
Design, Synthesis and Biological Evaluation of New Piperazin-4-yl-(acetyl-thiazolidine-2,4-dione) Norfloxacin Analogues as Antimicrobial Agents

 , , , ,
, , , ,  , , , ,
, , , ,
Abstract
:1. Introduction
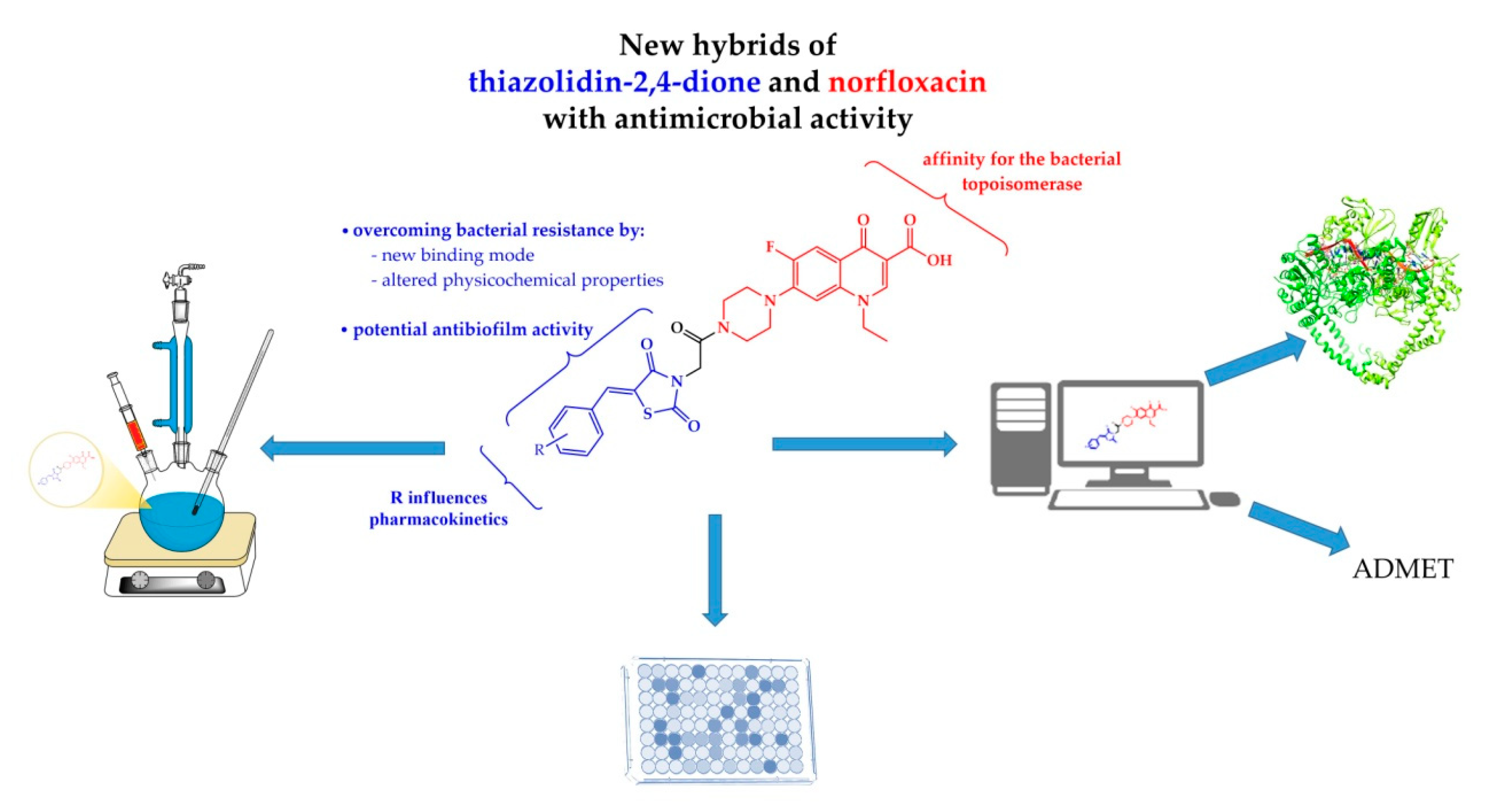
2. Results and Discussion
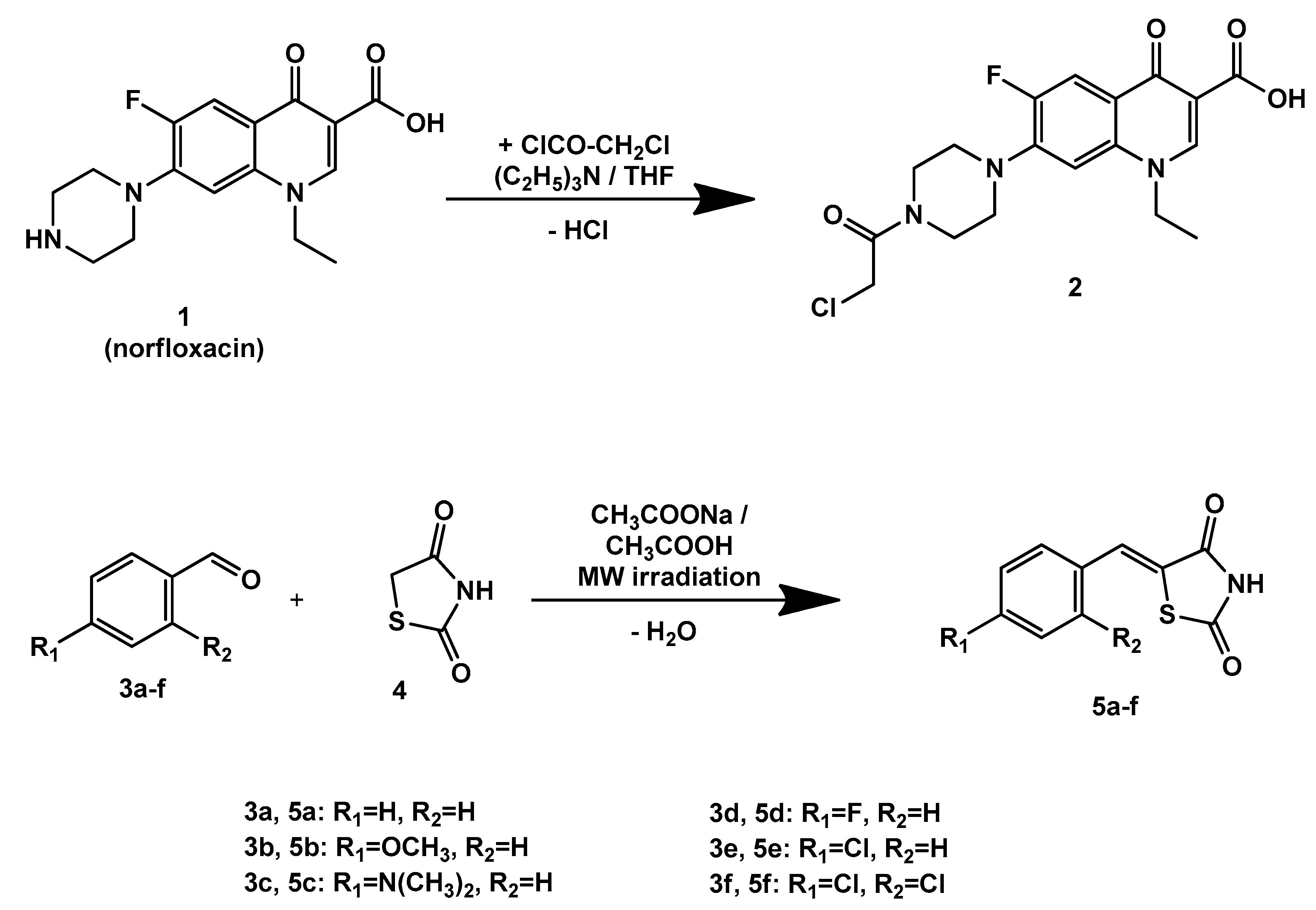
2.1. Chemistry
2.2. Biological Assays
2.2.1. Antimicrobial Activity—Initial In Vitro Qualitative Screening Study
2.2.2. Antimicrobial Activity—In Vitro Quantitative Assay
2.2.3. Anti-Biofilm Activity Assay
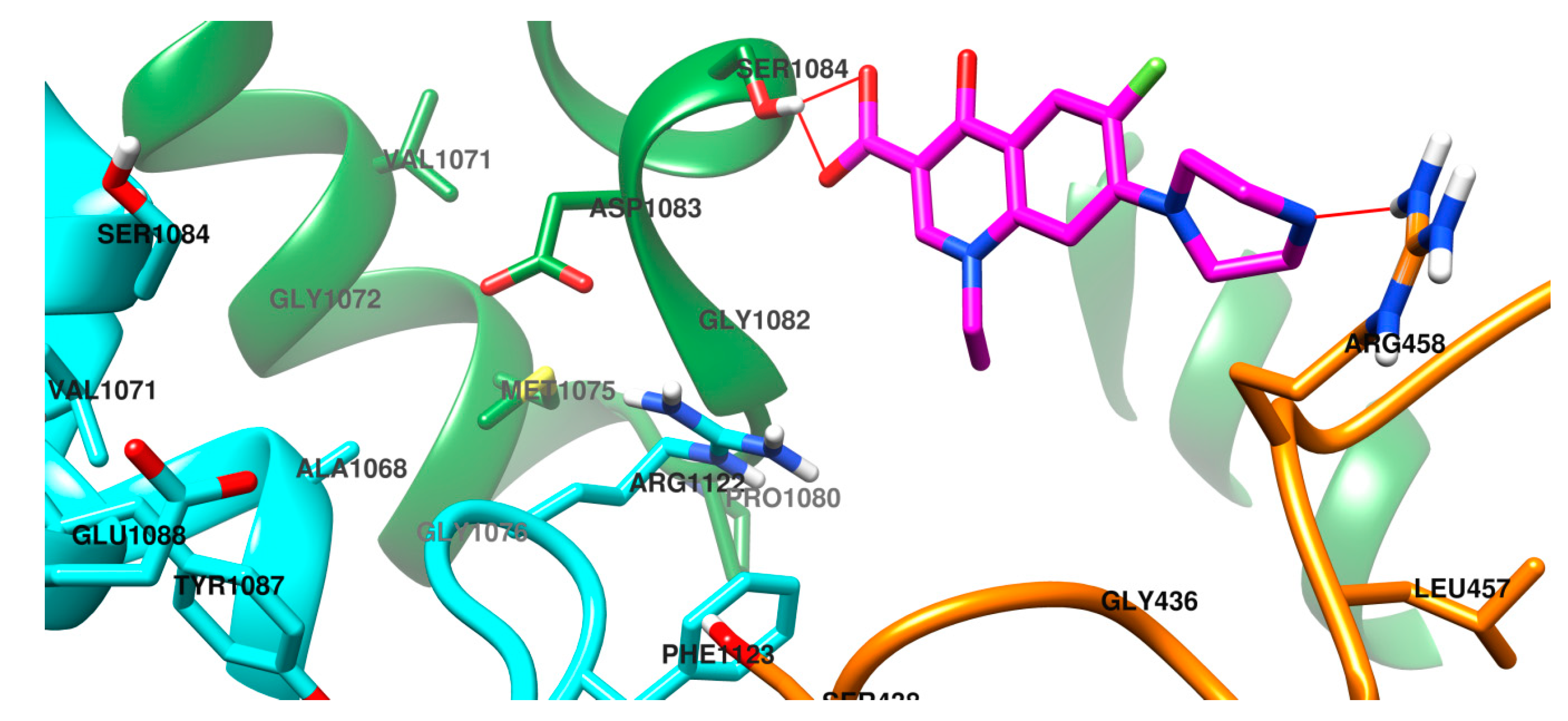
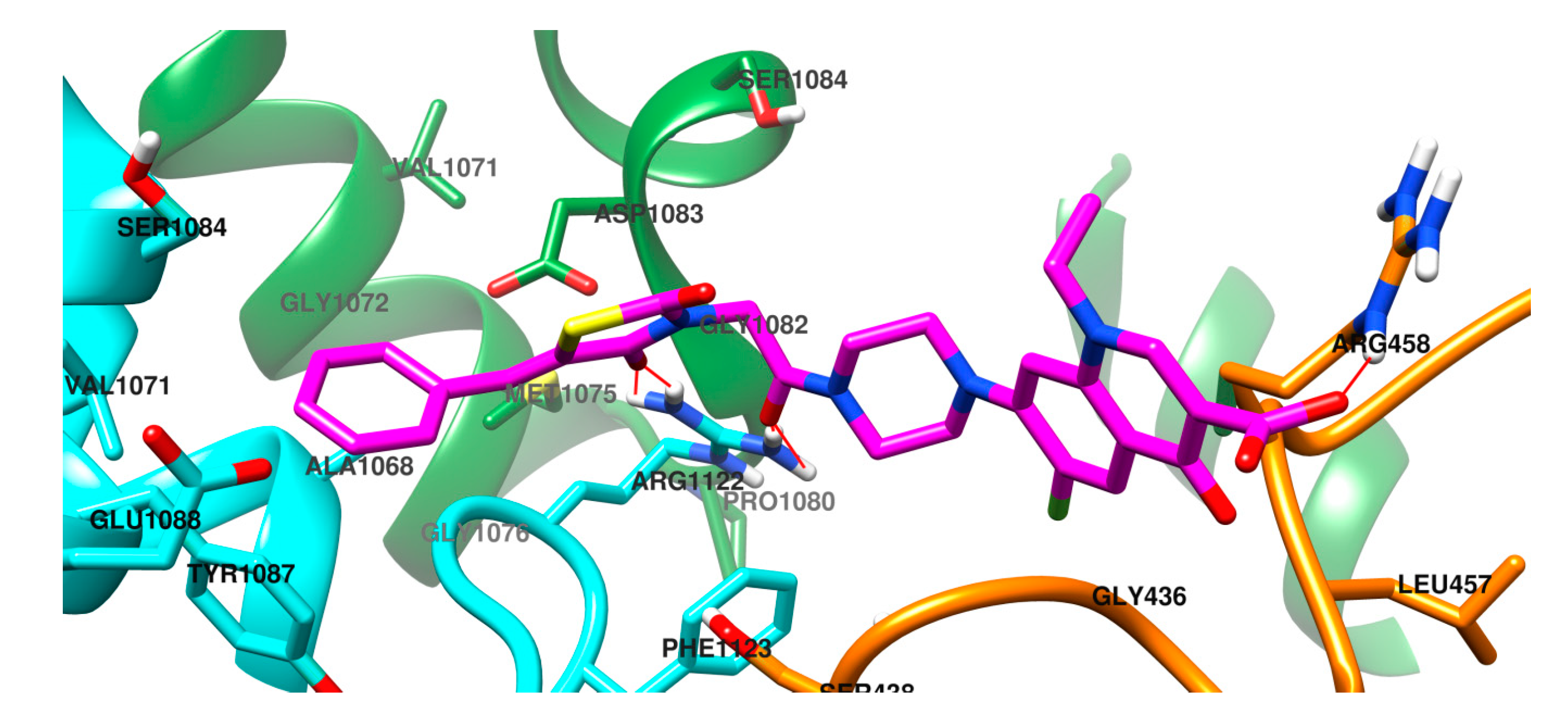
2.3. Molecular Docking Study
2.4. In Silico ADMET Study
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. General Procedure for the Synthesis of the Final Compounds 6, 7a–e
3.4. Biological Assays
3.4.1. Antimicrobial Activity—Initial In Vitro Qualitative Screening Study
3.4.2. Antimicrobial Activity—In Vitro Quantitative Assay
3.4.3. Anti-Biofilm Activity Assay
3.5. Molecular Docking Study
3.6. In silico ADMET study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Patrick, G. Agents that act on nucleic acid transcription and replication: Quinolones and fluoroquinolones. In An introduction to Medicinal Chemistry; Oxford University Press: Oxford, UK, 2017; pp. 476–478. [Google Scholar]
- Suaifan, G.A.R.Y.; Mohammed, A.A.M. Fluoroquinolones structural and medicinal developments (2013–2018): Where are we now? Bioorg. Med. Chem. 2019, 27, 3005–3060. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Malik, M.; Hong, Y.; Li, L.; Drlica, K. Quinolones. In Reference Module in Biomedical Sciences; Elsevier: Newark, NJ, USA, 2014; pp. 1–7. [Google Scholar]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- Mustaev, A.; Malik, M.; Zhao, X.; Kurepina, N.; Luan, G.; Oppegard, L.M.; Hiasa, H.; Marks, K.R.; Kerns, R.J.; Berger, J.M.; et al. Fluoroquinolone-Gyrase-DNA Complexes. J. Biol. Chem. 2014, 289, 12300–12312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Cuprys, A.; Pulicharla, R.; Brar, S.K.; Drogui, P.; Verma, M.; Surampalli, R.Y. Fluoroquinolones metal complexation and its environmental impacts. Coord. Chem. Rev. 2018, 376, 46–61. [Google Scholar] [CrossRef]
