin derivatives of l- and d-amino acids: Synthesis and evaluation of their antiviral and antitumoral activity. Molecules 2006, 11, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 14. [Google Scholar] [CrossRef]
- Hibasami, H.; Iwase, H.; Yoshioka, K.; Takahashi, H. Glycyrrhetic acid (a metabolic substance and aglycon of glycyrrhizin) induces apoptosis in human hepatoma, promyelotic leukemia and stomach cancer cells. Int. J. Mol. Med. 2006, 17, 215–219. [Google Scholar] [CrossRef]
- Satomi, Y.; Nishino, H.; Shibata, S. Glycyrrhetinic acid and related compounds induce g1 arrest and apoptosis in human hepatocellular carcinoma hepg2. Anticancer Res. 2005, 25, 4043–4047. [Google Scholar]
- Lee, C.S.; Yang, J.C.; Kim, Y.J.; Jang, E.R.; Kim, W.; Myung, S.C. 18 beta-glycyrrhetinic acid potentiates apoptotic effect of trichostatin a on human epithelial ovarian carcinoma cell lines. Eur. J. Pharmacol. 2010, 649, 354–361. [Google Scholar] [CrossRef]
- Yang, J.C.; Myung, S.C.; Kim, W.; Lee, C.S. 18 beta-glycyrrhetinic acid potentiates hsp90 inhibition-induced apoptosis in human epithelial ovarian carcinoma cells via activation of death receptor and mitochondrial pathway. Mol. Cell Biochem. 2012, 370, 209–219. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. P53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Haas, M. Frequent mutations in the p53 tumor suppressor gene in human leukemia t-cell lines. Mol. Cell Biol. 1990, 10, 5502–5509. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors. |
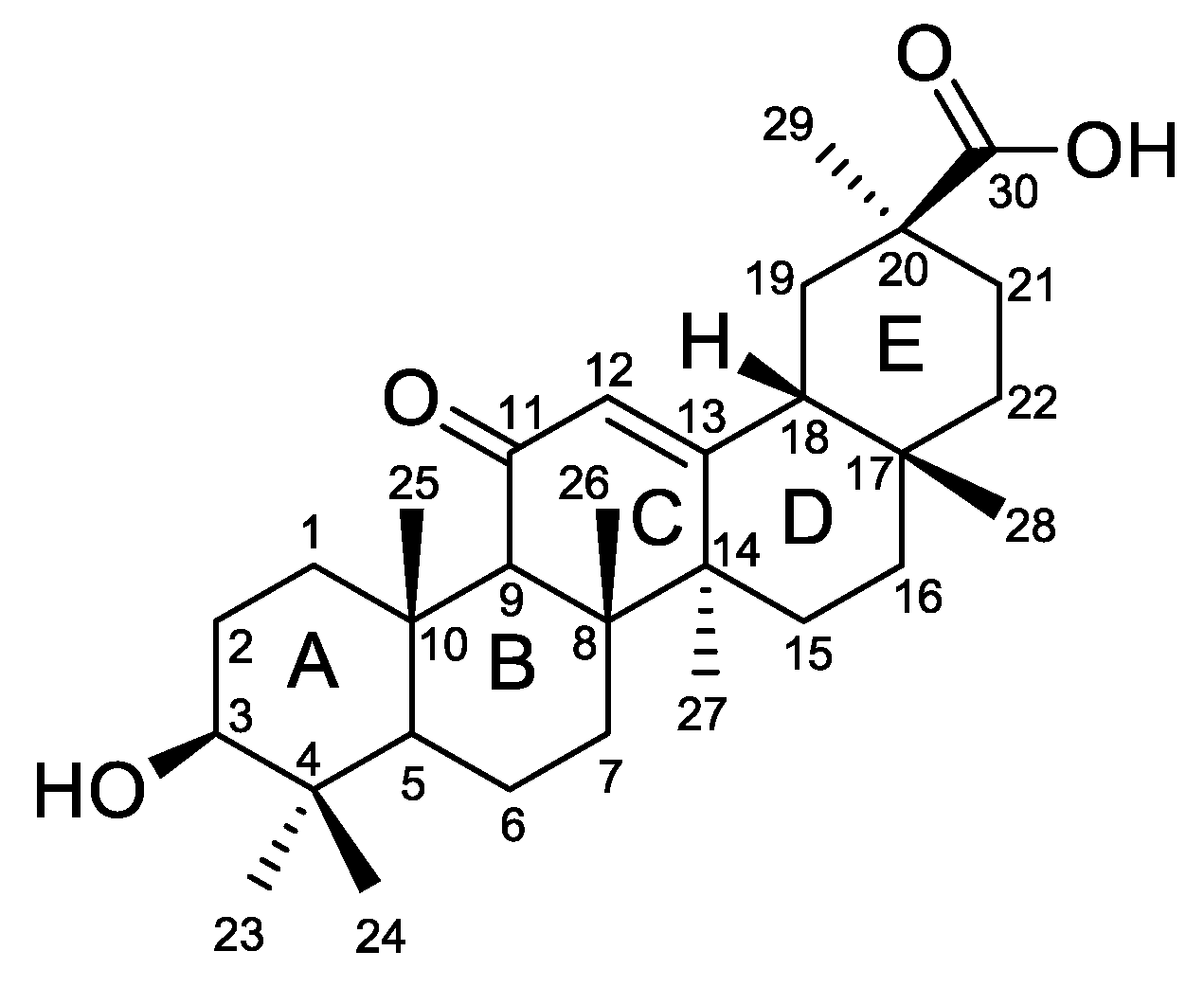
Figure 1.
Chemical structure of glycyrrhetinic acid 1.
Scheme 1.
Reagents and conditions: (a) zinc dust, concentrated HCl, dioxane, r.t.; (b) CH3I, K2CO3, DMF, r.t.; (c) Jones reagent, acetone, 0 °C; (d) ethyl formate, NaOMe, benzene, r.t., N2; and (e) CDI, CBMI or CDT, dry THF, reflux, N2.
Scheme 1.
Reagents and conditions: (a) zinc dust, concentrated HCl, dioxane, r.t.; (b) CH3I, K2CO3, DMF, r.t.; (c) Jones reagent, acetone, 0 °C; (d) ethyl formate, NaOMe, benzene, r.t., N2; and (e) CDI, CBMI or CDT, dry THF, reflux, N2.

Scheme 2.
Reagents and conditions: (a) Jones reagent, acetone, 0 °C; (b) ethyl formate, NaOMe, benzene, r.t., N2; and (c) CDI or CDT, dry THF, reflux, N2.
Scheme 2.
Reagents and conditions: (a) Jones reagent, acetone, 0 °C; (b) ethyl formate, NaOMe, benzene, r.t., N2; and (c) CDI or CDT, dry THF, reflux, N2.
Scheme 3.
Reagents and conditions: (a) TsCl, Et3N, CH2Cl2, r.t.; (b) NaN3, acetone, r.t.; and (c) methyl propiolate, CuI, THF, 65 °C.
Scheme 3.
Reagents and conditions: (a) TsCl, Et3N, CH2Cl2, r.t.; (b) NaN3, acetone, r.t.; and (c) methyl propiolate, CuI, THF, 65 °C.