- Towle, T.R.; Kulkarni, C.A.; Oppegard, L.M.; Williams, B.P.; Picha, T.A.; Hiasa, H.; Kerns, R.J. Design, synthesis, and evaluation of novel N -1 fluoroquinolone derivatives: Probing for binding contact with the active site tyrosine of gyrase. Bioorg. Med. Chem. Lett. 2018, 28, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Lentz, S.R.C.; Chheda, P.R.; Oppegard, L.M.; Towle, T.R.; Kerns, R.J.; Hiasa, H. The C7-aminomethylpyrrolidine group rescues the activity of a thio-fluoroquinolone. Biochimie 2019, 160, 24–27. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Wang, L.-L.; Battini, N.; Bheemanaboina, R.R.Y.; Zhang, S.-L.; Zhou, C.-H. Design and synthesis of aminothiazolyl norfloxacin analogues as potential antimicrobial agents and their biological evaluation. Eur. J. Med. Chem. 2019, 167, 105–123. [Google Scholar] [CrossRef]
- Hu, Y.-Q.; Zhang, S.; Xu, Z.; Lv, Z.-S.; Liu, M.-L.; Feng, L.-S. 4-Quinolone hybrids and their antibacterial activities. Eur. J. Med. Chem. 2017, 141, 335–345. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Girgis, A.S.; Tiwari, A.D.; Honkanadavar, H.H.; Thomas, S.J.; Samir, A.; Kalmouch, A.; Alamry, K.A.; Khan, K.A.; Ibrahim, T.S.; et al. Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates. Eur. J. Med. Chem. 2018, 143, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-quinoline/quinolone hybrids as potential anti-bacterial agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, P.; Yang, H.; Miao, Q.; Ma, L.; Lu, G. Recent developments of quinolone-based derivatives and their activities against Escherichia coli. Eur. J. Med. Chem. 2018, 157, 1223–1248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xu, P.; Wan, C.; Wang, Z. Evening versus morning dosing regimen drug therapy for hypertension. Cochrane Database Syst. Rev. 2011, CD004184. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.-J.; Lv, Z.-S.; Gao, F.; Wang, Y.; Zhang, F.; Bai, L.; Deng, J.-L. Fluoroquinolone-isatin hybrids and their biological activities. Eur. J. Med. Chem. 2019, 162, 396–406. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Sączewski, J.; Konopacka, A.; Waleron, K.; Lejnowski, D.; Ciura, K.; Tomašič, T.; Skok, Ž.; Savijoki, K.; Morawska, M.; et al. Synthesis and biological evaluation of hybrid quinolone-based quaternary ammonium antibacterial agents. Eur. J. Med. Chem. 2019, 179, 576–590. [Google Scholar] [CrossRef]