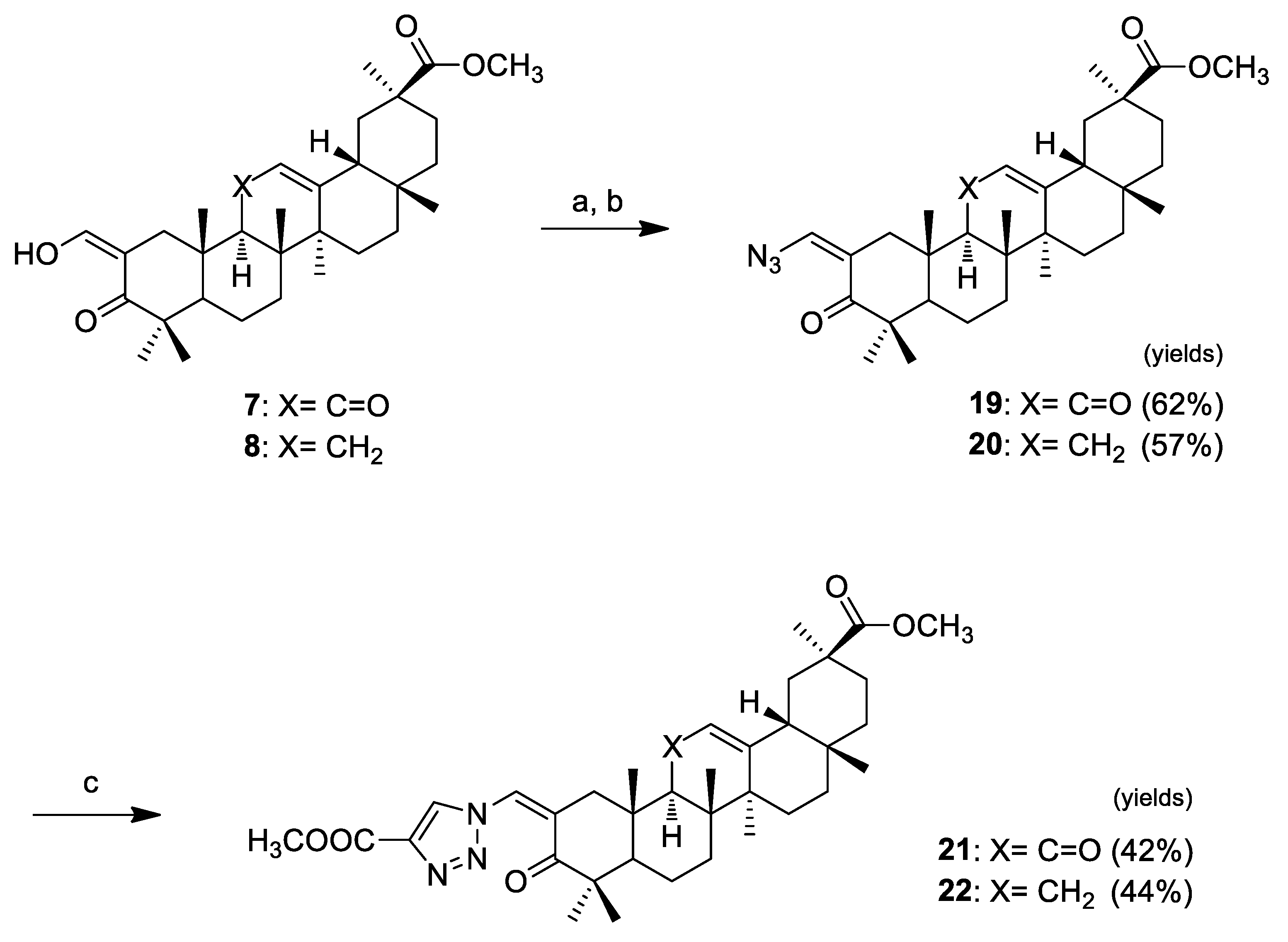
Figure 2.
Effect of compound 10 on cell viability. Jurkat cells were treated with 1.1 μM compound 10 for different time periods ((A): 3, 6, 12, and 24 h; (B): 24, 48, and 72 h). Cell numbers were determined by Trypan Blue counting assays. Results are presented as means ± SD of three independent experiments.
Figure 2.
Effect of compound 10 on cell viability. Jurkat cells were treated with 1.1 μM compound 10 for different time periods ((A): 3, 6, 12, and 24 h; (B): 24, 48, and 72 h). Cell numbers were determined by Trypan Blue counting assays. Results are presented as means ± SD of three independent experiments.

Figure 3.
Effect of compound 10 on cell cycle distribution. Cell cycle analysis of Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 3 (A), 6 (B), and 24h (C). After treatment, cells were stained with PI and DNA content analyzed by flow cytometry. A representative histogram is shown for each time of incubation and condition. Results are presented as means ± SD of three independent experiments.
Figure 3.
Effect of compound 10 on cell cycle distribution. Cell cycle analysis of Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 3 (A), 6 (B), and 24h (C). After treatment, cells were stained with PI and DNA content analyzed by flow cytometry. A representative histogram is shown for each time of incubation and condition. Results are presented as means ± SD of three independent experiments.

Figure 4.