- Scaiola, A.; Leibundgut, M.; Boehringer, D.; Caspers, P.; Bur, D.; Locher, H.H.; Rueedi, G.; Ritz, D. Structural basis of translation inhibition by cadazolid, a novel quinoxolidinone antibiotic. Sci. Rep. 2019, 9, 5634. [Google Scholar] [CrossRef]
- Rashid, M.-U.; Dalhoff, A.; Bäckström, T.; Björkhem-Bergman, L.; Panagiotidis, G.; Weintraub, A.; Nord, C.E. Ecological impact of MCB3837 on the normal human microbiota. Int. J. Antimicrob. Agents 2014, 44, 125–130. [Google Scholar] [CrossRef]
- Oniga, S.; Palage, M.; Araniciu, C.; Marc, G.; Oniga, O.; Vlase, L.; Prisăcari, V.; Valica, V.; Curlat, S.; Uncu, L. Design, synthesis, molecular docking, and antibacterial activity evaluation of some novel norfloxacin analogues. Farmacia 2018, 66, 1048–1058. [Google Scholar] [CrossRef]
- Marc, G.; Araniciu, C.; Oniga, S.; Vlase, L.; Pîrnău, A.; Duma, M.; Măruțescu, L.; Chifiriuc, M.; Oniga, O. New N-(oxazolylmethyl)-thiazolidinedione Active against Candida albicans Biofilm: Potential Als Proteins Inhibitors. Molecules 2018, 23, 2522. [Google Scholar] [CrossRef]
- Feldman, M.; Shenderovich, J.; Lavy, E.; Friedman, M.; Steinberg, D. A Sustained-Release Membrane of Thiazolidinedione-8: Effect on Formation of a Candida/Bacteria Mixed Biofilm on Hydroxyapatite in a Continuous Flow Model. Biomed Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, M.; Ginsburg, I.; Al-Quntar, A.; Steinberg, D. Thiazolidinedione-8 Alters Symbiotic Relationship in C. albicans-S. mutans Dual Species Biofilm. Front. Microbiol. 2016, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Naim, M.J.; Alam, M.J.; Ahmad, S.; Nawaz, F.; Shrivastava, N.; Sahu, M.; Alam, O. Therapeutic journey of 2,4-thiazolidinediones as a versatile scaffold: An insight into structure activity relationship. Eur. J. Med. Chem. 2017, 129, 218–250. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.C.; Patel, S.K.S.; Kang, Y.C.; Lee, J.-K. Quorum sensing inhibitors as antipathogens: biotechnological applications. Biotechnol. Adv. 2019, 37, 68–90. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Alam, A.; Rani, M.; Ehtesham, N.Z.; Hasnain, S.E. Biofilms: Survival and defense strategy for pathogens. Int. J. Med. Microbiol. 2017, 307, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Oniga, S.; Araniciu, C.; Palage, M.; Stoica, C.; Chifiriuc, M.C.; Marutescu, L. Synthesis and bioevaluation of the antimicrobial features of some new thiazolyl- Azoles. Rev. Chim. 2016, 67, 426–429. [Google Scholar]
- Rémy, B.; Mion, S.; Plener, L.; Elias, M.; Chabrière, E.; Daudé, D. Interference in Bacterial Quorum Sensing: A Biopharmaceutical Perspective. Front. Pharmacol. 2018, 9, 203. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Pîrnău, A.; Vlase, L.; Vodnar, D.C.; Duma, M.; Tiperciuc, B.; Oniga, O. 3,5-Disubstituted Thiazolidine-2,4-Diones: Design, Microwave-Assisted Synthesis, Antifungal Activity, and ADMET Screening. SLAS Discov. Adv. Life Sci. R&D 2018, 23, 807–814. [Google Scholar] [Green Version]
- Chadha, N.; Bahia, M.S.; Kaur, M.; Silakari, O. Thiazolidine-2,4-dione derivatives: programmed chemical weapons for key protein targets of various pathological conditions. Bioorg. Med. Chem. 2015, 23, 2953–2974. [Google Scholar] [CrossRef]
- Costa-Orlandi, C.; Sardi, J.; Pitangui, N.; de Oliveira, H.; Scorzoni, L.; Galeane, M.; Medina-Alarcón, K.; Melo, W.; Marcelino, M.; Braz, J.; et al. Fungal Biofilms and Polymicrobial Diseases. J. Fungi 2017, 3, 22. [Google Scholar] [CrossRef]
- Dufrêne, Y.F. Sticky microbes: forces in microbial cell adhesion. Trends Microbiol. 2015, 23, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Oniga, S.; Araniciu, C.; Palage, M.; Popa, M.; Chifiriuc, M.-C.; Marc, G.; Pirnau, A.; Stoica, C.; Lagoudis, I.; Dragoumis, T.; et al. New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules 2017, 22, 1827. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.; Zanfirescu, A.; Olaru, O.T.; Nicorescu, I.M.; Nitulescu, G.M.; Margina, D. Structural analysis of sortase A inhibitors. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Oniga, O.; Marc, G.; Palage, M.D.; Marutescu, L.; Chifiriuc, M.C.; Stoica, C.I.; Ionut, I.; Oniga, S.D. Anti-biofilm activity evaluation and molecular docking study of some 2(3-pyridyl)-thiazolyl-1,3,4-oxadiazolines. Farmacia 2018, 66, 627–634. [Google Scholar] [CrossRef]