Induction of apoptosis by compound 10. Flow cytometric quantification of apoptosis in Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 6 (A) and 24 h (B). After treatment, cells were stained with annexin V-FICT/PI and analyzed by flow cytometry. A representative dot plot is shown for each time of incubation and condition; the right quadrants of each diagram (annexin+/PI− and annexin+/PI+) represent apoptotic cells. The percentage of early (dark gray bar) and late (light gray bar) apoptotic cells in each condition is represented as a bars diagram, calculated from dot plots. Results are presented as means ± SD of three independent experiments.
Figure 4.
Induction of apoptosis by compound 10. Flow cytometric quantification of apoptosis in Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 6 (A) and 24 h (B). After treatment, cells were stained with annexin V-FICT/PI and analyzed by flow cytometry. A representative dot plot is shown for each time of incubation and condition; the right quadrants of each diagram (annexin+/PI− and annexin+/PI+) represent apoptotic cells. The percentage of early (dark gray bar) and late (light gray bar) apoptotic cells in each condition is represented as a bars diagram, calculated from dot plots. Results are presented as means ± SD of three independent experiments.
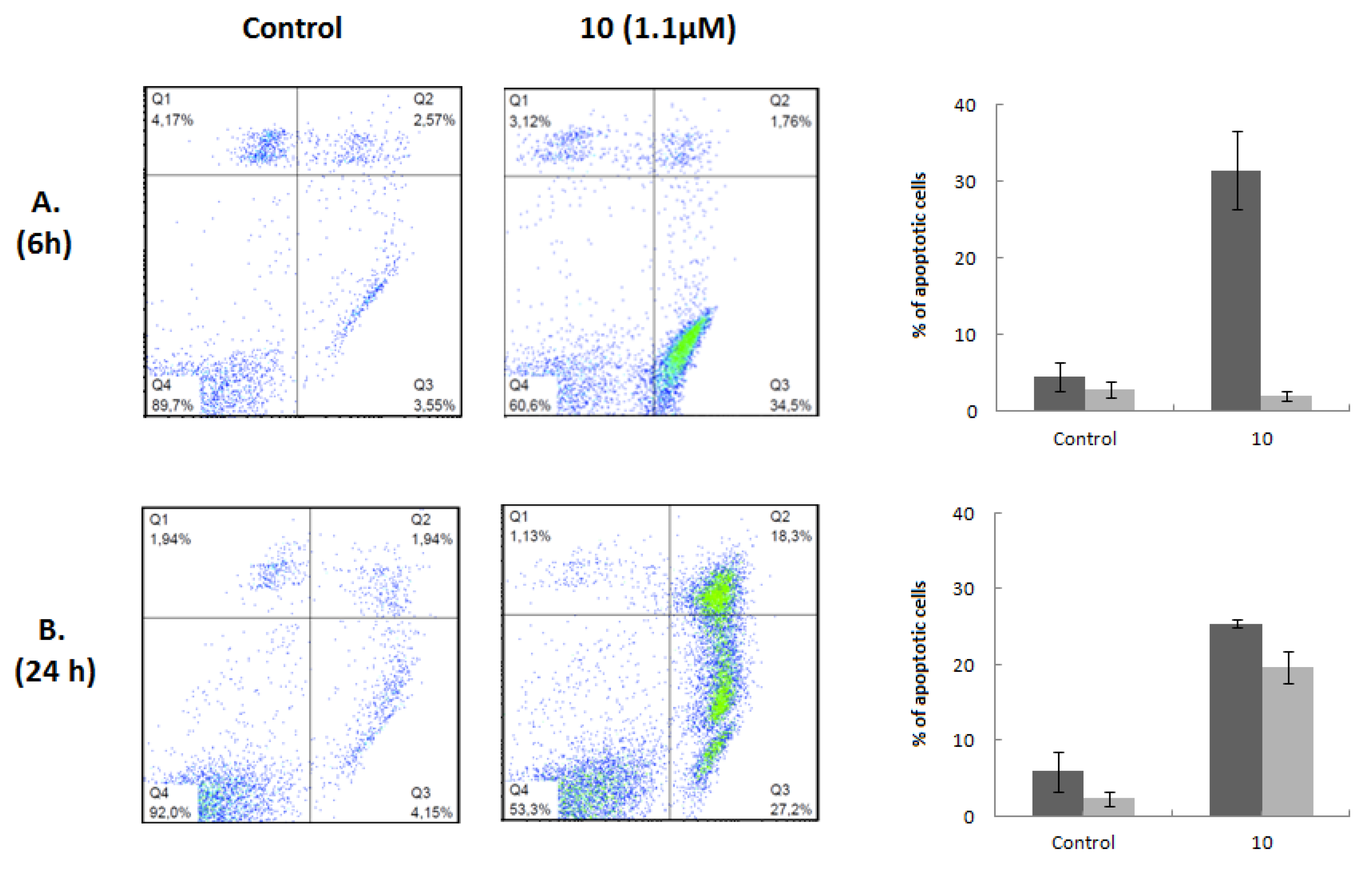
Figure 5.