- McCarthy, H.; Rudkin, J.K.; Black, N.S.; Gallagher, L.; O’Neill, E.; O’Gara, J.P. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2015, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef]
- Fu, G.; Wu, J.; Liu, W.; Zhu, D.; Hu, Y.; Deng, J.; Zhang, X.-E.; Bi, L.; Wang, D.-C. Crystal structure of DNA gyrase B′ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 2009, 37, 5908–5916. [Google Scholar] [CrossRef]
- Jakopin, Ž.; Ilaš, J.; Barančoková, M.; Brvar, M.; Tammela, P.; Sollner Dolenc, M.; Tomašič, T.; Kikelj, D. Discovery of substituted oxadiazoles as a novel scaffold for DNA gyrase inhibitors. Eur. J. Med. Chem. 2017, 130, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Dahlgren, D.; Lennernäs, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef]
- Brillault, J.; De Castro, W.V.; Harnois, T.; Kitzis, A.; Olivier, J.-C.; Couet, W. P-Glycoprotein-Mediated Transport of Moxifloxacin in a Calu-3 Lung Epithelial Cell Model. Antimicrob. Agents Chemother. 2009, 53, 1457–1462. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, N.; Fromm, M.F. The role of the transporter P-glycoprotein for disposition and effects of centrally acting drugs and for the pathogenesis of CNS diseases. Eur. Arch. Psychiatry Clin. Neurosci. 2006, 256, 281–286. [Google Scholar] [CrossRef] [PubMed]
- De Lange, E.C.M.; Marchand, S.; Van den Berg, D.J.; Van der Sandt, I.C.J.; Ve Boer, A.G.; Delon, A.; Bouquet, S.; Couet, W. In vitro and in vivo investigations on fluoroquinolones; Effects of the P-glycoprotein efflux transporter on brain distribution of sparfloxacin. Eur. J. Pharm. Sci. 2000, 12, 85–93. [Google Scholar] [CrossRef]
- Mentese, M.Y.; Bayrak, H.; Uygun, Y.; Mermer, A.; Ulker, S.; Karaoglu, S.A.; Demirbas, N. Microwave assisted synthesis of some hybrid molecules derived from norfloxacin and investigation of their biological activities. Eur. J. Med. Chem. 2013, 67, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Qandil, A.; Al-Zoubi, L.; Al-Bakri, A.; Amawi, H.; Al-Balas, Q.; Alkatheri, A.; Albekairy, A. Synthesis, Antibacterial Evaluation and QSAR of α-Substituted-N4-Acetamides of Ciprofloxacin and Norfloxacin. Antibiotics 2014, 3, 244–269. [Google Scholar] [CrossRef] [PubMed]
- Marc, G.; Oniga, S.D.; Pirnau, A.; Duma, M.; Vlase, L.; Oniga, O. Rational Synthesis of Some New para-Aminobenzoic Acid Hybrids with Thiazolidin-2,4-diones with Antimicrobial Properties ADMET and molecular docking evaluation. Rev. Chim. 2019, 70, 769–775. [Google Scholar]
- Yang, D.-H.; Chen, Z.-C.; Chen, S.-Y.; Zheng, Q.-G. A convenient synthesis of 5-benzylidenethiazolidine-2,4-diones under microwave irradiation without solvent. J. Chem. Res. 2003, 330–331. [Google Scholar] [CrossRef]
- Shelke, K.F.; Sapkal, S.B.; Kakade, G.K.; Sadaphal, S.A.; Shingate, B.B.; Shingare, M.S. Alum catalyzed simple and efficient synthesis of 5-arylidene-2,4-thiazolidinedione in aqueous media. Green Chem. Lett. Rev. 2010, 3, 17–21. [Google Scholar] [CrossRef]
- Matuschek, E.; Brown, D.F.J.; Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect. 2014, 20, O255–O266. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Activity | Antifungal Activity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gram-Positive | Gram-Negative | ||||||||
| Compound | S. aureus | L. monocytogenes | B. cereus | E. faecalis | E. coli | E. coli | S. enteritidis | C. albicans | C. parapsilosis |
| ATCC 6538P | ATCC 13932 | ATCC 11778 | ATCC 29212 | ATCC 10536 | ATCC 25922 | ATCC 13076 | ATCC 90028 | ATCC 22019 | |
| 1-Nor | 37 | 28 | 28 | 27 | 31 | 26 | 38 | - | - |
| 2 | 23 | 17 | 19 | 17 | 26 | 23 | 29 | 0 | 0 |
| 6 | 22 | 17 | 16 | 16 | 23 | 18 | 19 | 8 | 0 |
| 7a | 20 | 0 | 17 | 16 | 29 | 26 | 32 | 9 | 0 |
| 7b | 15 | 0 | 14 | 13 | 24 | 23 | 25 | 19 | 9 |
| 7c | 11 | 0 | 10 | 0 | 23 | 21 | 19 | 0 | 0 |
| 7d | 10 | 0 | 8 | 0 | 15 | 15 | 16 | 8 | 9 |
| 7e | 16 | 0 | 11 | 0 | 17 | 14 | 0 | 11 | 0 |
| 7f | 14 | 0 | 11 | 12 | 22 | 23 | 22 | 19 | 10 |
| Ketoconazole | - | - | - | - | - | - | - | 25 | 20 |
| DMSO | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Compound | Gram-Negative Bacteria | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E. coli ATCC 25922 | E. coli ATCC 10536 | S. typhimurium ATCC 14028 | S. enteritidis ATCC 13076 | P. aeruginosa ATCC 27853 | ||||||
| MIC | MIC50 | MIC | MIC50 | MIC | MIC50 | MIC | MIC50 | MIC | MIC50 | |
| 1-Nor | 0.0625 | 0.0312 | 0.125 | 0.0625 | 0.125 | 0.0625 | 0.0625 | 0.0312 | 1 | 0.5 |
| 2 | 0.5 | 0.125 | 1 | 0.25 | 2 | 1 | 1 | 0.5 | 2 | 1 |
| 6 | 2 | 1 | 8 | 2 | 32 | 8 | 8 | 4 | 16 | 8 |