Morphological changes in Jurkat cells. Morphology of Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 6 h was examined using a phase-contrast microscope (A) and fluorescence microscopy after Hoechst 33342 staining (B).
Figure 5.
Morphological changes in Jurkat cells. Morphology of Jurkat cells untreated (Control) or treated with 1.1 μM compound 10 for 6 h was examined using a phase-contrast microscope (A) and fluorescence microscopy after Hoechst 33342 staining (B).
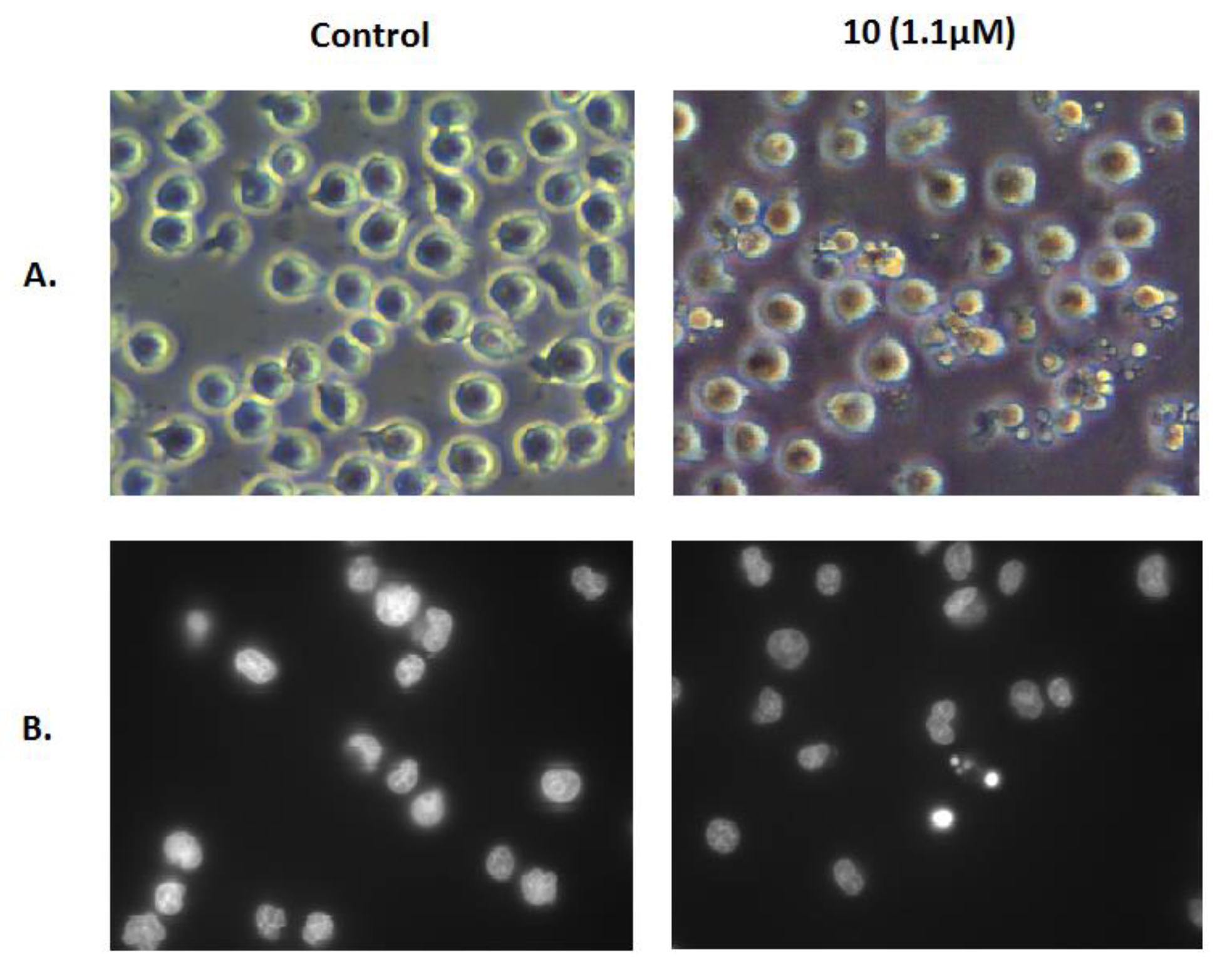
Figure 6.
Effect of compound 10 on the activation of caspases 3, 8, and 9 in Jurkat cells after 6 h of treatment; α-actin was used as a loading control.
Figure 6.
Effect of compound 10 on the activation of caspases 3, 8, and 9 in Jurkat cells after 6 h of treatment; α-actin was used as a loading control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antiproliferative activities of GA, its derivatives, and cisplatin against HT-29 and A549 cell lines.
Table 1.
Antiproliferative activities of GA, its derivatives, and cisplatin against HT-29 and A549 cell lines.
| Compound | Cell line (IC50, µM) 1 | |
|---|---|---|
| HT-29 | A549 | |
| 1 | 115.7 ± 1.6 | 110.5 ± 3.9 |
| 2 | 88.1 ± 1.4 | N.D. |
| 3 | 19.6 ± 0.6 | N.D. |
| 4 | 18.5 ± 0.9 | N.D. |
| 5 | 46.3 ± 2.3 | N.D. |
| 6 | 63.9 ± 1.1 | N.D. |
| 7 | 14.3 ± 0.3 | 18.5 ± 1.5 |
| 8 | 14.0 ± 0.2 | 17.0 ± 0.6 |
| 9 | 11.5 ± 0.5 | 11.1 ± 0.1 |
| 10 | 3.3 ± 0.2 | 2.8 ± 0.2 |
| 11 | 9.4 ± 0.7 | 10.3 ± 0.6 |
| 12 | 3.6 ± 0.1 | 3.1 ± 0.1 |
| 13 | 31.2 ± 1.5 | 24.7 ± 0.9 |
| 14 | 12.1 ± 0.2 | 12.3 ± 0.6 |
| 15 | 92.3 ± 1.5 | N.D. |
| 16 | 60.3 ± 2.0 | 54.4 ± 2.6 |
| 17 | 22.4 ± 0.5 | 23.1 ± 0.8 |
| 18 | 21.8 ± 1.6 | 24.5 ± 0.6 |
| 19 | 38.5 ± 0.6 | 48.5 ± 0.8 |
| 20 | 36.3 ± 1.4 | 44.3 ± 3.6 |
| 21 | 11.0 ± 0.8 | 10.6 ± 0.1 |
| 22 | 8.9 ± 0.5 | 7.9 ± 0.4 |
| Cisplatin | 6.1 [41] | 12.6 ± 0.8 [42] |
1 The cell lines were treated with different concentrations of each compound for 72 h. IC50 values were determined by 3-(4,5-dimethylthiozol-2-yl)-3,5-dipheryl tetrazolium bromide (MTT) assay and are expressed as means ± SD (standard deviation) of three independent experiments. IC50 is the concentration that inhibits 50% of cellular growth. N.D.: not determined.
Table 2.
Antiproliferative activities of GA, its heterocyclic derivatives, and cisplatin against several cancer cell lines and the nontumoral BJ cell line.
Table 2.