| 7a | 0.5 | 0.125 | 0.5 | 0.25 | 2 | 1 | 1 | 0.5 | 2 | 1 |
| 7b | 0.25 | 0.125 | 4 | 1 | 2 | 0.5 | 2 | 0.5 | 2 | 0.5 |
| 7c | 2 | 0.5 | 4 | 0.5 | 8 | 4 | 4 | 2 | 16 | 8 |
| 7d | 1 | 0.25 | 4 | 1 | 8 | 2 | 8 | 4 | 128 | 64 |
| 7e | 2 | 0.5 | 2 | 0.5 | 16 | 4 | 8 | 2 | 128 | 32 |
| 7f | 1 | 0.5 | 2 | 0.5 | 2 | 1 | 2 | 1 | 8 | 2 |
| Compound | Antimicrobial Activity | Antifungal Activity | ||||
|---|---|---|---|---|---|---|
| Gram-Positive | Gram-Negative | |||||
| S. aureus | E. faecium | E. coli | P. aeruginosa | C. albicans | C. parapsilosis | |
| ATCC 25923 | DSM 13950 | ATCC 25922 | ATCC 27853 | ATCC 10231 | ATCC 22019 | |
| 1-Nor | 4.9 | 156.2 | 156.2 | 4.9 | 312.5 | 2500 |
| 2 | 4.9 | 78.1 | 78.1 | 78.1 | 312.5 | 312.5 |
| 6 | 4.9 | 9.8 | 39.0 | 312.5 | 312.5 | 2500.0 |
| 7a | 19.5 | 625.0 | 156.2 | 625.0 | 156.2 | 156.2 |
| 7b | 19.5 | 78.1 | 156.2 | 78.1 | 312.5 | 78.1 |
| 7c | 39.0 | 156.2 | 156.2 | 78.1 | 312.5 | 312.5 |
| 7d | 4.9 | 156.2 | 39.0 | 625.0 | 312.5 | 312.5 |
| 7e | 9.8 | 78.1 | 78.1 | 312.5 | 312.5 | 312.5 |
| 7f | 4.9 | 625.0 | 156.2 | 78.1 | 312.5 | 312.5 |
| Berberine | 78.1 | 156.2 | 625.0 | 625.0 | 312.5 | 312.5 |
| Compound | Binding Affinity (kcal/mol) | Inhibition Constant (µM) | Cluster with the Best Conformation | Clusters | ||
|---|---|---|---|---|---|---|
| Average Binding Affinity (kcal/mol) | Number of Conformations | With Multiple Conformations | Total | |||
| 1-Nor | −6.35 | 22.15 | −6.20 | 6 | 7 | 7 |
| 2 | −6.16 | 30.53 | −5.76 | 18 | 4 | 10 |
| 6 | −6.66 | 13.13 | −5.91 | 13 | 7 | 17 |
| 7a | −7.82 | 1.85 | −7.59 | 18 | 6 | 21 |
| 7b | −7.78 | 1.98 | −7.41 | 6 | 6 | 24 |
| 7c | −7.71 | 2.23 | −7.42 | 6 | 5 | 27 |
| 7d | −7.89 | 1.65 | −6.98 | 13 | 6 | 29 |
| 7e | −7.63 | 2.55 | −7.54 | 15 | 6 | 24 |
| 7f | −8.16 | 1.04 | −7.91 | 9 | 8 | 25 |
| Compound | MW | RB | HBA | HBD | Surface | LogP | Water Solubility | ||
|---|---|---|---|---|---|---|---|---|---|
| Total | Polar | mg/mL | Class | ||||||
| 1-Nor | 319 | 3 | 5 | 2 | 131.68 | 74.57 | 1.04 | 16.2 | Very |
| 2 | 395 | 5 | 5 | 1 | 159.09 | 82.85 | 1.28 | 0.168 | Soluble |
| 6 | 476 | 6 | 7 | 1 | 191.08 | 145.53 | 0.05 | 0.191 | Soluble |
| 7a | 564 | 7 | 7 | 1 | 231.81 | 145.53 | 1.21 | 0.003 | Moderately |