Antiproliferative activities of GA, its heterocyclic derivatives, and cisplatin against several cancer cell lines and the nontumoral BJ cell line.
| Compound | Cell line (IC50, µM) 1 | |||||||
|---|---|---|---|---|---|---|---|---|
| MIAPaca2 | HeLa | A375 | MCF7 | HepG2 | SH-SY5Y | Jurkat | BJ | |
| 1 | 101.6 ± 1.6 | 107.2 ± 2.5 | 112.2 ± 2.6 | 97.8 ± 3.9 | 125.1 ± 9.1 | 109.7 ± 2.5 | 105.8 ± 5.0 | 165.0 ± 7.1 |
| 9 | 14.2 ± 0.9 | 12.4 ± 1.2 | 10.4 ± 1.0 | 6.4 ± 0.3 | 14.3 ± 0.3 | 6.0 ± 0.2 | 3.2 ± 0.1 | N.D. |
| 10 | 3.3 ± 0.2 | 2.2 ± 0.1 | 2.0 ± 0.2 | 3.0 ± 0.2 | 3.1 ± 0.1 | 1.7 ± 0.15 | 1.1 ± 0.06 | 6.9 ± 0.07 |
| 11 | 10.8 ± 0.2 | 10.9 ± 1.0 | 7.1 ± 0.3 | 5.6 ± 0.3 | 13.5 ± 0.4 | 5.6 ± 0.2 | 2.4 ± 0.1 | N.D. |
| 12 | 3.3 ± 0.2 | 2.6 ± 0.2 | 2.3 ± 0.2 | 3.2 ± 0.3 | 3.5 ± 0.2 | 2.2 ± 0.2 | 1.3 ± 0.12 | N.D. |
| 13 | 28.7 ± 2.6 | 28.9 ± 1.7 | 26.5 ± 0.9 | N.D. | N.D. | N.D. | N.D. | N.D. |
| 14 | 13.4 ± 0.5 | 11.8 ± 0.7 | 10.3 ± 1.0 | N.D. | N.D. | N.D. | N.D. | N.D. |
| 17 | 17.8 ± 1.1 | 22.2 ± 0.8 | 17.9 ± 0.2 | N.D. | N.D. | N.D. | N.D. | N.D. |
| 18 | 15.2 ± 0.5 | 17.9 ± 0.6 | 13.4 ± 1.0 | N.D. | N.D. | N.D. | N.D. | N.D. |
| 21 | 12.0 ± 1.0 | 10.6 ± 0.1 | 7.2 ± 0.5 | 6.0 ± 0.3 | 11.8 ± 0.4 | 3.7 ± 0.1 | 1.7 ± 0.10 | N.D. |
| 22 | 6.9 ± 0.3 | 5.4 ± 0.3 | 4.9 ± 0.1 | 5.2 ± 0.2 | 9.0 ± 0.1 | 3.2 ± 0.1 | 1.5 ± 0.12 | N.D. |
| Cisplatin | 5.0 ± 1.0 [46] | 2.3 ± 0.3 [43] | 3.1 ± 1.0 [42] | 19.1 ± 4.5 [43] | 2.9 [41] | 0.7 ± 0.1 [44] | 1.9 [45] | 10.1 ± 2.0 [43] |
1 The cell lines were treated with different concentrations of each compound for 72 h. IC50 values were determined by XTT assay in Jurkat and SH-SY5Y cells and by MTT assay in all the other cell lines. Results are expressed as means ± SD of three independent experiments. IC50 is the concentration that inhibits 50% of cellular growth. N.D.: not determined.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alho, D.P.S.; Salvador, J.A.R.; Cascante, M.; Marin, S. Synthesis and Antiproliferative Activity of Novel Heterocyclic Glycyrrhetinic Acid Derivatives. Molecules 2019, 24, 766. https://doi.org/10.3390/molecules24040766
AMA Style
Alho DPS, Salvador JAR, Cascante M, Marin S. Synthesis and Antiproliferative Activity of Novel Heterocyclic Glycyrrhetinic Acid Derivatives. Molecules. 2019; 24(4):766. https://doi.org/10.3390/molecules24040766
Chicago/Turabian StyleAlho, Daniela P. S., Jorge A. R. Salvador, Marta Cascante, and Silvia Marin. 2019. "Synthesis and Antiproliferative Activity of Novel Heterocyclic Glycyrrhetinic Acid Derivatives" Molecules 24, no. 4: 766. https://doi.org/10.3390/molecules24040766