| 7b | 594 | 8 | 8 | 1 | 243.29 | 154.76 | 0.91 | 0.002 | Moderately |
| 7c | 607 | 8 | 7 | 1 | 250.30 | 148.77 | 1.11 | 0.002 | Moderately |
| 7d | 582 | 7 | 8 | 1 | 235.98 | 145.53 | 1.58 | 0.002 | Moderately |
| 7e | 599 | 7 | 7 | 1 | 242.12 | 145.53 | 1.68 | 0.001 | Moderately |
| 7f | 633 | 7 | 7 | 1 | 252.42 | 145.53 | 2.14 | <0.001 | Poorly |
| Compound | Absorption | BBBP | PgpS | CYP Inhibition | |||||
|---|---|---|---|---|---|---|---|---|---|
| Caco2 Permeability | Intestinal Absorption | Class | 1A2 | 2C19 | 2C9 | 2D6 | |||
| 1-Nor | 1.254 | 83.73 | High | No | Yes | No | No | No | No |
| 2 | 1.270 | 96.52 | High | No | No | No | No | No | No |
| 6 | 0.984 | 52.33 | Low | No | Yes | No | No | Yes | No |
| 7a | 0.823 | 61.39 | Low | No | No | No | No | Yes | No |
| 7b | 0.747 | 62.85 | Low | No | No | No | No | Yes | No |
| 7c | 0.841 | 58.97 | Low | No | No | No | No | Yes | No |
| 7d | 0.774 | 64.77 | Low | No | No | No | No | Yes | No |
| 7e | 0.727 | 62.56 | Low | No | No | No | No | Yes | No |
| 7f | 0.722 | 63.74 | Low | No | No | No | No | Yes | No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marc, G.; Araniciu, C.; Oniga, S.D.; Vlase, L.; Pîrnău, A.; Nadăș, G.C.; Novac, C.Ș.; Matei, I.A.; Chifiriuc, M.C.; Măruțescu, L.; et al. Design, Synthesis and Biological Evaluation of New Piperazin-4-yl-(acetyl-thiazolidine-2,4-dione) Norfloxacin Analogues as Antimicrobial Agents. Molecules 2019, 24, 3959. https://doi.org/10.3390/molecules24213959
Marc G, Araniciu C, Oniga SD, Vlase L, Pîrnău A, Nadăș GC, Novac CȘ, Matei IA, Chifiriuc MC, Măruțescu L, et al. Design, Synthesis and Biological Evaluation of New Piperazin-4-yl-(acetyl-thiazolidine-2,4-dione) Norfloxacin Analogues as Antimicrobial Agents. Molecules. 2019; 24(21):3959. https://doi.org/10.3390/molecules24213959
Chicago/Turabian StyleMarc, Gabriel, Cătălin Araniciu, Smaranda Dafina Oniga, Laurian Vlase, Adrian Pîrnău, George Cosmin Nadăș, Cristiana Ștefania Novac, Ioana Adriana Matei, Mariana Carmen Chifiriuc, Luminița Măruțescu, and et al. 2019. "Design, Synthesis and Biological Evaluation of New Piperazin-4-yl-(acetyl-thiazolidine-2,4-dione) Norfloxacin Analogues as Antimicrobial Agents" Molecules 24, no. 21: 3959. https://doi.org/10.3390/molecules24213959